CRISPR for functional genomics | Quizlet
1/19
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
20 Terms
why do some people get ill and others not?
- has to do with genetic and environmental risk factors
- two humans are at least 99.9% genetically identical
- remaining 0.1% are genetic variants that influence risk of disease
- also has to do with differences in environmental exposures
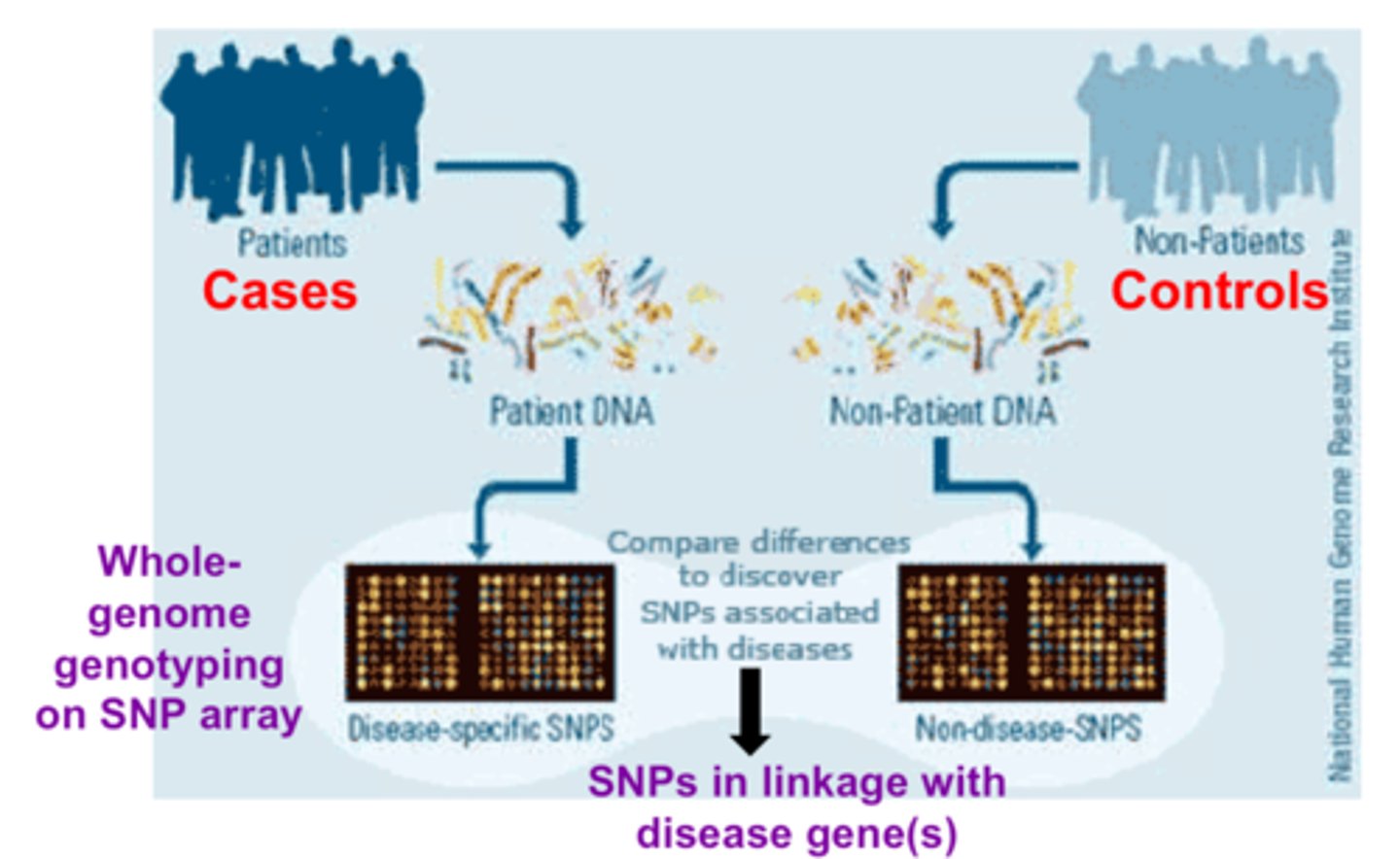
GWAS (Genome-wide association studies)
study in which thousands or millions of variable sites tested for association with human diseases
- cases are compared to controls to see what genetic variants are associated with disease
what is the main issue with GWAS studies?
that the risk factors are IDed but it remains unknown why they are associated
Quantitative trait locus analysis (QTL)
-used to identify/locate possible genes involved in polygenic (quantitative) traits
-looking for an association of a marker gene on a particular chromosome with variation in a specific trait
- allows to look at layers underneath the genetic variant (e.g., methylation/ gene expression/ proteins and metabolites/ the microbiome) to determine what the exact disease phenotype is
- can also be used to find out why non-coding parts of the of the DNA are associated with disease
genetic variants often act cell type- and context-specific
- by taking a biopsy, cells can be taken and isolated. various things can be investigated using single-cell technology
- look at e.g., proteins/ histones
eQTL
An expression QTL, i.e. a genetic variant that quantitatively affects the expression of a gene
- reveals expression of gene variants
- now: all cells can be analysed at once and see whether they are active upon signalling
- requires international consortia as they are expensive to do + data is sparse => meta-analysis allows for an increased sample size + better detection of the effects
downside of QTLs
analysis cannot distinguish between causal genetic variant and other SNPs within the same linkage disequilibrium (LD) block
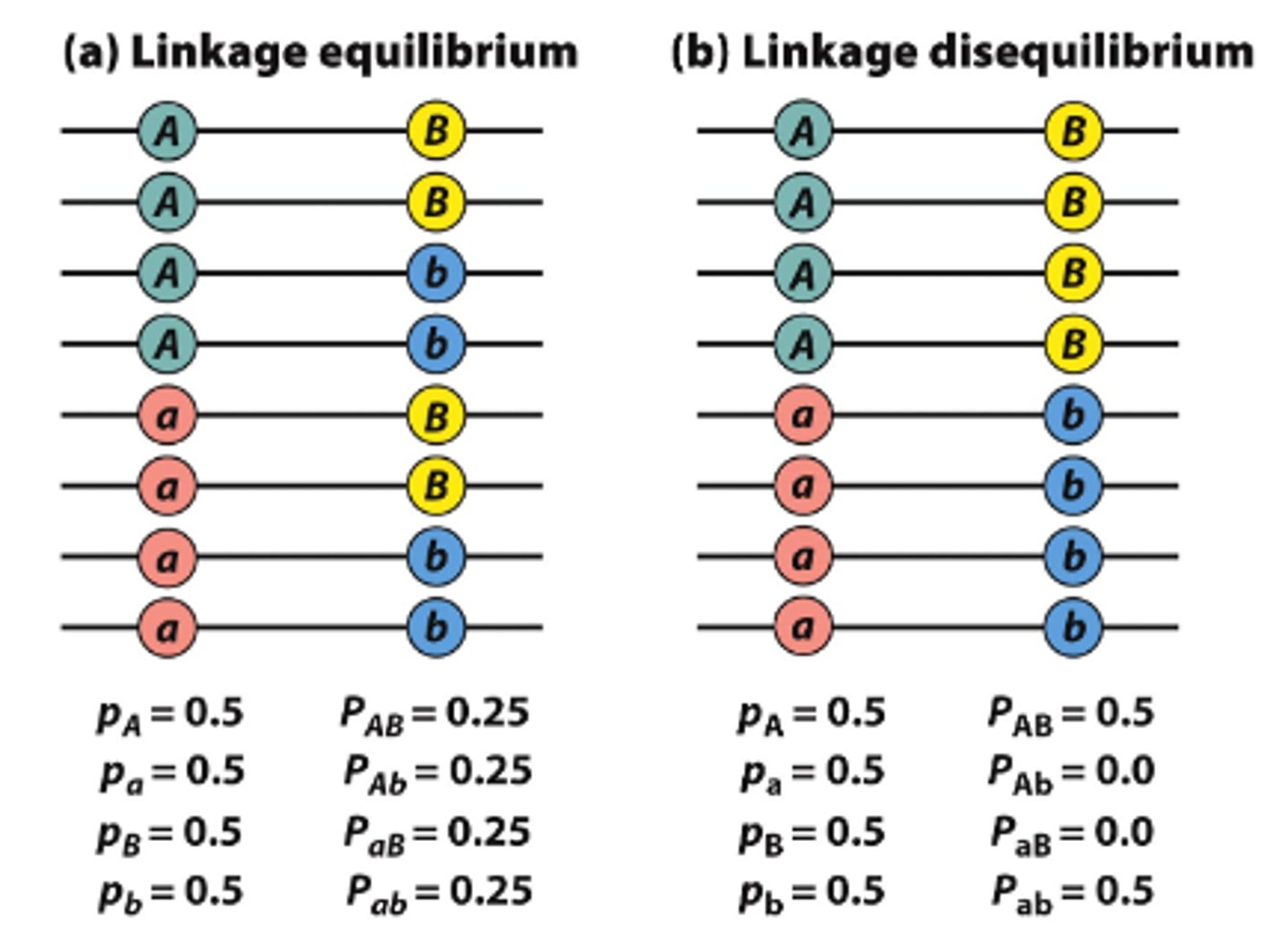
Linkage disequilibrium
When a pair of alleles from two loci are inherited together in the same gamete more/less often than random chance would expect
- are represented within blocks and slightly different based on genetic background
massively parallel reporter assay (MPRA)
- shows the regulatory potential of DNA
- measures the activity of thousands of DNA elements in a single experiment
- tells whether there is regulatory potential, not what it affects! CRISPR can be used for that
massively parallel reporter assay (MPRA) procedure
1. create a DNA element reporter library (plasmid + DNA element + barcode to ID plasmid)
2. the cells are transfected into the cells and genomic integration is checked
3. the DNA element activity is measured using high-throughput sequencing
=> if a DNA element is inserted twice, the observed RNA count is 4
base editors
Create single base pair substitutions
- PAM-less variants exist; only 3 types of point mutations can be made, which are 42% of all disease-associated genetic variants
prime editors
edit larger pieces of DNA using pegRNA (sgRNA + RT template + PBS)/ Reverse transcriptase/ nCas9
- can make most of the mutations except of copy number loss/ gain
- 89% of all disease-associated genetic variants can be made
CGBE
1. dCas9 + deaminase deaminates C and turns it into a U
2. MMR turns G into an A
3. DNA replication and repair makes it C:G
=> capable of doing a transversion!!
Adenosine base editor (ABE)
1. dCas9 + deaminase deaminates A into I
2. I is read as G by DNA polymerase
3. MMR fixes the mistake by turning G into C
4. DNA is replicated + further repair turns A:T into G:C
cytosine base editor
C:G => T:A
1. dCas9 + deaminase deaminates C and turns it into U
2. MMR fixes the mismatch and turns G into A
3. further DNA replication and repair turns C:G into T:A
CRISPR prime editing
1. nCas9 + RT bind to the target sequence using the pegRNA (sgRNA + RT template + PBS)
2. PBS initiates RT, RT synthesises new cDNA strand (complementary to NTS) with the edit
3. either 3' flap (with edit) or 5' flap (w/o edit)
4. use extra sgRNA to make another nick to ensure that the 5' flap gets cleaved off and that the edit is incorporated into the DNA
which CRISPR tool to select for functional genomics screens?
- base editors are preferred as high scalability and efficiency is required due to a large sample size (1000s of genetic variants have to be tested)
- either detect off-targets and remove them (most off-targets are local bystander edits) or reduce the occurrence through imperfect sgRNAs)
imperfect sgRNAs
- used to reduce off-targets
- sgRNA can be designed to mismatch the most likely off-target site => the first edit s the preferred edit, afterwards, the sgRNA cannot bind efficiently anymore as there are 2 mismatches now
high-throughput CRISPR screens
1. arrayed: 96well plate, every well is transfected with a different guide. creates a lot of opportunities
2. pooled: cells are transfected at once, genetically-encoded sgRNAs can be read out by sequencing (lentiviral delivery)
- cannot look at their expression levels anymore
3. multiplexed sgRNAs: multiple predefine sgRNAs as one single pertubation (multiple guides in 1 plasmid)
pooled CRISPR screen with high-dimensional read-out
- cell survival is coupled with a RNA sequence
- cells with only one sgRNA are selected!
- gel beads are creating mini testing tubes
- a bead will capture all of the RNA from 1 cel
- the Polydt will capture mRNA
- allows for determination which cell had which sgRNA