A2 organic chemistry
1/90
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
91 Terms
Why are arenes highly stable, and why do they undergo electrophilic substitution instead of addition reactions?
Stability of Arenes:
Arenes, such as benzene, have delocalized π-electrons spread over the ring, giving extra stability.
The aromatic system remains intact during reactions, preventing disruption of this stabilisation.
Electrophilic Substitution:
Electrophiles attack the benzene ring, replacing hydrogen atoms without disturbing delocalisation.
Unlike alkenes, arenes do not undergo addition reactions because these would break aromatic stability.
How do arenes undergo halogenation, and how does the presence of alkyl groups affect substitution positions?
Halogenation Reaction:
Arenes react with Cl₂ or Br₂ in the presence of AlCl₃ or AlBr₃ catalysts to form halogenoarenes (aryl halides).
The catalyst polarises the halogen, making it a stronger electrophile that substitutes hydrogen on benzene.
Effect of Alkyl Groups (Methylbenzene Example):
Electron-donating alkyl groups (like -CH₃ in methylbenzene) activate the 2 and 4 positions, leading to directed substitution.
Excess halogen results in multiple substitutions at these positions.
How does the nitration of benzene and methylbenzene occur, and what is the effect of alkyl groups?
Nitration Reaction:
Benzene reacts with a mixture of concentrated HNO₃ and H₂SO₄ at 25–60°C.
Sulfuric acid acts as a catalyst, generating the NO₂⁺ electrophile, which substitutes a hydrogen atom on benzene.
Effect of Alkyl Groups (Methylbenzene Example):
Alkyl groups activate the 2 and 4 positions, leading to directed nitration at these locations.
What are Friedel–Crafts reactions, and how do alkylation and acylation modify benzene’s reactivity?
Friedel–Crafts Reactions:
Used to introduce alkyl (-R) or acyl (-CO-R) groups into benzene via electrophilic substitution.
Alkylation:
Reagents: CH₃Cl + AlCl₃ catalyst + heat.
Mechanism: Alkyl cation attacks benzene, replacing hydrogen with an alkyl group.
Example: Benzene reacts with chloropropane (CH₃CH₂CH₂Cl) to form propylbenzene.
Acylation:
Reagents: CH₃COCl + AlCl₃ catalyst + heat.
Mechanism: Acyl cation attacks benzene, replacing hydrogen with an acyl group.
Example: Methylbenzene reacts with propanoyl chloride to form an acyl benzene, with substitution at the 4-position due to the methyl (-CH₃) group.
How are alkyl side chains in arenes oxidised, and what are the final products?
Oxidation Reaction:
Hot alkaline KMnO₄ followed by dilute acid oxidises alkyl side-chains to carboxylic acids.
Benzene ring remains intact, and the side-chain is converted to a benzoic acid.
Example:
Ethylbenzene → Benzoic acid (after oxidation with KMnO₄ and H₂SO₄).
How does the hydrogenation of benzene and methylbenzene occur, and what effect does it have on aromaticity?
Hydrogenation Reaction:
Reagents: H₂ + Pt/Ni catalyst + heat.
Mechanism: Benzene loses its aromatic stability, converting to cyclohexane.
Effect on Methylbenzene:
Methylbenzene undergoes hydrogenation to form cycloethylbenzene.
Loss of aromaticity, but alkyl groups remain attached.
What are the key reactions of arenes, their conditions, and products?

What are the key steps in the electrophilic substitution mechanism of arenes, as exemplified by the formation of nitrobenzene and bromobenzene?
Step 1: Generation of the Electrophile
Arenes have a stable delocalised π-system, meaning electrophiles must be highly reactive.
Electrophiles used:
Halogenation: X⁺ (e.g., Br⁺)
Nitration: NO₂⁺
How Electrophiles Are Formed:
Halogenation: Br₂ reacts with AlBr₃ → Br⁺ electrophile.
Nitration: Conc. HNO₃ + Conc. H₂SO₄ → NO₂⁺ electrophile.
Step 2: Electrophilic Attack
Benzene donates a pair of electrons to form a covalent bond with the electrophile.
This disrupts aromaticity, leaving only four π-electrons and a positively charged intermediate.
Step 3: Restoring Aromaticity
Heterolytic cleavage of the C–H bond occurs.
Bonding electrons re-enter the benzene π-system, restoring aromaticity.
Example Reactions:
Bromination: Benzene + Br₂ + AlBr₃ → Bromobenzene.
Nitration: Benzene + HNO₃ + H₂SO₄ (reflux, 25–60°C) → Nitrobenzene.
How does delocalisation (aromatic stabilisation) explain why arenes undergo substitution instead of addition reactions?
Aromatic Stability:
Benzene has a delocalised π-system with extra stability.
Addition reactions break this delocalisation, leading to energy loss.
Electrophilic Substitution vs. Addition:
Substitution: Retains aromaticity (electrons restored via heterolytic cleavage).
Addition: Destroys aromaticity (π-electron system disrupted).
Example: Hydrogenation of Benzene
H₂ + Pt/Ni catalyst + heat → Cyclohexane (loses aromaticity, less stable).
How do reaction conditions determine whether halogenation occurs in the aromatic ring or the side-chain of arenes?
Halogenation in the Aromatic Ring:
Occurs when a halogen (Cl₂ or Br₂) is used with a halogen carrier catalyst (AlCl₃ or AlBr₃).
Involves electrophilic substitution, generating a halogen electrophile (e.g., Br⁺).
Example: Bromination of methylbenzene → Bromomethylbenzene.
Halogenation in the Side-Chain:
Occurs when the halogen is passed into boiling alkylarene in the presence of UV light.
Involves free-radical substitution, forming halogenated side-chains.
Example: Methylbenzene + Cl₂ (UV light) → Chloromethylbenzene.
Effect of Excess Halogen:
In the side-chain: Every hydrogen is replaced with a halogen atom.
In the ring: Multiple substitutions occur at 2 and 4 positions (if an activating group is present).
How do different substituents affect the position of electrophilic substitution in arenes?
Substituents Influence Reactivity:
Electron-donating groups (-NH₂, -OH, -R): Increase electron density, making the ring more reactive.
Electron-withdrawing groups (-NO₂, -COOH, -COR): Reduce electron density, making the ring less reactive.

How are halogenoarenes formed, and how does methylbenzene direct halogenation?
Formation of Halogenoarenes (Electrophilic Substitution Reaction)
Reactants: Arene (benzene or methylbenzene) + Cl₂ or Br₂ + anhydrous AlCl₃ or AlBr₃ catalyst.
Mechanism:
Halogen carrier catalyst (AlCl₃ or AlBr₃) generates the Cl⁺ or Br⁺ electrophile.
Electrophile attacks electron-rich benzene ring, disrupting delocalised π system.
A hydrogen atom is removed, restoring aromatic stability and forming halogenoarene.
Examples:
Benzene + Cl₂ (AlCl₃ catalyst) → Chlorobenzene.
Benzene + Br₂ (AlBr₃ catalyst) → Bromobenzene.
Halogenation of Methylbenzene (2,4-Directing Effect)
Methyl group (-CH₃) is electron-donating, making the ring more reactive.
Directs substitution at the 2 and 4 positions, forming 2-chloromethylbenzene & 4-chloromethylbenzene.
Excess Cl₂ → substitution at the 6 position also occurs.
Why are halogenoarenes less reactive than halogenoalkanes, and how does the carbon-halogen bond affect substitution?
Reactivity of Halogenoalkanes
Example: Chloroethane readily undergoes nucleophilic substitution.
Mechanism:
Nucleophile (OH⁻) attacks slightly positive carbon.
Forms a covalent bond, replacing the halogen.
Reason: C-Cl bond in halogenoalkanes is weaker, making it easier to break.
Reactivity of Halogenoarenes
Example: Chlorobenzene is highly unreactive under normal conditions.
Only reacts at extreme conditions (200°C & 200 atm pressure) in nucleophilic substitution.
Reason for Low Reactivity:
Lone pairs on Cl interact with benzene π-system, forming partial double-bond character.
This strengthens the C-Cl bond, making it hard to break.
Delocalisation over benzene adds extra stability, reducing reactivity.
Why do halogenoarenes have stronger carbon-halogen bonds compared to halogenoalkanes, making them less reactive?
Strength of the Carbon-Halogen Bond in Halogenoarenes
Lone pairs on halogen interact with benzene’s π-system, creating partial double-bond character.
This strengthens the C–X bond, making nucleophilic substitution more difficult.
Aryl halides (e.g., chlorobenzene) are unreactive under normal conditions due to this extra stability.

What are the key factors influencing halogenoarene formation and reactivity?
Formation of Halogenoarenes (Electrophilic Substitution):
Arene + Cl₂ or Br₂ + AlCl₃ or AlBr₃ catalyst → Halogenoarene.
Example: Benzene + Cl₂ (AlCl₃ catalyst) → Chlorobenzene.
Methylbenzene directs substitution to 2 and 4 positions, forming 2-chloromethylbenzene & 4-chloromethylbenzene.
Reactivity Differences Between Halogenoalkanes & Halogenoarenes:
Halogenoalkanes readily undergo nucleophilic substitution (e.g., chloroethane reacts with OH⁻).
Halogenoarenes have strong C–X bonds due to delocalisation, making substitution difficult.
Chlorobenzene requires extreme conditions (200°C, 200 atm) for nucleophilic substitution.
Why are acyl chlorides highly reactive, and how do they facilitate ester formation?
Structure & Reactivity:
Acyl chlorides contain the -COCl functional group.
The carbonyl carbon is electron-deficient, making it highly susceptible to nucleophilic attack.
The C-Cl bond breaks, releasing white fumes of HCl during the reaction.
Why Use Acyl Chlorides Instead of Carboxylic Acids for Ester Formation?
Acyl chlorides are more reactive, leading to faster ester formation.
Reactions go to completion, ensuring higher ester yields compared to carboxylic acid reactions.
How do acyl chlorides react with alcohols to form esters?
Mechanism (Addition-Elimination):
The -OH group in the alcohol acts as a nucleophile, attacking the electron-deficient carbonyl carbon.
The chlorine atom is substituted, forming an ester and releasing HCl gas (white fumes).
Example – Formation of Ethyl Ethanoate:
Reactants: Ethanoyl chloride (CH₃COCl) + Ethanol (CH₃CH₂OH).
Products: Ethyl ethanoate (CH₃COOCH₂CH₃) + HCl gas.
Equation: CH₃COCl + CH₃CH₂OH → CH₃COOCH₂CH₃ + HCl
Why do acyl chlorides require special conditions to react with phenols, and how does esterification occur?
Why Special Conditions?
Phenols are less reactive than alcohols, so heat and a base (e.g., NaOH) are needed.
The base deprotonates phenol, forming a phenoxide ion, which is more nucleophilic than phenol itself.
Mechanism:
Phenoxide ion attacks the carbonyl carbon of the acyl chloride.
The chlorine atom is substituted, forming an ester and releasing HCl gas.
Example – Formation of Phenyl Ethanoate:
Reactants: Ethanoyl chloride (CH₃COCl) + Phenol (C₆H₅OH) + NaOH.
Products: Phenyl ethanoate (CH₃COOC₆H₅) + HCl.
Equation: CH₃COCl + C₆H₅O⁻ → CH₃COOC₆H₅ + Cl⁻
How is phenol produced from phenylamine, and what reaction conditions are required?
Reaction Overview:
Phenol is prepared from phenylamine (C₆H₅NH₂) through diazotisation, followed by hydrolysis of the diazonium salt.
Reaction Steps:
Formation of Nitrous Acid (HNO₂):
Reagents: NaNO₂ + Dilute HCl.
Conditions: Below 10°C (ice bath used).
HNO₂ is unstable, so it must be prepared in situ.
Formation of Benzenediazonium Chloride:
Reagents: Phenylamine + HNO₂.
Conditions: Below 10°C.
Benzenediazonium chloride (C₆H₅N₂Cl) is highly unstable.
Formation of Phenol:
Reagents: Diazonium salt + H₂O.
Conditions: Heat applied → Thermal decomposition occurs.
Products: Phenol + N₂ gas (effervescence).
Equation: C₆H₅NH₂ + HNO₂ → C₆H₅N₂Cl → C₆H₅OH + N₂
How does phenol react with bases, and what product is formed?
Phenol with Aqueous NaOH:
Reagents: Phenol + NaOH (aq).
Reaction Type: Neutralisation (Acid-Base Reaction).
Product: Sodium phenoxide (C₆H₅O⁻Na⁺) + H₂O (soluble salt).
Phenol with Sodium Metal:
Reagents: Molten phenol + Na(s).
Reaction Type: Redox reaction.
Products: Sodium phenoxide (C₆H₅O⁻Na⁺) + H₂ gas (effervescence).
Equation: C₆H₅OH + Na → C₆H₅O⁻Na⁺ + ½H₂
How does phenol react with diazonium salts to form azo compounds?
Reaction Overview:
Phenol reacts with benzenediazonium chloride in alkaline conditions to form azo compounds, which are brightly colored and used as dyes.
Reaction Mechanism:
Phenol is dissolved in NaOH(aq) → Forms sodium phenoxide (C₆H₅O⁻Na⁺).
Benzenediazonium chloride (C₆H₅N₂Cl) is added to cold solution.
Electrophilic substitution occurs → Formation of yellow-orange azo compound.
Equation: C₆H₅O⁻Na⁺ + C₆H₅N₂Cl → C₆H₅-N=N-C₆H₅ + NaCl
How does phenol undergo nitration, and what products are formed?
Reaction with Dilute HNO₃ (Electrophilic Substitution):
Reagents: Phenol + Dilute HNO₃ (aq).
Conditions: Room temperature.
Products: 2-nitrophenol + 4-nitrophenol (mixture).
Reaction with Concentrated HNO₃:
Reagents: Phenol + Conc. HNO₃.
Products: 2,4,6-Trinitrophenol (Picric acid).
Equation: C₆H₅OH + HNO₃ → C₆H₄(OH)(NO₂) + H₂O
How does phenol react with bromine water, and what product is formed?
Reaction with Br₂ (Electrophilic Substitution):
Reagents: Phenol + Br₂ (aq).
Conditions: Room temperature.
Product: 2,4,6-Tribromophenol (White precipitate).
Observation: Bromine water is decolourised.
Equation: C₆H₅OH + 3Br₂ → C₆H₂Br₃OH + 3HBr
Why is phenol weakly acidic, and how does the stability of the phenoxide ion affect its acidity?
Phenol’s Acidity:
Although phenol contains an -OH group, it is more acidic than alcohols due to delocalisation.
Oxygen’s lone pair overlaps with the π-system of benzene, increasing electron density and acidic behavior.
Formation of Phenoxide Ion (Conjugate Base):
Phenol ionises: C₆H₅OH ⇌ C₆H₅O⁻ + H⁺.
Phenoxide ion is more stable than phenol due to delocalisation of negative charge over the ring.
Effect on Equilibrium Position:
Since phenoxide ion is highly stable, the equilibrium shifts right, meaning phenol readily loses a proton.
Higher proportion of dissociated phenoxide ions compared to water and ethanol → Phenol is a stronger acid.
How do the acidities of water, phenol, and ethanol compare, and what factors influence their relative strengths?
Explaining the Trend:
Phenol (Strongest Acid)
Phenoxide ion stabilised by delocalisation, reducing charge density on oxygen.
Equilibrium shifts right, meaning phenol readily loses H⁺.
Higher proportion of dissociated phenoxide ions → More acidic.
Water (Moderate Acid)
Hydroxide ion lacks delocalisation, making it less stable than phenoxide ion.
Equilibrium position is further left, meaning water retains more undissociated molecules.
Ethanol (Weakest Acid)
Ethoxide ion has electron-donating alkyl group, increasing charge density on oxygen.
Oxygen readily rebinds to H⁺, making equilibrium shift left.
Least dissociation → Least acidic.

Why does phenol undergo nitration and bromination more easily than benzene, and how do their reaction conditions differ?
Phenol’s Increased Reactivity:
The -OH group in phenol donates electron density into the benzene ring.
Oxygen’s lone pair overlaps with the π-system, increasing electron density and making phenol more susceptible to electrophilic attack.
Stronger activation of the ring compared to benzene.
Why Milder Conditions for Phenol?
Phenol is more reactive → No need for strong acids (H₂SO₄) or catalysts (AlBr₃).
Dilute HNO₃ sufficient for nitration at room temperature.
Bromine water (Br₂ (aq)) reacts readily without a catalyst.

How does the -OH group in phenol influence the positions of electrophilic substitution?
Activation of the Benzene Ring:
Electron donation from oxygen’s lone pair into the π-system increases reactivity.
Higher electron density → Easier electrophilic attack.
Hydroxyl (-OH) group directs substitution to the 2-, 4-, and 6-positions.
Example – Bromination of Phenol:
Br₂ (aq) reacts with phenol, forming 2,4,6-tribromophenol (white precipitate).
Substitution occurs at positions 2, 4, and 6 due to electron donation.
Equation: C₆H₅OH + 3Br₂ → C₆H₂Br₃OH + 3HBr
How do other phenolic compounds, like naphthol, undergo electrophilic substitution reactions similarly to phenol?
Structure of 1-Naphthol:
Contains a phenol (-OH) group attached to another benzene ring.
Behaves similarly to phenol, with directing effects at positions 2 and 4.
Substitution at the 6-position is not possible (bonded to second benzene ring).
Electrophilic Substitution in 1-Naphthol:
-OH activates the ring, increasing susceptibility to electrophiles.
Reaction follows similar patterns to phenol, directing electrophiles to positions 2 and 4.
How is benzoic acid produced from an alkylbenzene such as methylbenzene?
Oxidation of Alkylbenzenes
Reagents: Hot alkaline KMnO₄ followed by dilute acid (HCl).
Conditions: Reflux.
Observation: The purple Mn⁷⁺ ions are reduced to Mn⁴⁺, forming a brown MnO₂ precipitate.
Final Step: Acidification with dilute HCl protonates the organic product → Benzoic acid (C₆H₅COOH) is formed.
Equation: C₆H₅CH₃ + [O] → C₆H₅COOH
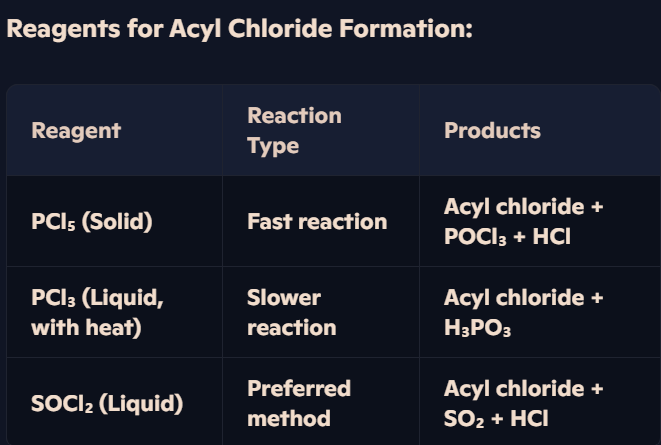
How do carboxylic acids react with PCl₃, PCl₅, or SOCl₂ to form acyl chlorides?
Formation of Acyl Chlorides (-COCl)
Carboxylic acids react with:
Solid PCl₅ → Acyl chloride + POCl₃ + HCl.
Liquid PCl₃ + Heat → Acyl chloride + H₃PO₃.
Liquid SOCl₂ → Acyl chloride + SO₂ + HCl.
Example – Formation of Ethanoyl Chloride:
CH₃COOH + PCl₅ → CH₃COCl + POCl₃ + HCl
How is methanoic acid (HCOOH) further oxidised, and what observations can be made?
Oxidation of Methanoic Acid (HCOOH):
Methanoic acid acts as a reducing agent and undergoes oxidation to CO₂.
Oxidising Agents Used:
Fehling’s solution: Cu²⁺ reduced to Cu₂O (red precipitate).
Tollens’ reagent: Ag⁺ reduced to metallic silver (mirror effect).
Acidified KMnO₄ or K₂Cr₂O₇: Mn⁷⁺ → Mn²⁺ (Purple → Colourless) / Cr⁶⁺ → Cr³⁺ (Orange → Green).
Equation: HCOOH + [O] → CO₂ + H₂O
How is ethanedioic acid (HOOCCOOH) oxidised, and what is the role of KMnO₄?
Oxidation of Ethanedioic Acid:
Reagent: Warm acidified KMnO₄.
Products: Carbon dioxide (CO₂) & water.
Observations: Mn⁷⁺ ions are reduced to Mn²⁺, causing the purple KMnO₄ solution to turn colourless.
Equation: HOOCCOOH + [O] → 2CO₂ + H₂O
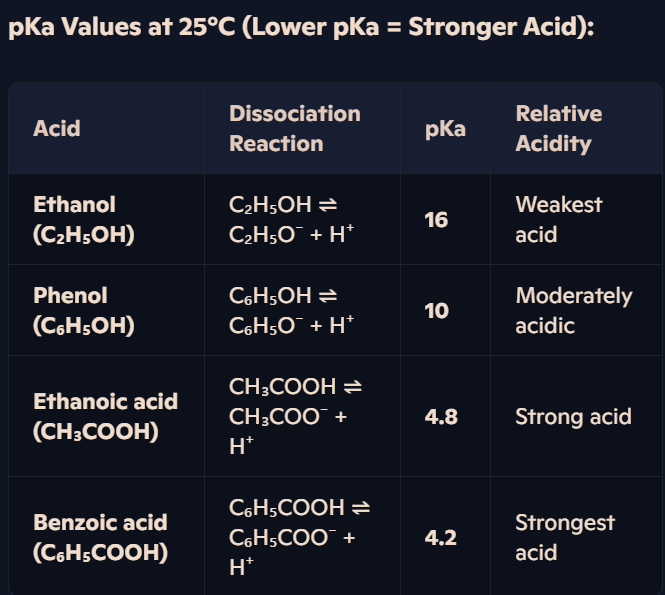
How do the acidities of carboxylic acids, phenols, and alcohols compare, and what factors influence their relative strengths?
Factors Affecting Acidity:
Strength of O-H Bond:
In carboxylic acids, the carbonyl (-C=O) group withdraws electron density, weakening the O-H bond.
Alcohols lack this electron-withdrawing effect, making them weaker acids.
Stability of Conjugate Base:
Carboxylate ions (-COO⁻) are highly stable due to charge delocalisation, making carboxylic acids stronger acids.
Phenoxide ions (C₆H₅O⁻) are stabilised but not as effectively as carboxylates → Weaker acidity than carboxylic acids.
Alkoxide ions (C₂H₅O⁻) are destabilised due to electron donation from the alkyl group → Ethanol is the weakest acid.
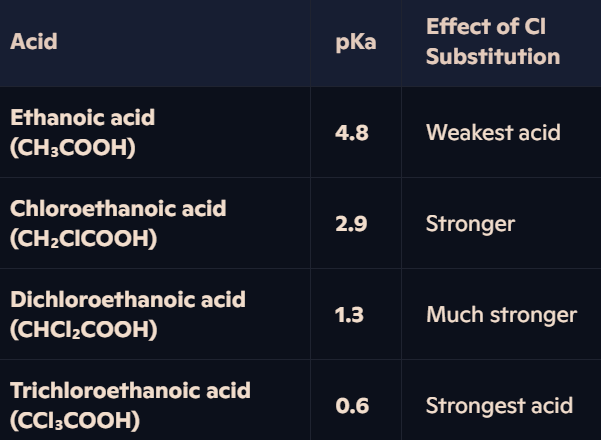
How do electron-withdrawing chlorine atoms affect the acidity of carboxylic acids?
Why More Chlorine = Stronger Acidity?
Chlorine atoms withdraw electron density from the O-H bond, making it weaker and facilitating proton loss.
Delocalisation of charge in the carboxylate ion is further extended by Cl atoms → Greater stability → Lower tendency to recombine with H⁺.
Example – Trichloroethanoic Acid (Strongest Acid):
Three electronegative Cl atoms withdraw electron density.
Highly stabilised carboxylate ion (-COO⁻) reduces attraction to H⁺ → Very strong acid.
What are esters, and why are acyl chloride reactions preferred for their synthesis?
Structure & Uses of Esters:
Esters have the -COOR functional group.
Used in perfumes, cosmetics, and solvents due to their characteristic smells.
Named based on their reactants:
Alcohol contributes the first part of the name.
Acyl chloride contributes the second part (e.g., Ethyl ethanoate = ethanol + ethanoyl chloride).
Why Use Acyl Chlorides Instead of Carboxylic Acids?
Acyl chlorides are more reactive → Faster ester formation.
Reaction goes to completion → No equilibrium mixture, maximum ester yield.
How does ethanol react with ethanoyl chloride to form ethyl ethanoate?
Reaction Details:
Reagents: Ethanol (C₂H₅OH) + Ethanoyl chloride (CH₃COCl).
Products: Ethyl ethanoate (CH₃COOCH₂CH₃) + HCl (white fumes).
Mechanism:
Nucleophilic attack by ethanol’s -OH group on the electron-deficient carbonyl carbon.
Chlorine (-Cl) atom is eliminated, forming HCl gas.
Ester bond (-COO-) is formed, producing ethyl ethanoate.
Equation: CH₃COCl + C₂H₅OH → CH₃COOCH₂CH₃ + HCl
How does phenol react with benzoyl chloride to form phenyl benzoate?
Reaction Details:
Reagents: Phenol (C₆H₅OH) + Benzoyl chloride (C₆H₅COCl).
Products: Phenyl benzoate (C₆H₅COOC₆H₅) + HCl.
Mechanism:
Phenol’s -OH group attacks the carbonyl carbon in benzoyl chloride.
Chlorine (-Cl) is eliminated, releasing HCl gas.
Phenyl benzoate (ester) is formed.
Equation: C₆H₅COCl + C₆H₅OH → C₆H₅COOC₆H₅ + HCl
How are acyl chlorides synthesized from carboxylic acids, and what reagents are required?
Acyl Chlorides & Their Reactivity:
Acyl chlorides contain the -COCl functional group.
They are more reactive than carboxylic acids, making them valuable intermediates in organic synthesis.
Example – Formation of Propanoyl Chloride:
CH₃CH₂COOH + SOCl₂ → CH₃CH₂COCl + SO₂ + HCl
How do acyl chlorides react with water, and what is the mechanism?
Reaction Overview:
Reagent: H₂O (room temperature).
Products: Carboxylic acid + HCl gas.
Observations: White fumes of HCl are released.
Mechanism:
Water’s lone pair attacks the carbonyl carbon.
Addition of H₂O molecule.
Elimination of HCl, forming the carboxylic acid.
Equation: CH₃CH₂COCl + H₂O → CH₃CH₂COOH + HCl
How do acyl chlorides react with alcohols and phenols to form esters?
Reaction with Alcohols:
Reagent: Alcohol (room temperature).
Products: Ester + HCl.
Example: Ethanol + Ethanoyl chloride → Ethyl ethanoate + HCl.
Equation: CH₃COCl + C₂H₅OH → CH₃COOCH₂CH₃ + HCl
Reaction with Phenols:
Reagent: Phenol + Base (NaOH, needed for phenoxide formation).
Products: Ester + NaCl.
Example: Phenol + Benzoyl chloride → Phenyl benzoate + NaCl.
Equation: C₆H₅COCl + C₆H₅O⁻ → C₆H₅COOC₆H₅ + NaCl
How do acyl chlorides react with ammonia and amines to form amides?
Reaction with Ammonia (NH₃):
Reagents: NH₃ (room temperature).
Products: Non-substituted amide + HCl gas.
Example: Ethanoyl chloride + NH₃ → Ethanamide + HCl.
Equation: CH₃COCl + NH₃ → CH₃CONH₂ + HCl
Reaction with Primary & Secondary Amines:
Reagent: Amine (room temperature).
Products: Substituted amide + Ammonium salt.
Example: Ethanoyl chloride + Methylamine → N-Methylethanamide + Methylammonium chloride.
Equation: CH₃COCl + CH₃NH₂ → CH₃CONHCH₃ + CH₃NH₃⁺Cl⁻
What is the general addition-elimination mechanism for acyl chloride reactions?
Step 1 – Addition of a Nucleophile:
Water, alcohol, phenol, ammonia, or amine attacks carbonyl carbon using a lone pair.
Electron transfer weakens C=O bond.
Step 2 – Elimination of a Small Molecule:
Leaving group (-Cl) is expelled, forming HCl gas or NaCl (with phenols).
Final Product: Carboxylic acid, ester, or amide.
Example – Hydrolysis of Acyl Chlorides:
Nucleophilic attack by water.
Formation of intermediate.
Elimination of HCl, forming carboxylic acid.
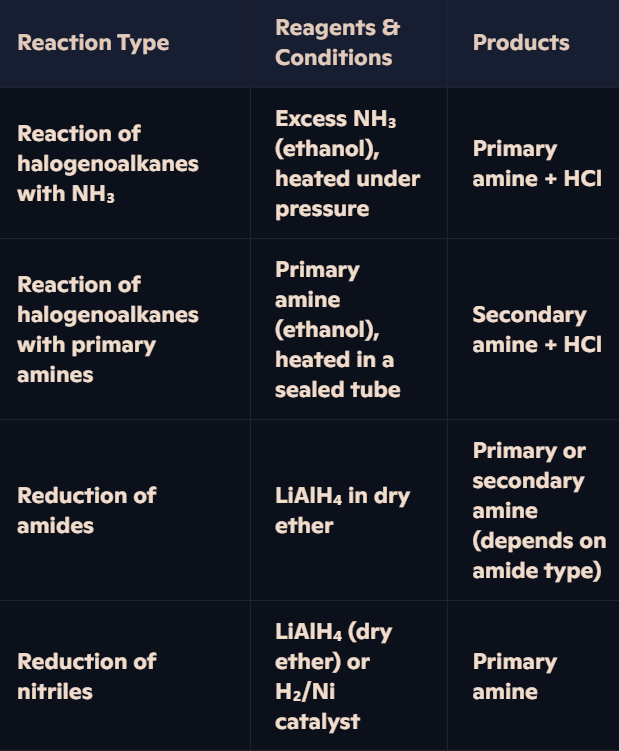
How are primary and secondary amines synthesized, and what reagents and conditions are required?
Example – Formation of Ethylamine (Primary Amine):
C₂H₅Cl + NH₃ → C₂H₅NH₂ + HCl
Example – Formation of Diethylamine (Secondary Amine):
C₂H₅Cl + C₂H₅NH₂ → (C₂H₅)₂NH + HCl
How do acyl chlorides react with ammonia and amines to form amides?
Reaction Overview:
Acyl chlorides contain the electronegative -COCl group, making the carbonyl carbon highly susceptible to nucleophilic attack.
Reaction Mechanism:
Nucleophile (NH₃ or amine) attacks carbonyl carbon using its lone pair.
Addition occurs, followed by elimination of HCl, forming the amide.

Why do amines act as bases, and what factors affect their basicity?
Basicity of Amines:
Amines contain nitrogen atoms with a lone pair of electrons, allowing them to accept protons (H⁺ ions).
Reaction with HCl (acid-base reaction): NH₃ + HCl → NH₄⁺Cl⁻ C₂H₅NH₂ + HCl → C₂H₅NH₃⁺Cl⁻
Factors Affecting Basicity:
Inductive Effect:
Alkyl groups (-R) donate electron density to nitrogen, making the lone pair more available for bonding with H⁺ → Increases basicity.
Example: Ethylamine is more basic than phenylamine.
Delocalisation Effect:
Aromatic rings (benzene) withdraw electron density, reducing availability of lone pair → Decreases basicity.
Example: Phenylamine is weaker than ethylamine.

How is phenylamine synthesized from benzene via nitration, reduction, and deprotonation?
Step 1 – Nitration of Benzene:
Reagents: Conc. HNO₃ + Conc. H₂SO₄, 25–60°C.
Mechanism: Electrophilic substitution, forming nitrobenzene (C₆H₅NO₂).
Step 2 – Reduction of Nitrobenzene:
Reagents: Hot Sn + Conc. HCl, reflux.
Mechanism: Reduction, forming phenylammonium chloride (C₆H₅N⁺H₃Cl⁻).
Step 3 – Deprotonation of Phenylammonium Ion:
Reagent: NaOH(aq), room temperature.
Mechanism: Deprotonation, forming phenylamine (C₆H₅NH₂).
Equation – Overall Reaction:
C₆H₆ → C₆H₅NO₂ → C₆H₅N⁺H₃Cl⁻ → C₆H₅NH₂
How does phenylamine react with Br₂(aq), and why does it undergo substitution more readily than benzene?
Reaction Overview:
Reagent: Br₂ (aq), room temperature.
Mechanism: Electrophilic substitution, forming 2,4,6-tribromophenylamine.
Why Does It React Easily?
Lone pair on nitrogen delocalises into benzene ring, increasing electron density.
Stronger activation of the ring, leading to faster bromination without a catalyst.
Equation: C₆H₅NH₂ + 3Br₂ → C₆H₂Br₃NH₂ + 3HBr
How does phenylamine react with HNO₂ (formed in situ) to produce a diazonium salt, and how is phenol formed upon warming?
Formation of Diazonium Salt:
Reagents: NaNO₂ + Dilute HCl, below 10°C.
Mechanism: Formation of unstable benzenediazonium chloride (C₆H₅N₂⁺Cl⁻).
Decomposition to Phenol:
Reagent: H₂O, warming.
Mechanism: Decomposition, forming phenol (C₆H₅OH) + N₂ gas.
Equation: C₆H₅NH₂ + HNO₂ → C₆H₅N₂⁺Cl⁻ → C₆H₅OH + N₂ + HCl
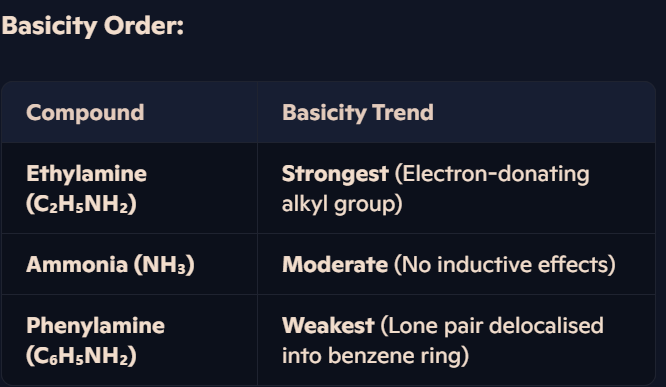
How do ammonia, ethylamine, and phenylamine compare in basicity, and what affects their ability to accept protons?
Ethylamine:
Alkyl group donates electron density, increasing availability of lone pair for proton bonding.
Stronger base than ammonia and phenylamine.
Ammonia:
No extra effects, lone pair freely available, making it moderately basic.
Phenylamine:
Lone pair delocalised into benzene ring, reducing availability for proton bonding.
Weakest base due to reduced electron density on nitrogen.
What are azo compounds, and how is the azo group identified?
Definition of Azo Compounds:
Azo compounds are organic compounds containing the azo group (-N=N-), which links two organic groups (R₁ and R₂).
General formula: R₁-N=N-R₂, where R₁ and R₂ are typically aromatic groups.
Identification of the Azo Group:
The azo group (-N=N-) is a functional group characterized by a nitrogen-nitrogen double bond.
It acts as a bridge between two aromatic rings, extending the delocalized π-electron system.
Stability of Azo Compounds:
The delocalization of electrons across the aromatic rings and the azo group makes azo compounds highly stable.
How is an azo compound formed from benzenediazonium chloride and phenol in NaOH(aq)?
Step 1 – Formation of Benzenediazonium Chloride (Diazotisation):
Reagents: Sodium nitrite (NaNO₂) + Dilute HCl, below 10°C.
Reaction:
Sodium nitrite reacts with HCl to form nitrous acid (HNO₂).
Nitrous acid reacts with phenylamine (C₆H₅NH₂) to form benzenediazonium chloride (C₆H₅N₂⁺Cl⁻).
Conditions: Temperature must be kept below 10°C to prevent decomposition of the diazonium salt.
Equation: NaNO₂ + HCl → HNO₂ + NaCl C₆H₅NH₂ + HNO₂ + HCl → C₆H₅N₂⁺Cl⁻ + 2H₂O
Step 2 – Coupling Reaction with Phenol:
Reagents: Benzenediazonium chloride + Phenol in NaOH(aq).
Mechanism:
The diazonium ion (C₆H₅N₂⁺) acts as an electrophile.
It substitutes into the 4th position of the benzene ring in phenol.
Alkaline conditions (NaOH) are required to deprotonate phenol, forming the azo compound.
Equation: C₆H₅N₂⁺Cl⁻ + C₆H₅OH → C₆H₅-N=N-C₆H₄OH + HCl
Product:
The product is an azo compound with the structure C₆H₅-N=N-C₆H₄OH.
Why are azo compounds used as dyes, and how can other azo dyes be formed?
Azo Compounds as Dyes:
Azo compounds are widely used as dyes due to their extended delocalized π-electron systems, which absorb visible light and give bright, stable colors.
The color depends on the substituents on the aromatic rings.
Formation of Other Azo Dyes:
Other azo dyes can be formed by coupling benzenediazonium chloride with different aromatic compounds.
Example: Coupling with dimethylaniline (C₆H₅N(CH₃)₂) instead of phenol forms a yellow azo dye.
Equation: C₆H₅N₂⁺Cl⁻ + C₆H₅N(CH₃)₂ → C₆H₅-N=N-C₆H₄N(CH₃)₂ + HCl
What are the key steps in the formation of azo compounds?
Key Steps:
Formation of Nitrous Acid (HNO₂):
Reagents: NaNO₂ + Dilute HCl.
Conditions: Below 10°C.
Diazotisation:
Reagents: Phenylamine + HNO₂ + HCl.
Product: Benzenediazonium chloride (C₆H₅N₂⁺Cl⁻).
Conditions: Below 10°C to prevent decomposition.
Coupling Reaction:
Reagents: Benzenediazonium chloride + Phenol in NaOH(aq).
Product: Azo compound (e.g., C₆H₅-N=N-C₆H₄OH).
Mechanism: Electrophilic substitution at the 4th position of phenol.
Applications:
Azo compounds are used as dyes due to their bright colors and stability.
Other dyes can be formed by coupling with different aromatic compounds, such as dimethylaniline.
How are amides produced from acyl chlorides, and what reagents and conditions are required?
Amides Formation Overview:
Amides are formed via condensation reactions between acyl chlorides and ammonia or amines.
Reaction with Ammonia:
Reagents: Acyl chloride + Ammonia (NH₃).
Conditions: Room temperature.
Products: Non-substituted amide + HCl gas.
Mechanism:
Nucleophilic attack by ammonia on the carbonyl carbon of the acyl chloride.
Elimination of HCl, forming the amide.
Equation: CH₃COCl + NH₃ → CH₃CONH₂ + HCl (Example: Ethanoyl chloride + Ammonia → Ethanamide + HCl)
Reaction with Primary Amines:
Reagents: Acyl chloride + Primary amine (R-NH₂).
Conditions: Room temperature.
Products: Substituted amide + HCl gas.
Mechanism:
Nucleophilic attack by the primary amine on the carbonyl carbon of the acyl chloride.
Elimination of HCl, forming the substituted amide.
Equation: CH₃COCl + CH₃NH₂ → CH₃CONHCH₃ + HCl (Example: Ethanoyl chloride + Methylamine → N-Methylethanamide + HCl)
How do amides undergo hydrolysis, and what are the products under acidic and alkaline conditions?
Hydrolysis Overview:
Amides can be hydrolyzed by refluxing with aqueous acid or alkali, breaking the amide bond (-CON- group).
Acidic Hydrolysis:
Reagents: Excess dilute acid (e.g., HCl).
Conditions: Reflux.
Products: Carboxylic acid + Ammonium salt (for non-substituted amides) or Amine salt (for substituted amides).
Example: CH₃CONH₂ + H₂O + HCl → CH₃COOH + NH₄Cl (Ethanamide → Ethanoic acid + Ammonium chloride)
Alkaline Hydrolysis:
Reagents: Excess dilute alkali (e.g., NaOH).
Conditions: Reflux.
Products: Carboxylate ion + Ammonia (for non-substituted amides) or Primary amine (for substituted amides).
Example: CH₃CONH₂ + NaOH → CH₃COO⁻Na⁺ + NH₃ (Ethanamide → Sodium ethanoate + Ammonia)
How are amides reduced, and what are the products?
Reduction Overview:
Amides can be reduced by LiAlH₄ (Lithium Aluminium Hydride), a strong reducing agent, to form amines.
Reagents & Conditions:
Reagent: LiAlH₄ in dry ether.
Conditions: Anhydrous.
Products:
Non-substituted amides: Primary amine + Water.
Substituted amides: Secondary amine + Water.
Example – Reduction of Ethanamide: CH₃CONH₂ + 4[H] → CH₃CH₂NH₂ + H₂O (Ethanamide → Ethylamine + Water)
Example – Reduction of N-Methylethanamide: CH₃CONHCH₃ + 4[H] → CH₃CH₂NHCH₃ + H₂O (N-Methylethanamide → N-Methylethylamine + Water)
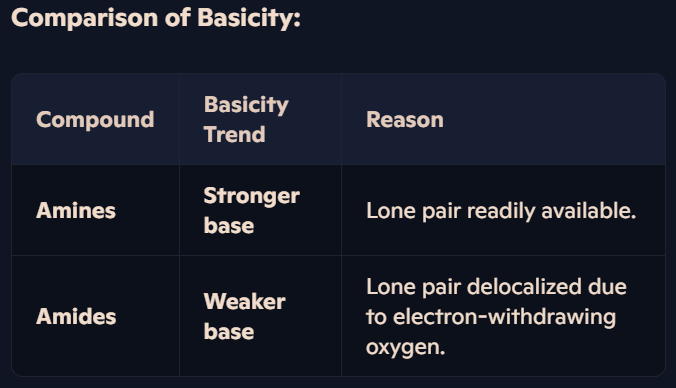
Why are amides less basic than amines, and how does the structure of amides affect their basicity?
Basicity of Amines:
Amines are basic because the nitrogen atom has a lone pair of electrons that can accept a proton (H⁺) to form a dative covalent bond.
Electron-donating groups (e.g., alkyl groups) increase the availability of the lone pair, making amines stronger bases.
Basicity of Amides:
Amides also contain a nitrogen atom with a lone pair of electrons.
However, the electron-withdrawing oxygen atom in the amide group (-CON-) reduces the electron density on the nitrogen atom.
The lone pair on nitrogen becomes less available for proton bonding, making amides much weaker bases than amines.
What are the acid/base properties of amino acids, and how do zwitterions form?
Structure of Amino Acids:
Amino acids contain two functional groups:
Amino group (-NH₂): Basic.
Carboxylic acid group (-COOH): Acidic.
General formula: RCH(NH₂)COOH, where R is the side chain.
Amphoteric Nature:
Amino acids are amphoteric, meaning they can act as both acids and bases:
As an acid: The -COOH group donates a proton (H⁺).
As a base: The -NH₂ group accepts a proton (H⁺).
Zwitterions:
A zwitterion is a molecule with both a positive charge (-NH₃⁺) and a negative charge (-COO⁻).
Formation:
The -COOH group donates a proton to the -NH₂ group within the same molecule.
Structure: RCH(NH₃⁺)COO⁻.
Zwitterions are soluble crystalline solids due to strong intermolecular forces.
What is the isoelectric point, and how does pH affect amino acids?
Isoelectric Point (pI):
The isoelectric point is the pH at which an amino acid exists as a neutral zwitterion (no net charge).
Effect of pH on Amino Acids:
At low pH (acidic):
The -COO⁻ group accepts an H⁺, forming -COOH.
The amino acid becomes positively charged.
At high pH (basic):
The -NH₃⁺ group donates an H⁺, forming -NH₂.
The amino acid becomes negatively charged.
At the isoelectric point:
The amino acid exists as a neutral zwitterion.
Buffering Action:
Amino acids act as buffers, resisting changes in pH by accepting or donating protons.
How are peptide bonds formed between amino acids?
Peptide Bond Formation:
A peptide bond is an amide bond formed between the -COOH group of one amino acid and the -NH₂ group of another.
Reaction Type: Condensation reaction (elimination of H₂O).
Formation of Dipeptides and Tripeptides:
Dipeptide Formation:
Two amino acids react to form a dipeptide.
Example: H₂N-CH(R)-COOH + H₂N-CH(R')-COOH → H₂N-CH(R)-CONH-CH(R')-COOH + H₂O
Tripeptide Formation:
A dipeptide reacts with another amino acid to form a tripeptide.
Polypeptides:
Long chains of amino acids joined by peptide bonds are called polypeptides.
How does electrophoresis separate amino acids and dipeptides at varying pH levels?
Principle of Electrophoresis:
Electrophoresis separates ions based on their charge and size in an electric field.
Setup:
A sample is placed between two electrodes (positive and negative).
Ions move towards the electrode with the opposite charge.
Factors Affecting Movement:
Charge of the Ion:
Positively charged ions move towards the negative electrode.
Negatively charged ions move towards the positive electrode.
Size of the Ion:
Larger ions move more slowly.
Smaller ions move more quickly.
Effect of pH on Movement:
The charge on amino acids depends on the pH of the solution:
At low pH (acidic): Amino acids are positively charged (-NH₃⁺).
At high pH (basic): Amino acids are negatively charged (-COO⁻).
At the isoelectric point: Amino acids do not move (neutral zwitterion).
Example – Mixture of Amino Acids at pH 7:
Lysine (positively charged): Moves towards the negative electrode.
Glycine (neutral): Remains stationary.
Glutamic acid (negatively charged): Moves towards the positive electrode.
What are the key points to remember about amino acids, peptide bonds, and electrophoresis?
Amino Acids:
Contain both -NH₂ (basic) and -COOH (acidic) groups.
Form zwitterions at the isoelectric point (neutral pH).
Act as buffers, resisting changes in pH.
Peptide Bonds:
Formed by condensation reactions between amino acids.
Result in the formation of dipeptides, tripeptides, and polypeptides.
Electrophoresis:
Separates amino acids and dipeptides based on charge and size.
Movement depends on pH:
Low pH: Positive ions move to the negative electrode.
High pH: Negative ions move to the positive electrode.
At the isoelectric point: No movement.
How are polyesters formed from a diol and a dicarboxylic acid or dioyl chloride?
Polyester Formation Overview:
Polyesters are formed by condensation polymerisation, where a small molecule (e.g., H₂O or HCl) is expelled.
Polyesters contain ester linkages (-COO-).
Reaction with Diol and Dicarboxylic Acid:
Reagents: Diol (2 -OH groups) + Dicarboxylic acid (2 -COOH groups).
Mechanism:
The -OH group of the diol reacts with the -COOH group of the dicarboxylic acid.
A water molecule (H₂O) is expelled.
Example: Formation of Polyethylene Terephthalate (PET): HO-CH₂CH₂-OH + HOOC-C₆H₄-COOH → [-O-CH₂CH₂-OCO-C₆H₄-CO-]ₙ + H₂O
Reaction with Diol and Dioyl Chloride:
Reagents: Diol + Dioyl chloride (2 -COCl groups).
Mechanism:
The -OH group of the diol reacts with the -COCl group of the dioyl chloride.
A molecule of HCl is expelled.
Example: HO-CH₂CH₂-OH + ClOC-C₆H₄-COCl → [-O-CH₂CH₂-OCO-C₆H₄-CO-]ₙ + HCl
How are polyesters formed from hydroxycarboxylic acids?
Hydroxycarboxylic Acids:
Hydroxycarboxylic acids contain both functional groups needed for polyester formation:
-OH group (alcohol).
-COOH group (carboxylic acid).
Polymerisation:
Mechanism:
The -OH group of one molecule reacts with the -COOH group of another.
A water molecule (H₂O) is expelled.
Example: Formation of a polyester from 3-hydroxybutanoic acid: HO-CH₂-CH(CH₃)-COOH → [-O-CH₂-CH(CH₃)-CO-]ₙ + H₂O
How are polyamides formed from a diamine and a dicarboxylic acid or dioyl chloride?
Polyamide Formation Overview:
Polyamides are formed by condensation polymerisation, where a small molecule (e.g., H₂O or HCl) is expelled.
Polyamides contain amide linkages (-CONH-), also called peptide bonds.
Reaction with Diamine and Dicarboxylic Acid:
Reagents: Diamine (2 -NH₂ groups) + Dicarboxylic acid (2 -COOH groups).
Mechanism:
The -NH₂ group of the diamine reacts with the -COOH group of the dicarboxylic acid.
A water molecule (H₂O) is expelled.
Example: Formation of Nylon 6,6: H₂N-(CH₂)₆-NH₂ + HOOC-(CH₂)₄-COOH → [-NH-(CH₂)₆-NHCO-(CH₂)₄-CO-]ₙ + H₂O
Reaction with Diamine and Dioyl Chloride:
Reagents: Diamine + Dioyl chloride (2 -COCl groups).
Mechanism:
The -NH₂ group of the diamine reacts with the -COCl group of the dioyl chloride.
A molecule of HCl is expelled.
Example: Formation of Kevlar: H₂N-C₆H₄-NH₂ + ClOC-C₆H₄-COCl → [-NH-C₆H₄-NHCO-C₆H₄-CO-]ₙ + HCl
How are polyamides formed from aminocarboxylic acids and amino acids?
Aminocarboxylic Acids:
Aminocarboxylic acids contain both functional groups needed for polyamide formation:
-NH₂ group (amine).
-COOH group (carboxylic acid).
Mechanism:
The -NH₂ group of one molecule reacts with the -COOH group of another.
A water molecule (H₂O) is expelled.
Example: Formation of Nylon 6: H₂N-(CH₂)₅-COOH → [-NH-(CH₂)₅-CO-]ₙ + H₂O
Amino Acids:
Amino acids are biological monomers with an aminocarboxylic acid structure.
Mechanism:
The -NH₂ group of one amino acid reacts with the -COOH group of another.
A water molecule (H₂O) is expelled.
Example: Formation of a dipeptide: H₂N-CH(R)-COOH + H₂N-CH(R')-COOH → H₂N-CH(R)-CONH-CH(R')-COOH + H₂O
How can the repeat unit of a condensation polymer be deduced from its monomers?
Steps to Deduce the Repeat Unit:
Identify the Monomers:
Look for functional groups (-OH, -COOH, -NH₂, -COCl).
Determine the Linkage:
Ester linkage (-COO-): Indicates a polyester.
Amide linkage (-CONH-): Indicates a polyamide.
Remove the Small Molecule:
Remove H₂O (from -OH and -COOH) or HCl (from -NH₂ and -COCl).
Combine the Remaining Parts:
Join the monomers to form the repeat unit.
Example – Polyester:
Monomers: HO-CH₂CH₂-OH and HOOC-C₆H₄-COOH.
Repeat Unit: [-O-CH₂CH₂-OCO-C₆H₄-CO-].
Example – Polyamide:
Monomers: H₂N-(CH₂)₆-NH₂ and HOOC-(CH₂)₄-COOH.
Repeat Unit: [-NH-(CH₂)₆-NHCO-(CH₂)₄-CO-].
How can the monomers of a condensation polymer be identified from its structure?
Steps to Identify Monomers:
Break the Polymer at the Linkages:
Ester linkage (-COO-): Indicates a diol and a dicarboxylic acid/dioyl chloride.
Amide linkage (-CONH-): Indicates a diamine and a dicarboxylic acid/dioyl chloride.
Add Back the Small Molecule:
Add H₂O (for -OH and -COOH) or HCl (for -NH₂ and -COCl).
Reconstruct the Monomers:
Recreate the original monomers from the fragments.
Example – Polyester:
Polymer: [-O-CH₂CH₂-OCO-C₆H₄-CO-]ₙ.
Monomers: HO-CH₂CH₂-OH and HOOC-C₆H₄-COOH.
Example – Polyamide:
Polymer: [-NH-(CH₂)₆-NHCO-(CH₂)₄-CO-]ₙ.
Monomers: H₂N-(CH₂)₆-NH₂ and HOOC-(CH₂)₄-COOH.
How can the type of polymerisation be predicted from the monomers?
Step 1 – Identify Functional Groups in the Monomers:
C=C Double Bond:
Indicates addition polymerisation.
The double bond opens up, allowing monomers to join.
Functional Groups for Condensation Polymerisation:
Polyamides:
Diamines (-NH₂) and dicarboxylic acids (-COOH) or dioyl chlorides (-COCl).
Aminocarboxylic acids (H₂N-CHR-COOH).
Polyesters:
Diols (-OH) and dicarboxylic acids (-COOH) or dioyl chlorides (-COCl).
Hydroxycarboxylic acids (HO-R-COOH).
Step 2 – Determine the Type of Polymerisation:
Addition Polymerisation:
Monomers contain C=C double bonds.
Forms (poly)alkenes or copolymers (if more than one monomer is used).
Condensation Polymerisation:
Monomers contain two functional groups that react to form a polymer.
A small molecule (e.g., H₂O or HCl) is expelled during the reaction.
Examples:
Addition Polymerisation: Ethene (CH₂=CH₂) forms polyethene.
Condensation Polymerisation:
Diol + Dicarboxylic acid → Polyester.
Diamine + Dicarboxylic acid → Polyamide.
What are the characteristics of addition polymerisation, and how can it be identified?
Characteristics of Addition Polymerisation:
Monomers contain C=C double bonds.
The double bond opens up, allowing monomers to join.
No small molecule is expelled.
The polymer backbone is a chain of carbon atoms.
Examples of Addition Polymers:
Polyethene:
Monomer: Ethene (CH₂=CH₂).
Polymer: Polyethene ([-CH₂-CH₂-]ₙ).
Polypropene:
Monomer: Propene (CH₂=CHCH₃).
Polymer: Polypropene ([-CH₂-CH(CH₃)-]ₙ).
Copolymers:
Formed from two or more different alkene monomers.
Example: Styrene-butadiene rubber (from styrene and butadiene).
Identifying Addition Polymers:
The polymer backbone is a plain carbon chain.
Side chains may include:
Benzene rings.
Nitrile groups (-CN).
Halogen atoms (-F, -Cl, -Br, -I).
What are the characteristics of condensation polymerisation, and how can it be identified?
Characteristics of Condensation Polymerisation:
Monomers contain two functional groups that react.
A small molecule (e.g., H₂O or HCl) is expelled.
The polymer backbone contains functional groups (e.g., ester or amide links).
Examples of Condensation Polymers:
Polyesters:
Monomers: Diol + Dicarboxylic acid or dioyl chloride.
Example: Polyethylene terephthalate (PET). HO-CH₂CH₂-OH + HOOC-C₆H₄-COOH → [-O-CH₂CH₂-OCO-C₆H₄-CO-]ₙ + H₂O
Polyamides:
Monomers: Diamine + Dicarboxylic acid or dioyl chloride.
Example: Nylon 6,6. H₂N-(CH₂)₆-NH₂ + HOOC-(CH₂)₄-COOH → [-NH-(CH₂)₆-NHCO-(CH₂)₄-CO-]ₙ + H₂O
Identifying Condensation Polymers:
The polymer backbone contains functional groups:
Ester links (-COO-) for polyesters.
Amide links (-CONH-) for polyamides.
How can the type of polymerisation be deduced from a given polymer section?
Step 1 – Examine the Polymer Backbone:
No Functional Groups:
Indicates addition polymerisation.
The backbone is a chain of carbon atoms.
Functional Groups Present:
Indicates condensation polymerisation.
Look for:
Ester links (-COO-) → Polyester.
Amide links (-CONH-) → Polyamide.
Step 2 – Identify the Monomers:
Addition Polymerisation:
Monomers contain C=C double bonds.
Example: Polyethene from ethene (CH₂=CH₂).
Condensation Polymerisation:
Monomers contain two functional groups.
Example: Nylon 6,6 from 1,6-diaminohexane and hexane-1,6-dioic acid.
Examples:
Polymer Section: [-CH₂-CH₂-]ₙ
Backbone: Plain carbon chain.
Type: Addition polymerisation.
Monomer: Ethene (CH₂=CH₂).
Polymer Section: [-NH-(CH₂)₆-NHCO-(CH₂)₄-CO-]ₙ
Backbone: Contains amide links (-CONH-).
Type: Condensation polymerisation.
Monomers: 1,6-diaminohexane and hexane-1,6-dioic acid.
What are the key differences between addition and condensation polymerisation?
Addition Polymerisation:
Monomers contain C=C double bonds.
No small molecule is expelled.
The polymer backbone is a plain carbon chain.
Examples: Polyethene, Polypropene.
Condensation Polymerisation:
Monomers contain two functional groups.
A small molecule (e.g., H₂O or HCl) is expelled.
The polymer backbone contains functional groups:
Ester links (-COO-) for polyesters.
Amide links (-CONH-) for polyamides.
Examples: Polyethylene terephthalate (PET), Nylon 6,6.
Why are poly(alkenes) chemically inert and difficult to biodegrade?
Structure of Poly(alkenes):
Poly(alkenes) are formed by addition polymerisation of alkene monomers.
The polymer chains are:
Non-polar (no reactive groups).
Saturated (no C=C double bonds).
Chemical Inertness:
The lack of reactive functional groups makes poly(alkenes) chemically inert.
They do not react with water, acids, bases, or enzymes, making them non-biodegradable.
Environmental Impact:
Poly(alkenes) accumulate in the environment, contributing to pollution.
Recycling options:
Melting and repurposing: Poly(alkenes) can be melted and reused, but they remain non-biodegradable.
Burning:
Complete combustion releases CO₂ and H₂O, contributing to global warming.
Incomplete combustion releases toxic gases like CO and soot.
How can polymers degrade by the action of light, and what are the limitations of photodegradation?
Mechanism of Photodegradation:
Polyesters and polyamides can degrade under UV light due to the presence of carbonyl groups (C=O) in their polymer chains.
Process:
Carbonyl groups absorb UV light from the electromagnetic spectrum.
UV energy weakens the bonds in the polymer chain, breaking it into smaller molecules.
Advantages:
Photodegradation allows polyesters and polyamides to break down naturally under sunlight.
Limitations:
Weaker Polymers:
When repurposed, photodegradable polymers may lose strength due to their susceptibility to UV light.
Landfill Conditions:
In landfill sites, polymers may not be exposed to UV light, preventing natural degradation.
How can polyesters and polyamides be biodegraded by acidic and alkaline hydrolysis?
Hydrolysis Overview:
Hydrolysis is the breakdown of molecules using water.
Polyesters and polyamides are biodegradable because their ester (-COO-) and amide (-CONH-) linkages can be hydrolyzed.
Acidic Hydrolysis:
Reagents: Dilute acid (e.g., HCl).
Conditions: Heat.
Products:
Polyamides: Dicarboxylic acid + Ammonium ions. Example: Hydrolysis of Kevlar: [-NH-C₆H₄-NHCO-C₆H₄-CO-]ₙ + H₂O + HCl → HOOC-C₆H₄-COOH + NH₄⁺
Polyesters: Diol + Dicarboxylic acid. Example: Hydrolysis of PET: [-O-CH₂CH₂-OCO-C₆H₄-CO-]ₙ + H₂O + HCl → HO-CH₂CH₂-OH + HOOC-C₆H₄-COOH
Alkaline Hydrolysis:
Reagents: Dilute alkali (e.g., NaOH).
Conditions: Heat.
Products:
Polyamides: Dicarboxylic acid salt + Diamines. Example: Hydrolysis of Kevlar: [-NH-C₆H₄-NHCO-C₆H₄-CO-]ₙ + H₂O + NaOH → Na⁺OOC-C₆H₄-COO⁻ + H₂N-C₆H₄-NH₂
Polyesters: Diol + Dicarboxylic acid salt. Example: Hydrolysis of PET: [-O-CH₂CH₂-OCO-C₆H₄-CO-]ₙ + H₂O + NaOH → HO-CH₂CH₂-OH + Na⁺OOC-C₆H₄-COO⁻
Advantages of Hydrolysis:
Polyesters and polyamides can be broken down in landfill sites, unlike poly(alkenes).
The products can be repurposed for other applications.
How do poly(alkenes), polyesters, and polyamides differ in terms of biodegradability?
Poly(alkenes):
Non-biodegradable:
Chemically inert due to non-polar, saturated chains.
Cannot be broken down by hydrolysis or enzymes.
Environmental Impact:
Accumulate in the environment.
Recycling options: Melting or burning (releases CO₂).
Polyesters and Polyamides:
Biodegradable:
Can be broken down by hydrolysis (acidic or alkaline).
Ester (-COO-) and amide (-CONH-) linkages are hydrolyzed.
Photodegradable:
Carbonyl groups absorb UV light, breaking the polymer chains.
Environmental Advantage:
Degrade naturally in landfill sites or under UV light.
What are the key points to remember about polymer biodegradability?
Poly(alkenes):
Formed by addition polymerisation.
Chemically inert and non-biodegradable.
Recycling options: Melting or burning (environmental concerns).
Polyesters and Polyamides:
Formed by condensation polymerisation.
Biodegradable by hydrolysis:
Acidic hydrolysis → Diol + Dicarboxylic acid (polyesters) or Dicarboxylic acid + Ammonium ions (polyamides).
Alkaline hydrolysis → Diol + Dicarboxylic acid salt (polyesters) or Dicarboxylic acid salt + Diamines (polyamides).
Photodegradable under UV light due to carbonyl groups.
How can functional groups in organic molecules be identified?
Alkenes (C=C):
Test: Add bromine water.
Observation: Orange bromine water decolorizes.
Alcohols (-OH):
Test: Add acidified potassium dichromate (VI) and heat.
Observation:
Primary/secondary alcohols: Orange solution turns green (oxidation).
Tertiary alcohols: No reaction.
Carboxylic Acids (-COOH):
Test: Add sodium carbonate or sodium hydrogen carbonate.
Observation: Effervescence due to CO₂ gas.
Phenols (-OH on benzene ring):
Test: Add bromine water.
Observation: White precipitate of 2,4,6-tribromophenol forms.
Aldehydes (-CHO):
Test: Add Tollens’ reagent or Fehling’s solution.
Observation:
Tollens’: Silver mirror forms.
Fehling’s: Blue solution forms a red precipitate.
Ketones (C=O):
Test: Add 2,4-DNPH.
Observation: Orange precipitate forms.
Amines (-NH₂):
Test: Add HCl gas.
Observation: White fumes of ammonium chloride form.
Acyl Chlorides (-COCl):
Test: Add water.
Observation: Vigorous reaction producing HCl fumes.
What are the properties and reactions of arenes?
Properties of Arenes:
Contain a benzene ring (aromatic structure).
Undergo electrophilic substitution reactions.
Reactions of Arenes:
Halogenation:
Reagents: Halogen (e.g., Cl₂) + AlCl₃ catalyst.
Product: Halogenoarene (e.g., chlorobenzene).
Nitration:
Reagents: Concentrated HNO₃ + Concentrated H₂SO₄.
Conditions: Heat to 50°C.
Product: Nitrobenzene.
Friedel-Crafts Alkylation:
Reagents: Alkyl halide (e.g., CH₃Cl) + AlCl₃ catalyst.
Product: Alkylbenzene (e.g., methylbenzene).
Friedel-Crafts Acylation:
Reagents: Acyl chloride (e.g., CH₃COCl) + AlCl₃ catalyst.
Product: Ketone (e.g., acetophenone).
Hydrogenation:
Reagents: H₂ gas + Ni or Pt catalyst.
Conditions: Heat under pressure.
Product: Cyclohexane (loss of aromaticity).
What are the properties and reactions of phenols?
Properties of Phenols:
Contain an -OH group attached to a benzene ring.
Weakly acidic (react with bases).
Reactions of Phenols:
Acid-Base Reaction:
Reagents: NaOH.
Product: Sodium phenoxide + Water.
Reaction with Sodium Metal:
Reagents: Na.
Product: Sodium phenoxide + H₂ gas.
Esterification:
Reagents: Acyl chloride (e.g., CH₃COCl).
Product: Ester (e.g., phenyl ethanoate) + HCl.
Nitration:
Reagents: Dilute HNO₃.
Product: Mixture of 2-nitrophenol and 4-nitrophenol.
Bromination:
Reagents: Bromine water.
Product: 2,4,6-tribromophenol (white precipitate).
What are the properties and reactions of carboxylic acids and their derivatives?
Carboxylic Acids (-COOH):
Acid-Base Reaction:
Reagents: NaOH or Na₂CO₃.
Product: Salt + Water (or CO₂ gas with Na₂CO₃).
Reduction:
Reagents: LiAlH₄ in dry ether.
Product: Primary alcohol.
Esterification:
Reagents: Alcohol + Concentrated H₂SO₄.
Product: Ester + Water.
Acyl Chlorides (-COCl):
Formation:
Reagents: Carboxylic acid + PCl₅, PCl₃, or SOCl₂.
Product: Acyl chloride + Byproducts (e.g., HCl, SO₂).
Reaction with Alcohols:
Reagents: Alcohol.
Product: Ester + HCl.
Reaction with Ammonia:
Reagents: NH₃.
Product: Amide + HCl.
Reaction with Water:
Reagents: H₂O.
Product: Carboxylic acid + HCl.
How can the properties and reactions of organic molecules be predicted?
Step 1 – Identify Functional Groups:
Use chemical tests to identify functional groups (e.g., bromine water for alkenes, Tollens’ reagent for aldehydes).
Step 2 – Predict Properties:
Polarity:
Polar functional groups (e.g., -OH, -COOH) increase solubility in water.
Acidity/Basicity:
Acidic groups: -COOH, phenols.
Basic groups: -NH₂.
Step 3 – Predict Reactions:
Oxidation:
Alcohols → Aldehydes → Carboxylic acids (with acidified K₂Cr₂O₇).
Reduction:
Aldehydes → Primary alcohols (with NaBH₄ or LiAlH₄).
Substitution:
Halogenoalkanes undergo nucleophilic substitution with OH⁻, CN⁻, or NH₃.
Esterification:
Alcohols + Carboxylic acids → Esters (with concentrated H₂SO₄).
How can multi-step synthetic routes be devised for preparing organic molecules?
Step 1 – Identify Functional Groups:
Determine the functional groups in the starting molecule and the target molecule.
Identify the reactions required to interconvert these functional groups.
Step 2 – Compare Carbon Chains:
Check if the number of carbon atoms in the starting molecule matches the target molecule.
If the chain needs to be lengthened, introduce a nitrile group via nucleophilic substitution.
Step 3 – Plan Reaction Pathways:
Use known reactions to convert the starting molecule into intermediates and then into the target molecule.
Ensure reagents and conditions are appropriate for each step.
Step 4 – Optimize Efficiency:
Minimize the number of steps and avoid unnecessary intermediates.
Consider yield and purity of the target molecule.
Example – Synthesis of Ethyl Ethanoate from Ethene:
Step 1: Ethene → Ethanol (Hydration: Steam + H₂SO₄, heat).
Step 2: Ethanol → Ethanoic Acid (Oxidation: K₂Cr₂O₇ + H₂SO₄, reflux).
Step 3: Ethanoic Acid → Ethyl Ethanoate (Esterification: Ethanol + H₂SO₄).
What are the key interconversions between aliphatic functional groups?
Key Reactions:
Alkene → Alcohol:
Reagents: Steam + H₂SO₄.
Conditions: Heat.
Reaction Type: Hydration.
Alcohol → Alkene:
Reagents: Al₂O₃ or concentrated acid.
Conditions: Heat.
Reaction Type: Elimination/Dehydration.
Alcohol → Haloalkane:
Reagents: NaX + H₂SO₄.
Conditions: Heat under reflux.
Reaction Type: Nucleophilic substitution.
Haloalkane → Alcohol:
Reagents: NaOH (aq).
Conditions: Heat under reflux.
Reaction Type: Nucleophilic substitution.
Primary Alcohol → Aldehyde:
Reagents: K₂Cr₂O₇ + H₂SO₄.
Conditions: Distillation.
Reaction Type: Oxidation.
Aldehyde → Primary Alcohol:
Reagents: NaBH₄ + H₂O.
Reaction Type: Reduction.
Carboxylic Acid → Ester:
Reagents: Alcohol + H₂SO₄.
Reaction Type: Esterification.
What are the key interconversions between aromatic functional groups?
Key Reactions:
Benzene → Nitrobenzene:
Reagents: Concentrated HNO₃ + Concentrated H₂SO₄.
Conditions: Heat to 50°C.
Reaction Type: Nitration (Electrophilic substitution).
Nitrobenzene → Aminobenzene (Phenylamine):
Reagents: Sn + HCl.
Reaction Type: Reduction.
Benzene → Bromobenzene:
Reagents: Br₂ + FeBr₃.
Reaction Type: Bromination (Electrophilic substitution).
Benzene → Phenylethanone:
Reagents: CH₃COCl + AlCl₃.
Reaction Type: Acylation (Electrophilic substitution).
Phenylethanone → 1-Phenylethanol:
Reagents: NaBH₄.
Reaction Type: Reduction.
How can a synthetic route be analyzed in terms of reactions, reagents, and by-products?
Step 1 – Identify Reaction Types:
Determine the type of reaction for each step (e.g., oxidation, reduction, nucleophilic substitution).
Step 2 – List Reagents and Conditions:
Specify the reagents and conditions required for each step.
Step 3 – Identify By-Products:
Note any by-products formed during the reactions (e.g., H₂O, HCl, CO₂).
Example – Synthesis of Ethyl Ethanoate from Ethene:
Step 1: Ethene → Ethanol.
Reaction Type: Hydration.
Reagents: Steam + H₂SO₄.
Conditions: Heat.
By-Products: None.
Step 2: Ethanol → Ethanoic Acid.
Reaction Type: Oxidation.
Reagents: K₂Cr₂O₇ + H₂SO₄.
Conditions: Reflux.
By-Products: None.
Step 3: Ethanoic Acid → Ethyl Ethanoate.
Reaction Type: Esterification.
Reagents: Ethanol + H₂SO₄.
Conditions: Heat.
By-Products: H₂O.
What are the key points to remember about synthetic routes and their analysis?
Designing Synthetic Routes:
Identify functional groups in the starting and target molecules.
Compare carbon chains and introduce nitrile groups if needed.
Use known reactions to interconvert functional groups.
Optimize the pathway for efficiency and yield.
Analyzing Synthetic Routes:
Identify the type of reaction for each step.
Specify reagents and conditions.
Note possible by-products.
Key Reaction Types:
Oxidation: Alcohol → Aldehyde → Carboxylic acid.
Reduction: Aldehyde → Alcohol.
Nucleophilic Substitution: Haloalkane → Alcohol or Nitrile.
Esterification: Carboxylic acid → Ester.