H2 Receptor Antagonists and PPIs
1/22
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
23 Terms
What is a peptic ulcer?
What causes it?
Risk factors that predispose someone to it?
What does one feel like?
- Localised erosion of the mucosal membrane (you will see white spots)- pain, irritation; caused by
exposed surface to stomach acid
- Causes: stress, alcohol, NSAIDs and H.pylori
- Potentiated by excess acid in the stomach - heartburn, indigestion, acid reflux
- Burning sensation - discomfort to excruciating pain, hole in abdomen,
mistaken as a heart attack
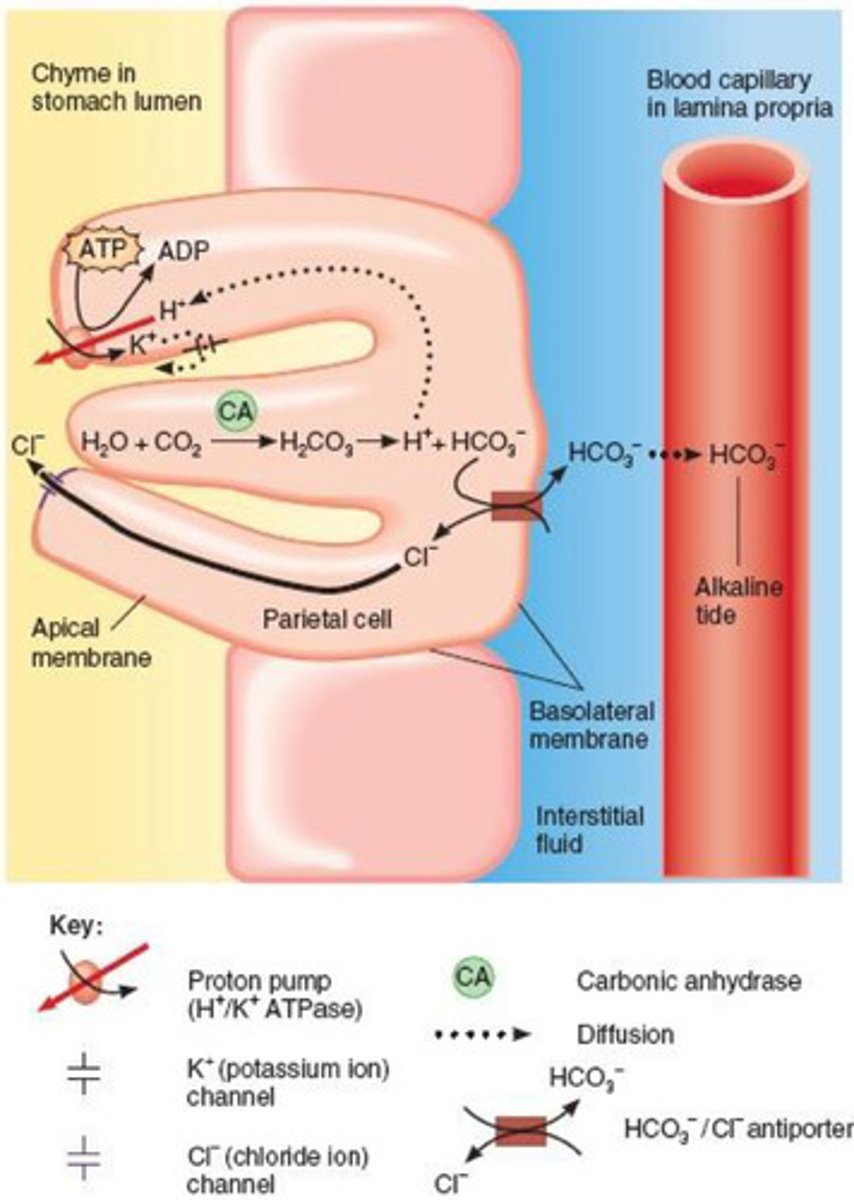
What is the mechanism for how gastric acid is secreted ?
- CO 2 and water combine (via carbonic anhydrase) to produce carbonate which
dissociates into bicarbonate and a proton – this proton is released via H-K-
ATPase pump
- Bicarbonate is actively excreted at the basal side of the cell and is exchanged
for chloride ions
- At the apical side of the cell, potassium ions go in and are exchanged for
protons which come out
- These chloride ions and protons make acid (in the chyme in the stomach lumen)- the H-K ATPase pump releases
the H+
- The HCl acid deactivates ingested bacteria and denatures ingested proteins
to allow for efficient protease reactions
What are the types of cells found in gastric glands in the walls of the stomach?
Several types of cells are found in the gastric glands: parietal, chief, mucus-
secreting, hormone-secreting cells (Gastrin) (some drugs target parietal cells)
What pump does the parietal cells have that is crucial in gastric secretion?
Parietal cells contain a H-K-ATPase pump which lets H+ ions out
How do transmembrane proteins in stomach cells secrete protons?
Via active transport using ATP
What is the concentration and pH of protons in the gastric juice?
can be as high as 0.15
mol/L, giving gastric juices pH <1
What is the concentration of protons within stomach cells ?
40 nmol/L
note: These cells are rich in mitochondria and consume high levels of energy
What are the sites for pharmaceutical manipulation to deal with the excessive acids?
- Antacids are alkaline substances which quickly neutralise acid in the stomach (short term relief as body keep making the acid)
- H2 antagonists reduce the production of histamine in the stomach
- Gastric PPIs inhibit enzymes stimulating acid secretion (therefore inhibit
proton release)
- Antimicrobial agents destroy bacteria which may cause ulcers (for acid being caused by bacteria)
DRUG PROFILE : ANTACIDS
Function
MOA
Examples
Actives
-Rapid pain relief
- Come in gums, lozenges, tablets, powders and liquids: short-term relief only
removing excess acid doesn’t solve underlying problem
- Effectiveness depends on their form and their ability to neutralise the acid
- Sodium carbonate, calcium carbonate, aluminium or magnesium salts
e.g. Gaviscon or Alka-Seltzer
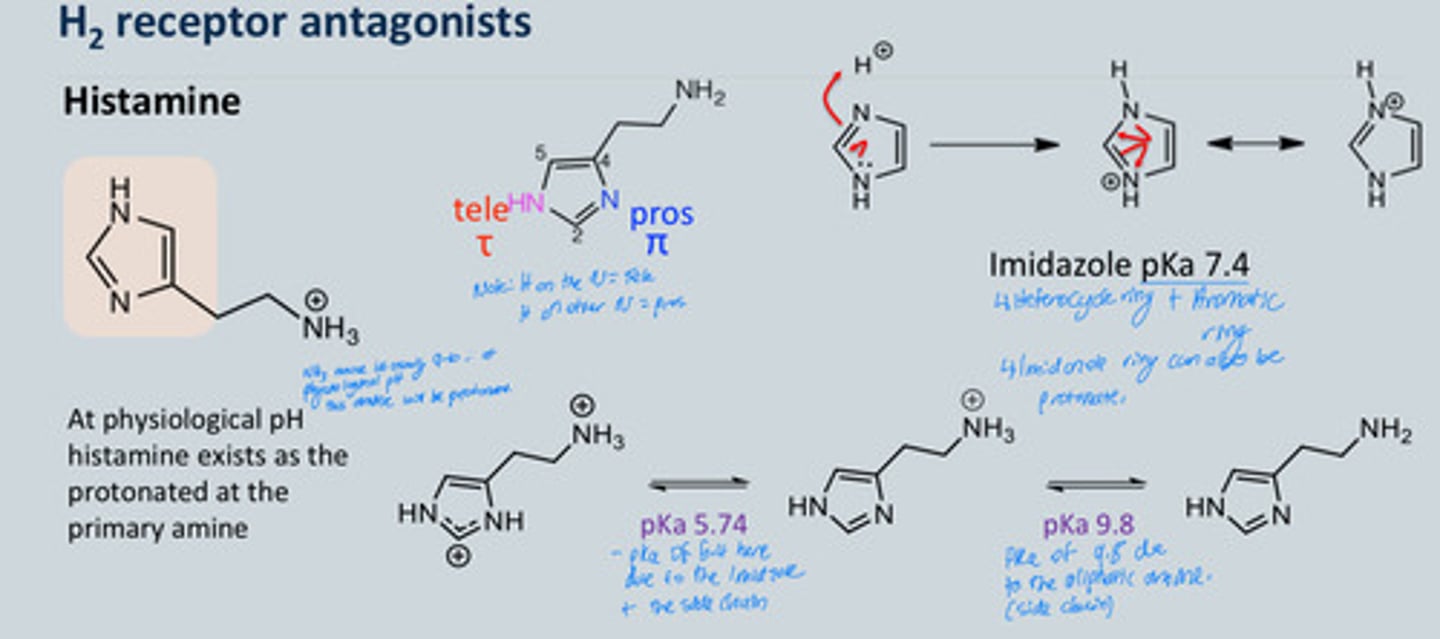
Histamine chemical structure and pka's - what causes the interaction with the H2 receptor specifically?
- At physiological pH, histamine exists as
protonated at the primary amine—the H on the ring can either be in the tele
position or the pros position- this matters because in one way it interacts with
the receptor and doesn’t at the other position
- pKa changes based on side chain e.g., 5.94 on diagram
- About 80% of histamine mono cation (one charge) exists in aqueous solution that binds to
the receptor
- The tele tautomer (H on the N = tele) allows binding with the receptor but not in
the pros position
- Tautomerism is important for H2 interaction and not with H 1 interaction
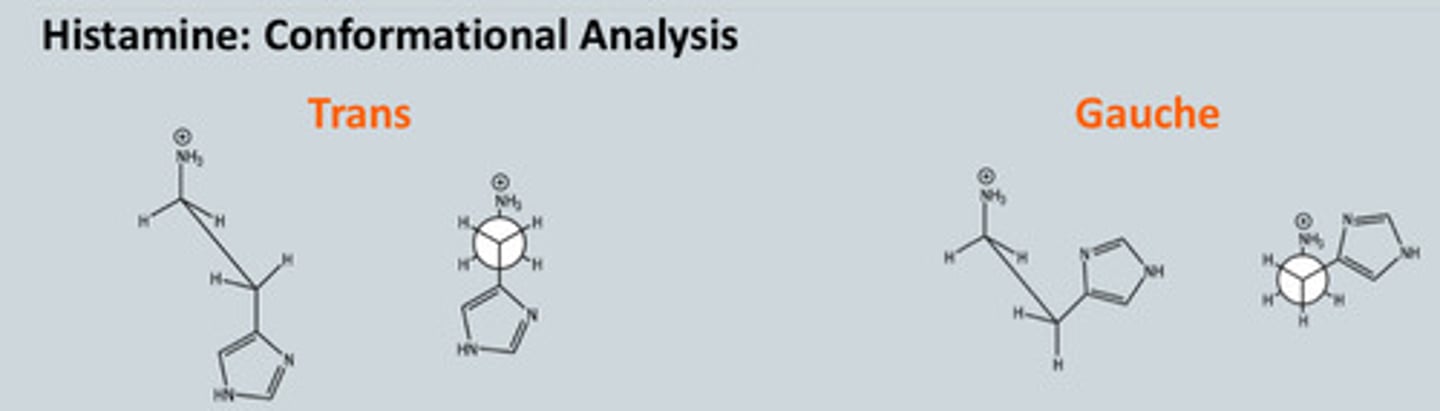
Describe the conformation analysis of Histamine
- The trans conformer has less steric hindrance but the gauche conformer
is stabilised by an ion-dipole interaction because we have ion and aromatic
ring
- Both conforms exist in solution
- It is believed that both H1 and H2 receptors bind to the trans conformer- this is
based on the observation that alpha and beta methyl histamine are unable to
assume the trans conformation and are weak agonists at both the H1 and H2
receptors
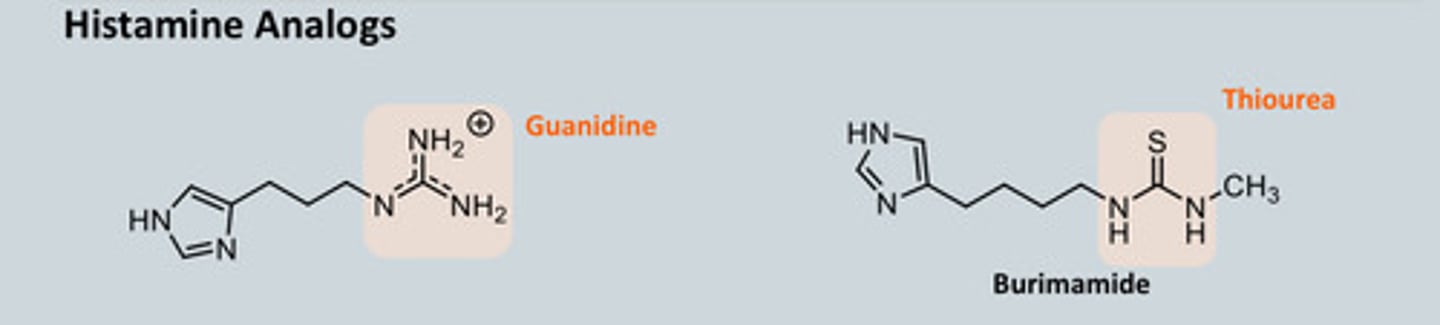
Describe how we synthesised Histamine Analogues
-Aim is to block the H2 receptor! (As histamine usually binds to the H2 receptor to release gastric acid)
- The Guanidine analogue was synthesised and tested as an antagonist but
was found to be an agonist instead which stimulated acid secretion rather
than blocking
- Replace Guanidine with Thiourea = guanidine is really basic which picks up
a proton but thiourea is less basic
- Replacing C=N with C=S gives Thiourea = the pKa drops to 7.5 and this
compound acts as an antagonist
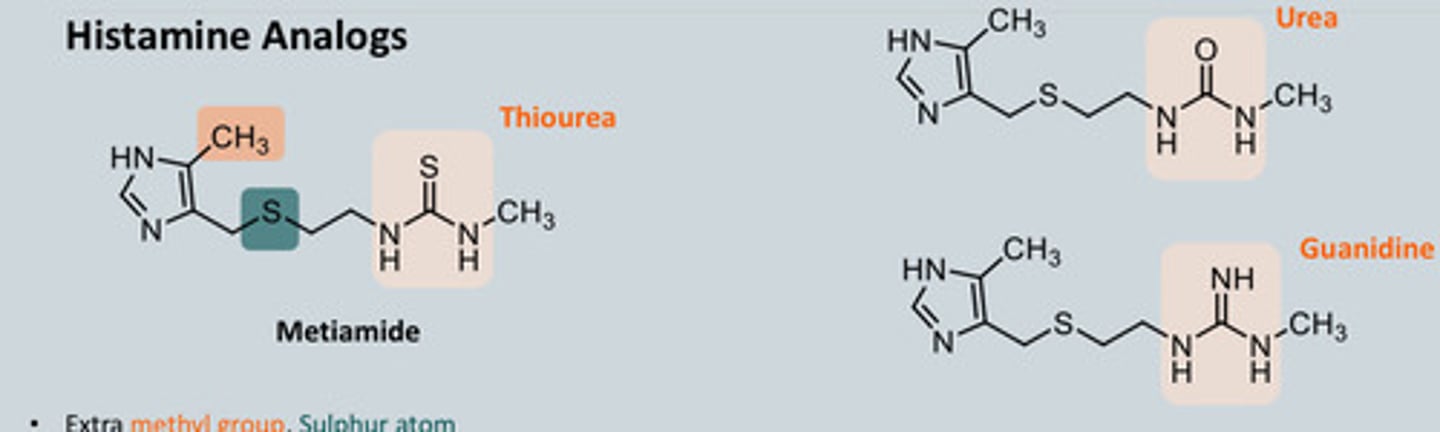
- But H in wrong position so now make Metiamide which has methyl
group which pushes electrons into the ring and the S has an inductive
effect on the N of the ring as it is more electronegative than C = Electron Withdrawing Group (EWG)
induction effect
- Metiamide 10x more active than Burimamide but had some toxicity due to
thiourea moiety - decrease in No. of WBC's = more susceptiple to infections ... we needed to still develop this analogue
- We tried replacing the Thiourea group of metiamide with Urea and Guanidine (which no longer showed agonist effect as this guanidine will be fully protonated) but neither was as effective as metiamide
Now what do we do? - Key thing is to look at pKa values
Memitiamide structure
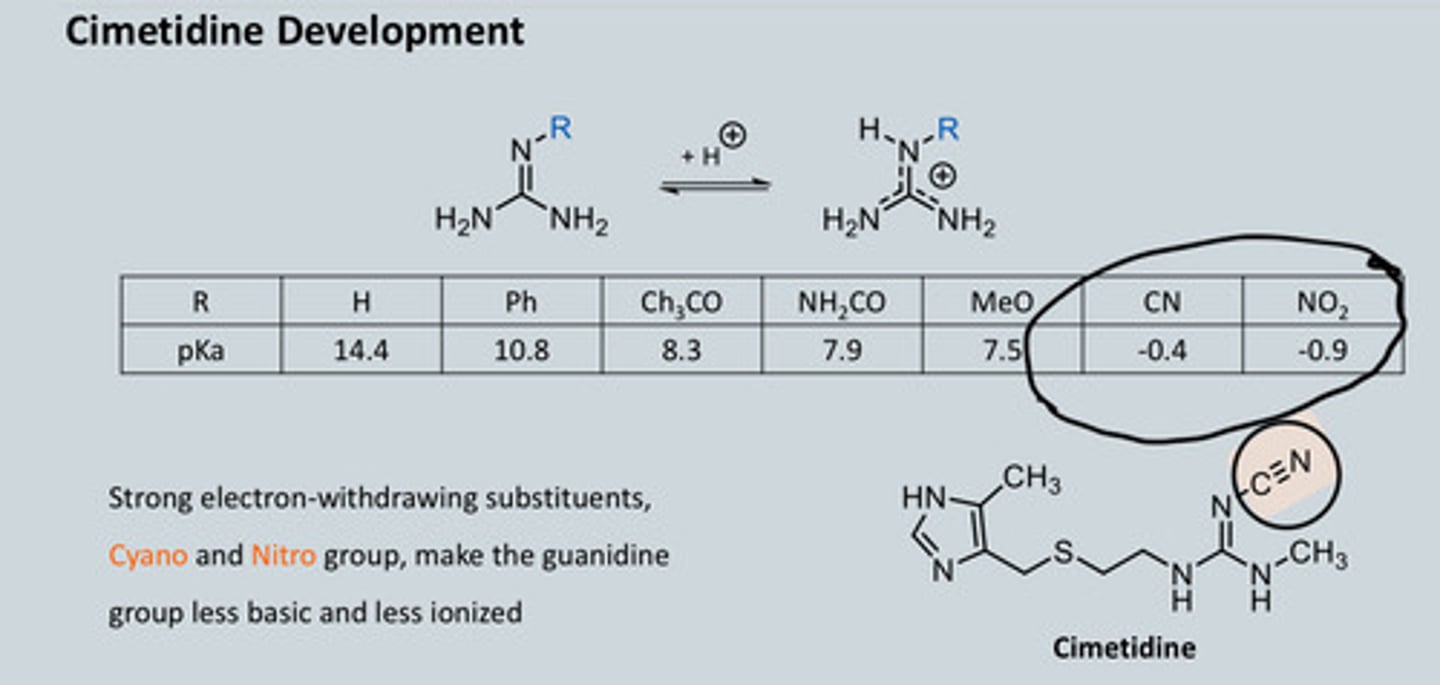
Describe how we synthesised Histamine Analogues part 2. Cimetidine Development
- CN (triple bond) is EWG = gives pKa of 0
- To change the properties of a drug, look at
functional group and see what happens if
they are changed
- Want something less basic as possible
- Keep rest of molecule the same but add -CN bond = Cimetidine
- Nitro group, NO 2 , is also EWG
-Strong electron-withdrawing substituents, Cyano and Nitro group, make the guanidine group less basic and less ionized
Ranitidine and Famotidine Drug profile
Ranitidine:
• Furan derivative, an isostere of the imidazole with an electrons on the oxygen, with 50% bioavailability
• 4 -10 times more potent than cimetidine with fewer side effects
• Longer duration of action and weaker CYP inhibitor
Famotidine:
• Thiazole derivative • 40 - 60times more potent than Cimetidine and weak inhibitor of CYP
• 40 – 50% bioavailability due to incomplete absorption
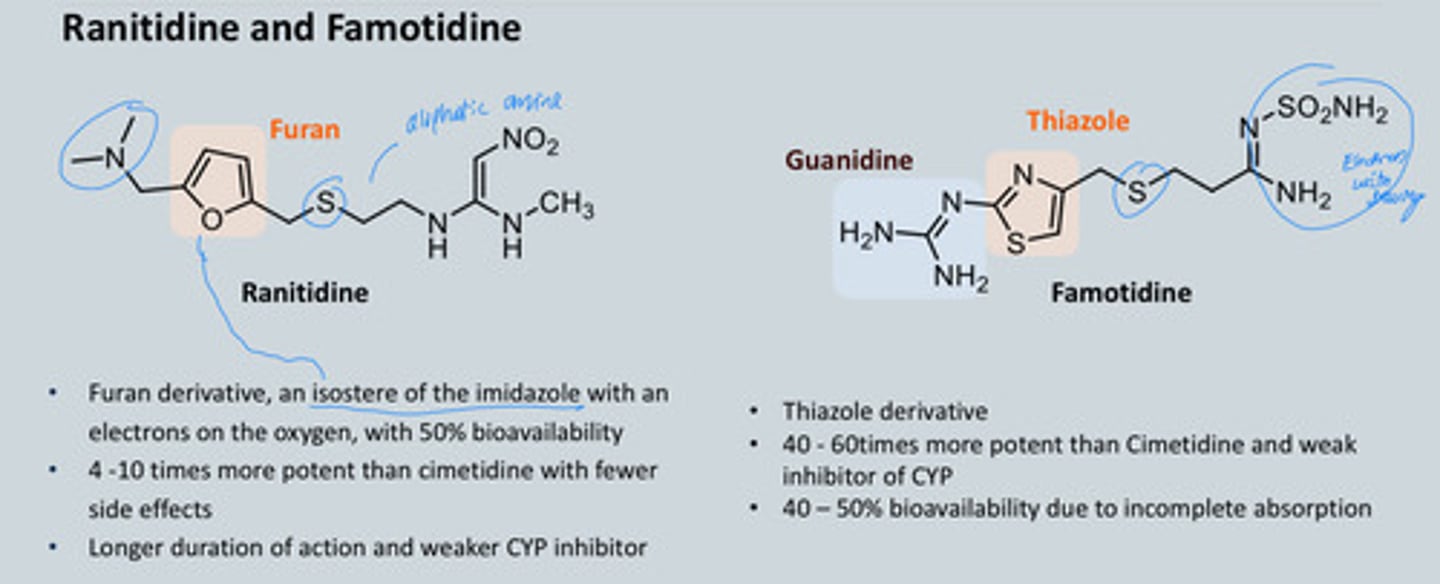
Why did Ranitidine get withdrawn?
Impurity in the synthesis = toxicity
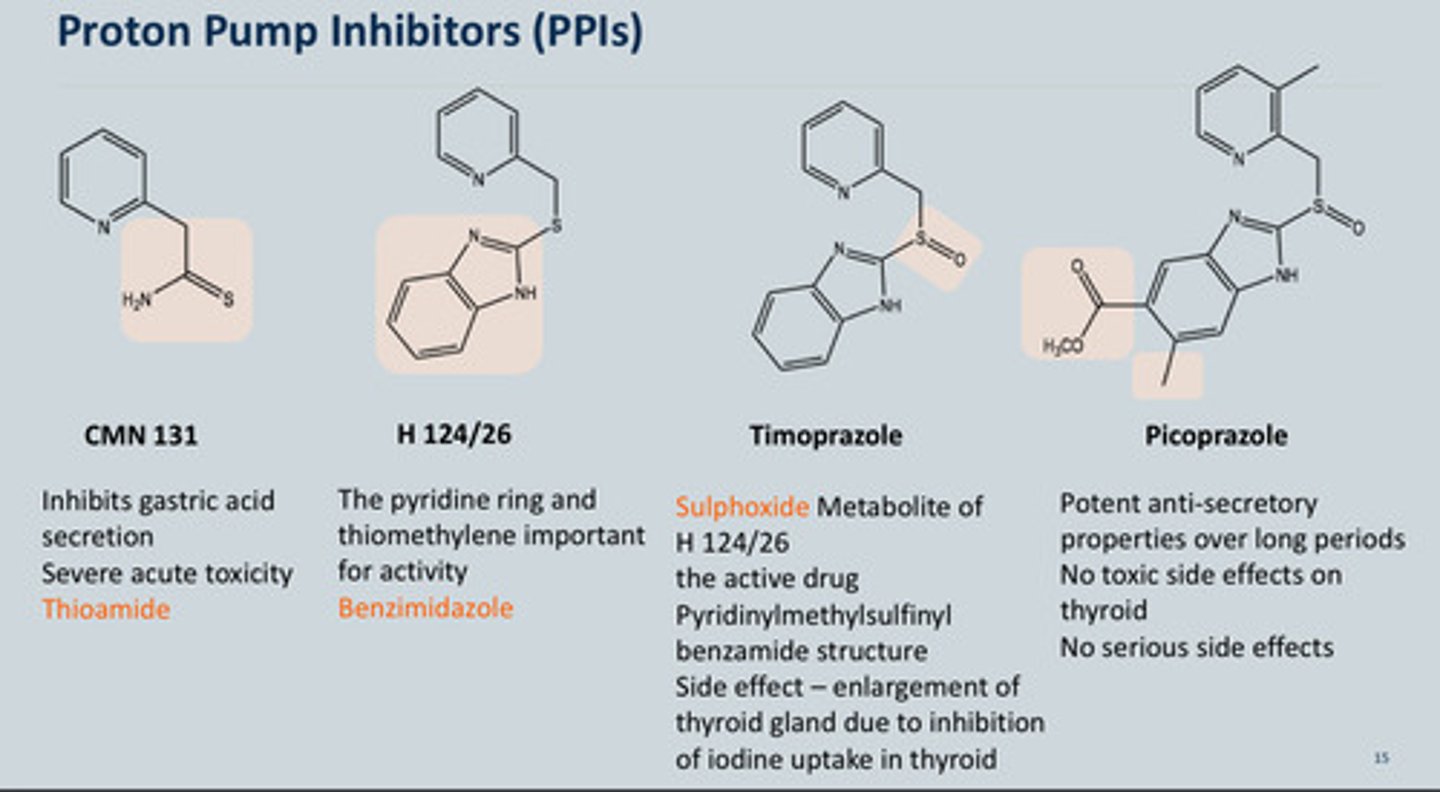
How were Proton pump inhibitors development
-Initially wanted to replace the Thioamide group in CMN 131 due to the Severe acute toxicity
-Replaced with a Benzimidazole of H 124/26
-Timoprazole is a metabolite of H 124/26 which has a Sulphoxide that can also be used as a PPI but it has a Side effect – enlargement of thyroid gland due to inhibition of iodine uptake in thyroid - not good
-Final one is Picoprazole with the addition of a methyly and a CH3OCO group on the aromatic ring, these had:
Potent anti-secretory properties over long periods No toxic side effects on thyroid No serious side effects
OMEPRAZOLE DRUG PROFILE: genral when and where in the body is it active and does it work
- Omeprazole is a weak base that accumulates in acidic areas. Substituents were added to its pyridine ring to adjust the pKa. his adjustment helps maximize accumulation in parietal cells (the cells that produce stomach acid), increasing its effectiveness
- Can be anywhere in body but will remain inert; it needs to be in the stomach
and needs the acid- when acid protonated the drug starts to change
- We initially thought that due to Lifelong toxicological studies at very high doses in rats revealed the endocrine
tumours in the stomach - we thought they were toxic
- This halted clinical studies until it was shown that tumours were due to very
high doses causing severe suppression
- Now we only use 20 mg dose
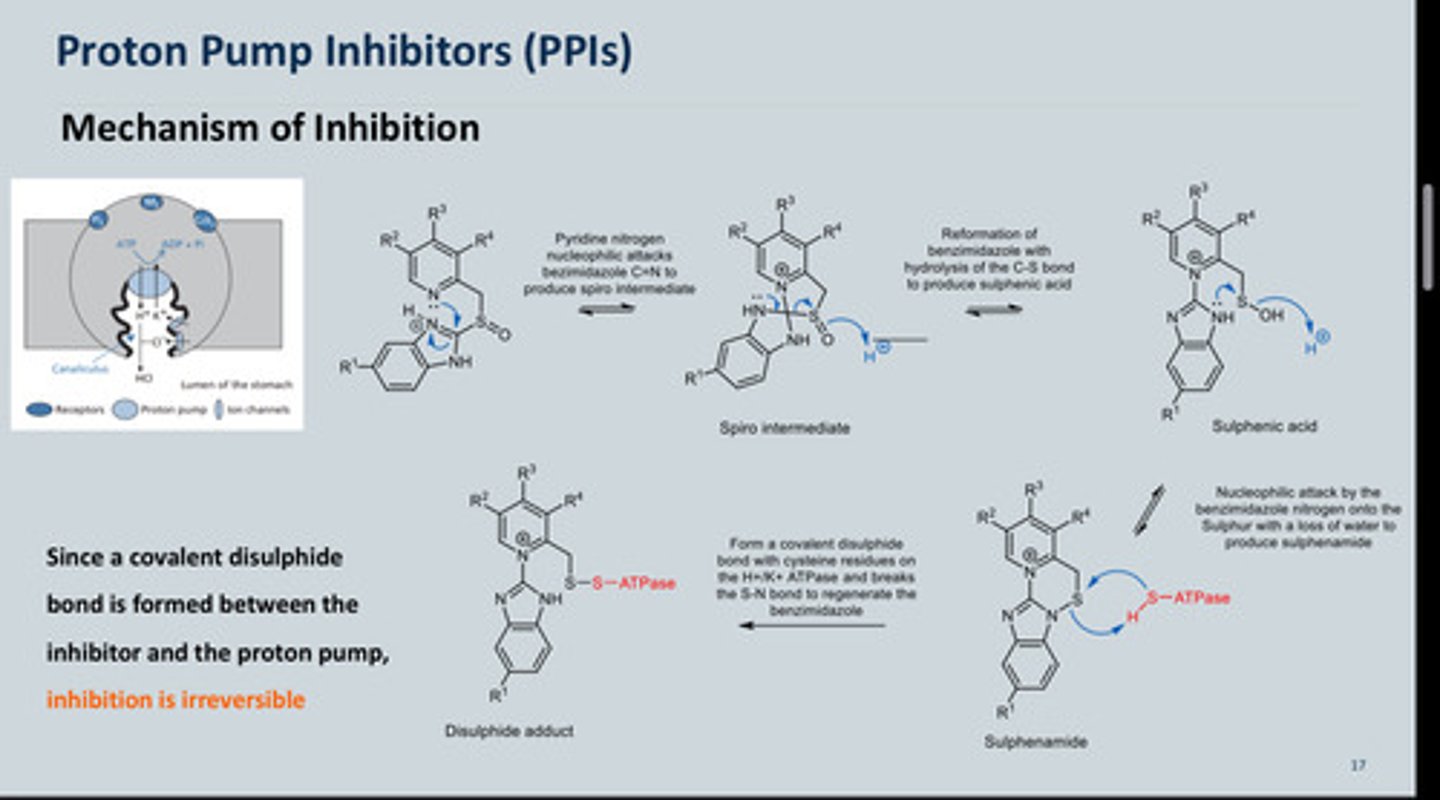
MOA of omeprazole
- Omeprazole is a prodrug and is protonated within the acidic canaliculi of the
parietal cells into the active form, a sulfenamide
- Accumulates in the stomach, once protonated the N Nu-attacks the C of C=N
= spiro intermediate
- Spiro intermediate opens up = break the bond between C-S and the S=O
picks up a proton as there
are many available forming Sulphenic acid
- The N will attack the S and
kick out OH but OH will pick
up a proton while leaving
- None of the intermediates
will inhibit acid secretion at
this point
- The ATPase pump has an -SH group which reacts with the S on (4) = now
ATPase pump will be stuck onto molecule (5) so cannot release acid = acid secretion is
inhibited
- This sulfenamide (the active compound) reacts with thiol groups in the
enzyme forming a disulphide link which inactivates the enzyme- this high
specificity of action is due to several factors
- N has to be protonated for mechanism to initiate (irreversible inhibition of the
ATPase pump)
What are the properties of Omeprazole - why does it have high specificity of action?
- Omeprazole is a weak base (pKa 4) - all PPI's are weak bases - therefore concentrates in acidic canaliculi
of the parietal cells
- The low pH causes the conversion into the active species close to the target
enzyme
- At higher pHs found in the body, omeprazole has good stability
- Commercial products are enteric coated to prevent gastric decomposition
Fun fact: Omeprazole has no optically active centre
What determines the rate of enzyme inhibition in PPI's
the rate of sulphenamide formation
What Two factors determine the extent to which PPI's accumulate in the canalicular lumen?
The pKa and the hydrophobicity of the PPIs
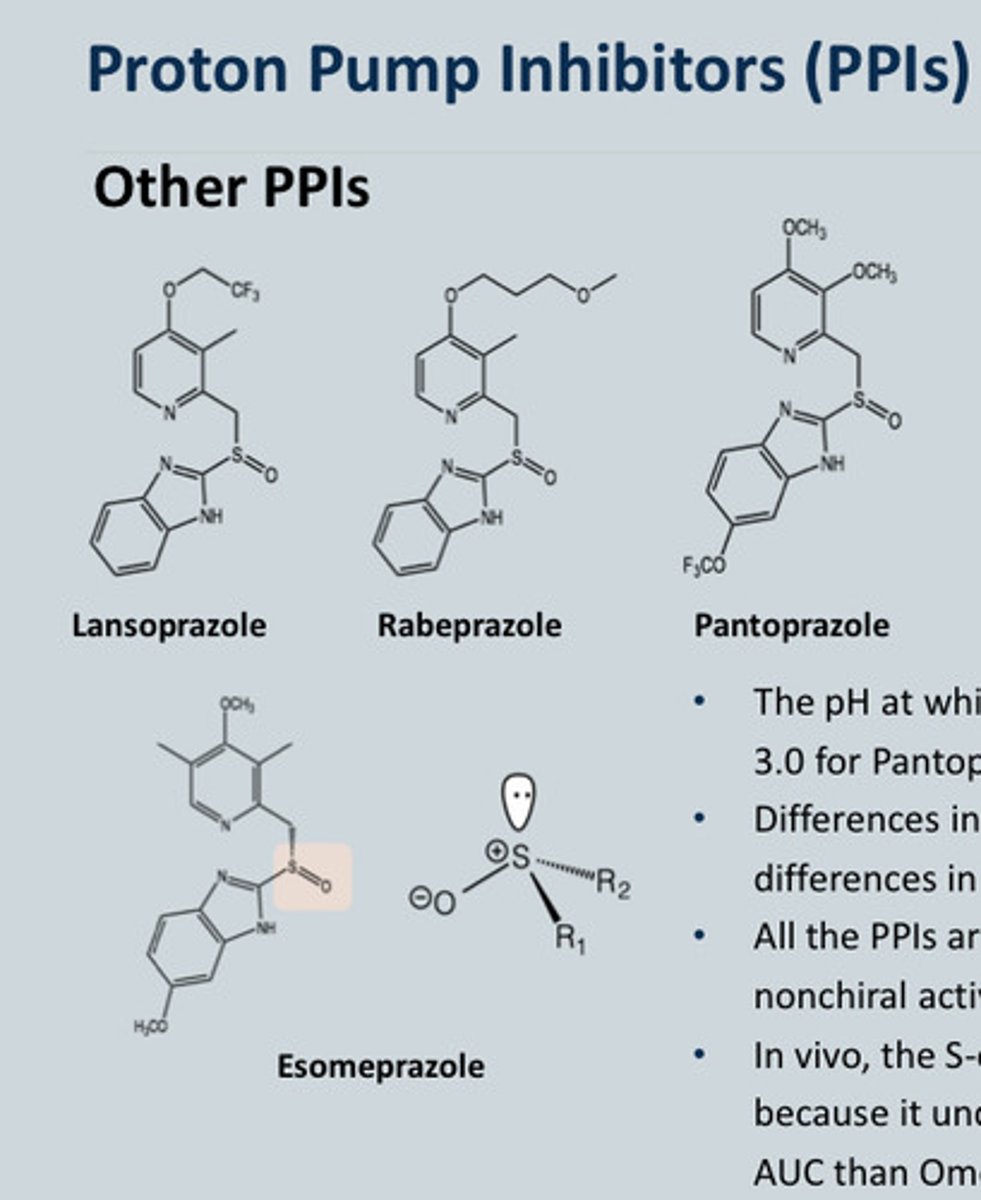
List some other PPI's and what are their properties - Why did Esomeprazole get licensed
-Pantoprazole
-Lansoprazole
-Rabeprazole
-Esomeprazole
Properties:
• The pH at which half of the maximum rate of activation occurs is 3.0 for Pantoprazole, 4.0 for Omeprazole and 5.0 for Lansoprazole.
• Differences in structure result in differences in pharmacokinetics, however, differences in oral bioavailability are not clinically important.
• All the PPIs are chiral because of the Sulphur, both isomers are converted into the nonchiral active species at the same rate.
Omeprazole vs Esomeprazole:
• In vivo, the S-omeprazole (esomeprazole) produced higher plasma concentrations because it undergoes less metabolism by CYP 2C19 and thus produces 70% higher AUC than Omeprazole
-Esomeprazole is the chiral compound of omeprazole (Shown on diagram)