BIOL0510: Prions
1/37
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
38 Terms
What are prions? Define them and explain what they are
name: proteinaceous infectious particles (PrP)
what: Prp are misfolded, resulting in protein aggregation/clumping that leads to neurodegeneration of the CNS
What are the 4 major properties of prions?
don’t have nucleic acids
they’re only proteins
resistant to heat, chemicals, and decomposition
can cause infectious, genetic, or sporadic disorders
disease results from the accumulation of PrPsc protein (infectious isomer of normal PrPC)
What are TSEs?
transmissible spongiform encephalopathy: infectious diseases (T) leading to brain vacuolation, aka holes, (S) and a lack of immune response (E)
What makes TSEs and other neurodegenerative diseases similar?
they both are characterized by disease-causing plaques in the brain
What makes TSEs different from other neurodegenerative diseases?
TSEs
are infectious
caused by prions
Other diseases
non-infectious
caused by unique proteins
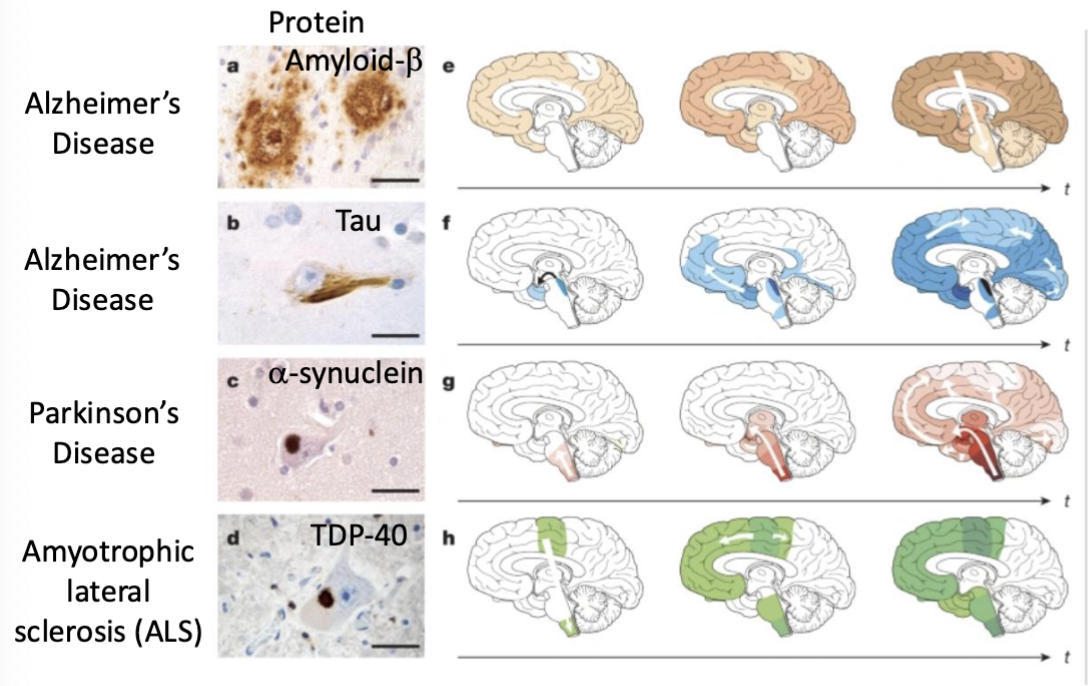
What causes Alzheimer’s, Parkinson’s, and Amyotrophic lateral sclerosis (ALS)?
Alzheimer’s: amyloid beta and tau misfolding
Parkinson’s: alpha-synuclein
Amyotrophic lateral sclerosis: TDP-40
Explain what makes neurodegenerative diseases distinct from each other
different proteins/mutations arise
leads to different protein misfolding, affecting different areas of the brain
different symptoms manifest
Summarize Scrapie (what, organism, route of infection, discovery, symptoms)
what: the first discovered prion disease
organism: sheep
route of infection: N/A
discovery: 1750s
symptoms: scraping, bite their feet and legs, eventually die, brain vacuolation (holes)
How did scientists in 1939 test and transmit scrapie?
experimentation: inoculated brain or spinal cord tissue from an affected sheep into 2 healthy sheep
result: sheep developed scrapie 1-2 years after inoculation
What caused the scrapie outbreak in 1939? How was it treated?
a vaccination against looping illness
treatment: formaldehyde (but it wasn’t strong enough to kill the prions)
Summarize Kuru (what, organism, route of infection, discovery, symptoms, survival)
what: Fore tribe disease common among women and children
organism: humans
route of infection: cannibalism (eating brain and spinal cord tissue)
acquired prions
discovery: 1950s
symptoms: shivers, ataxia, weight loss
survival: 9-24 months
What did Gajdusek do?
noticed cannibalism in Fore tribe, brain vacuolation in dead members
infected chimpanzees with Kuru and waited for them to die around 20-24 months later
proved prions were infectious
What did Hadlow do?
the vet that made the connection between brain vacuolation in Kuru and Scrapie
What did Alper do?
the radiologist who proved that prion disease lacks nuclei acids by showing that scrapie was not sensitive to UV radiation (everything with DNA is sensitive to UV)
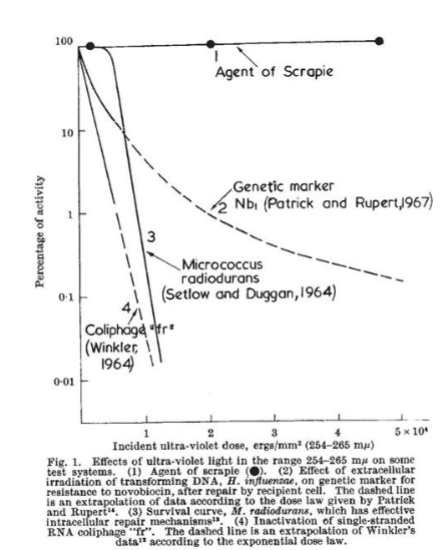
What 2 things did Pruisner do and how?
showed how resilient prions are
how: added DNAse, RNAse, viral inactivators, and proteases to brain slurry samples —> inoculated mice —> disease still occurred
won Nobel for discovering and purifying PrPsc protein
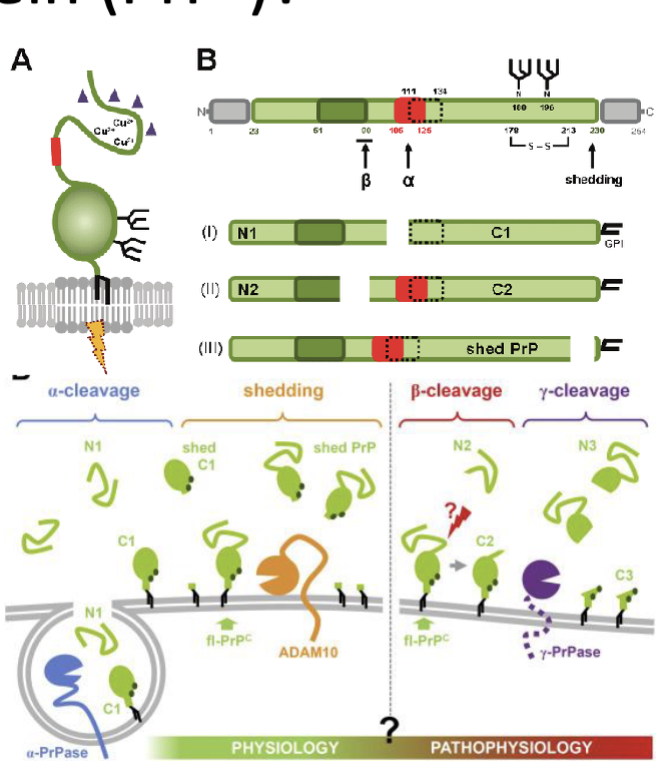
What is Prp? (length, parts, linkage, location)
250 amino acids
2 parts: Prp/PrpC
linked via GPI anchor to outside of membrane
found in all mammals (on human Chr 20)
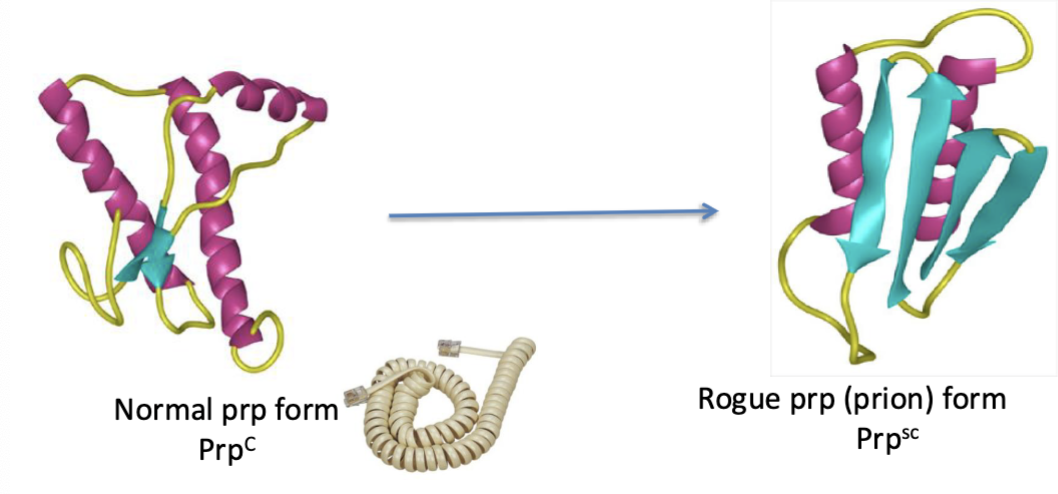
What is the normal function of cellular prion protein? (PrpC)
expressed in a variety of organs and tissues
high expression levels in the CNS and PNS
resides extracellularly in lipid rafts, where it is attached to a glycosyl phosphoinosityl anchor (GPI)
undergoes endocytosis and cleavage
knockout mice show unclear function
What type of infection are prion infections considered?
SLOW; unconventional agents
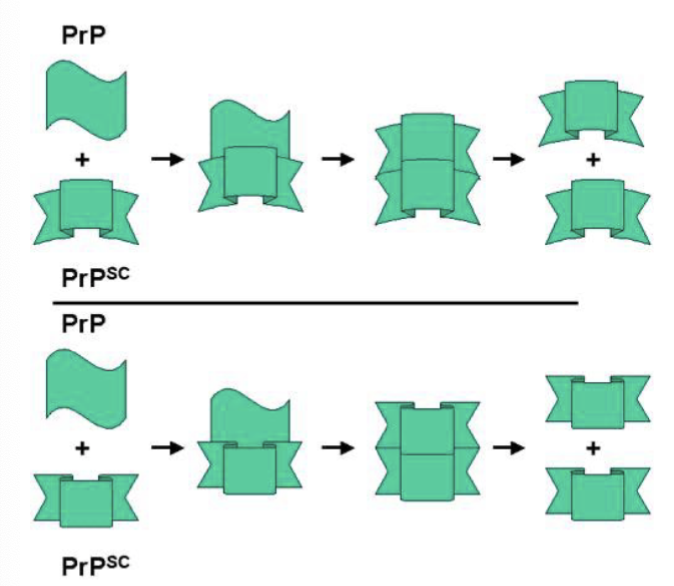
What is the prion theory?
prions spread silently across a person’s brain for years without causing any symptoms
the conversion of PrP to PrPsc increases
prions aren’t destroyed by the cell; they accumulate, eventually causing cell death
prions move on to another neuron to begin the cycle again
once PrPsc concentration is high enough, noticable symptoms strike, and the patient rapidly declines
Describe the timeline of prion disease (5 steps)
prions form (3 ways)
prion recruitment: prion infects other PrPc to make more prions
amyloid plaques form: prion aggregates, creating amyloid fibers —> amyloid plaques
neurons are killed (brain holes form): amyloid plaques destroy neurons, creating holes in the brain (vacuolation)
clinical symptoms present: anxiety, dementia, loss of motor function
What are the 3 methods for forming prions? Categorize the 7 human prion diseases respectively
spontaneous/idiopathic: folding issues arise overtime
ex. classical CJD, VPSPr
acquired: prion acquired from other organism’s tissues
ex. Kuru, vCJD
genetic/familial: genetic mutation increases likelihood of misfolding
ex. Fatal Familial Insomnia, (genetic)CJD, GSS
What are amyloid plaques?
misfolded, insoluble, protease-resistant protein aggregates w/ distinct staining properties
cause over 20 neurodegenerative diseases
some proteins have greater potential to misfold
What proteins are most likely to aggregate for each popular neurodegenerative diseases?
the amyloids for each disease also aggregate in different areas in the brain, perhaps explaining the different initial symptoms associated with each disease (ex. parkinson’s = motor problems, alzheimer’s = memory loss)
What are the 5 human prion diseases we are studying?
Kuru
Creutzfeldt-Jakob disease
Fatal Familial Insomnia
Gerstmann-Straussler-Scheinker syndrome
VPSPr
Why are there different forms of prion diseases?
different mutations within Prp will convert the normal conformation into unique structural forms of PrpSC, initiated in different locations in the brain
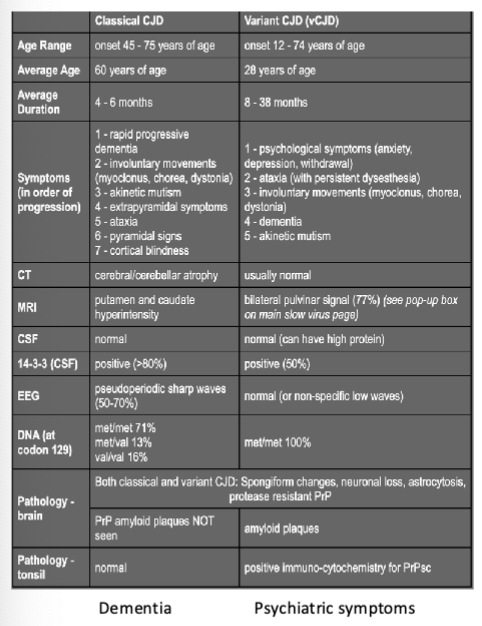
Summarize Classical Creutzfeldt-Jakob Disease (what, organism, route of infection, discovery, symptoms, survival)
what: most common prion disease
organism: humans
route of infection:
sporadic/idiopathic
genetic predisposition
eating beef with mad cow disease (vCJD: 20-30 y/o’s, 8-38 month survivial)
iatrogenic (hospital acquired via brain surgery tools)
discovery: 1920, Austria
symptoms:
classical: dementia
variant: pyschological
both: anxiety/depression, dementia, ataxia, coma
survival: 4 months
What are the three major differences in classical versus variant CJD?
duration
age range
main symptoms
Summarize fatal familial insomnia (organism, route of infection, symptoms, survival time)
organism: humans
route of infection: genetic (codon 178 mutation)
symptoms: plaques form in the hypothalamus, affects circadian rhythm, deadly insomnia
survival time: 18 months
Summarize Gerstmann-Straussler-Sheinker Syndrome (GSS) (what, organism, route of infection, symptoms, and survivial)
what: subclass of CJD
organism: humans
route of infection: genetic, autosomal dominant (a single copy of this mutation on an autosomal chromosome will cause GSS)
50% of patients have codon 102 mutation
disrupts and removes GPI anchor
symptoms: motor issues, anxiety, dementia
survival: 5 years
Summarize variably protease-sensitive prionopathy (VPSPr) (what, organism, route of infection, discovery)
what: newly discovered prion disease
organism: humans
route of infection: sporadic OR somatic (body cell) mutation in codon 129
discovery: 2006
What are the animal prion diseases we discussed?
scrapie
chronic wasting disease
Bovine spongiform encephalopathy (mad cow disease)
Summarize Bovine Spongiform Encepalopathy (BSE) (what, organism, route of infection, discovery, symptoms)
what: TSE in cows
organism: cows
route of infection: cow feed
acquired
discovery: 1980s
symptoms: nervousness, aggression; eventually inability to stand
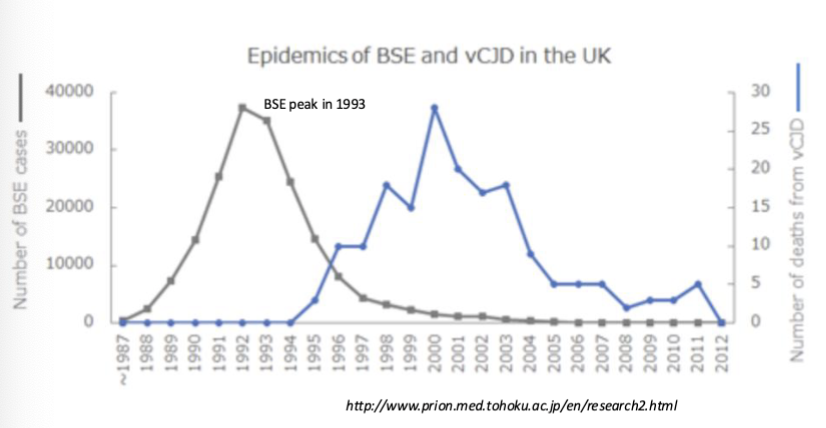
What did farmers do that unintentionally spread BSE and, eventually, vCJD?
added ruminants (brain material) to cow feed
cows to develop Mad cow disease
people that ate the cows developed vCJD years later
Summarize chronic wasting disease (organism, route of infection, discovery, symptoms)
organism: deer
route of infection: feces, body fluids, aerosols
acquired (HIGHLY CONTAGIOUS and COMMON)
discovery: 1967
symptoms: motor issues, weight loss
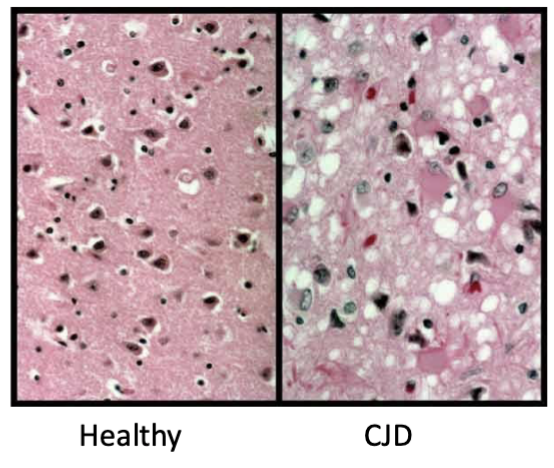
How are prion diseases diagnosed definitively? What is observed?
from looking directly at brain tissue post-mortem
observation: brain vacuolation
List 6 ways non-definitive prion disease diagnoses made, and the catch-22 associated with each
clinical presentation of familial history (most common)
EEG (changes in brain activity)
cerebrospinal fluid tests (shake samples to amplify prions)
brain biopsy (MIGHT catch a prion)
MRI (only certain diseases show up)
peripheral lymphoid tissue sample (look for PrPsc, somtimes works)
What makes fungi prions special?
contain proteins that can readily change conformation based on the organism’s needs/environment
What 2 characteristics are associated with fungal prions?
widespread (many fungi have them)
they’re non-pathogenic to fungi, yet are still infectious
at least 1 conformation is self-propagating (flips other proteins)