Chapter 4: protein methods
1/122
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
123 Terms
how can proteins be purified?
- performed by subjecting an impure mixture of starting material to a series of separations based on physical properties such as size and charge
- requires a test, or assay, that determines whether the protein of interest is present
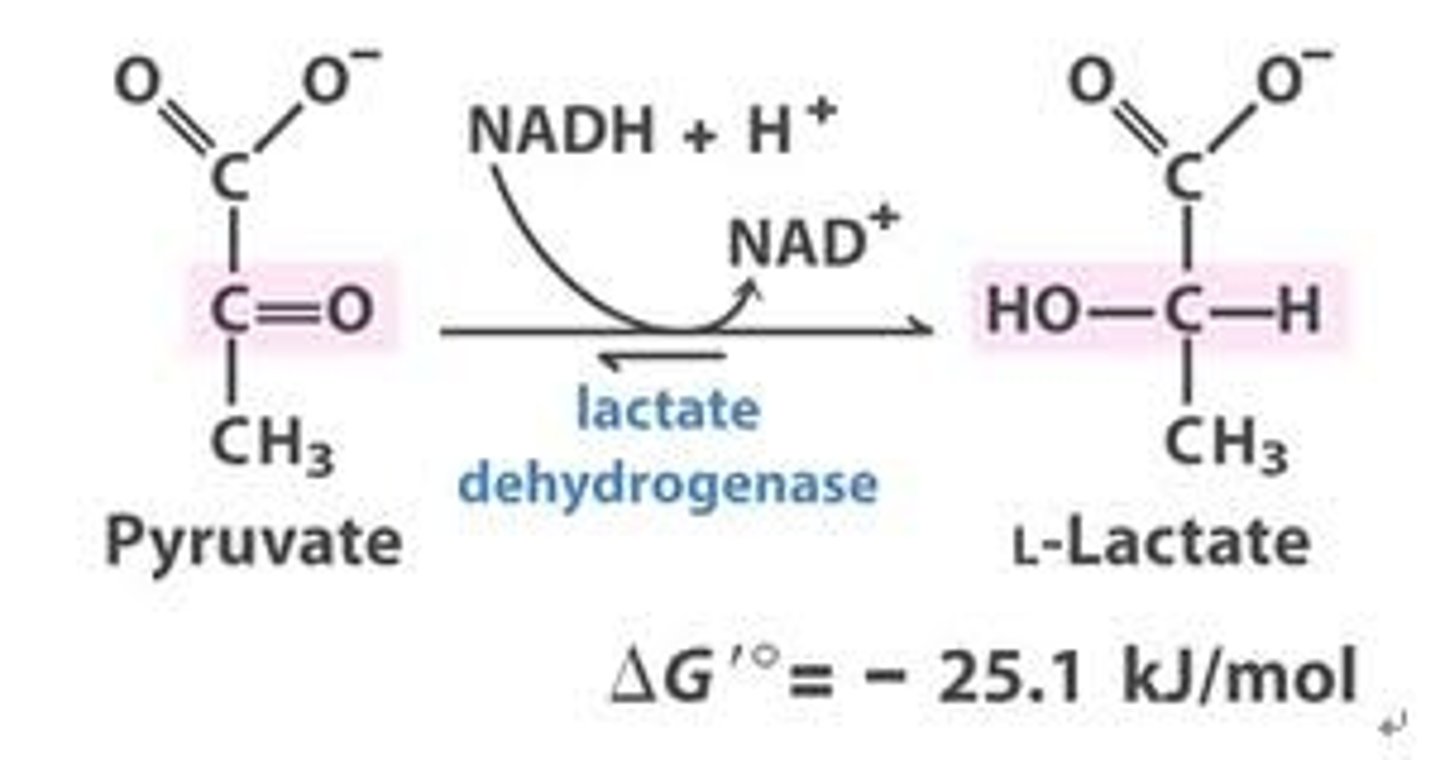
assay for lactate dehydrogenase
An assay for the enzyme lactate dehydrogenase detects NADH spectrophotometrically
how do you analyse a purification scheme?
the amount of protein in a mixture being assayed must be known
The overall goal of the purification is to maximize the specific activity
specific activity
the ratio of enzyme activity to the amount of protein mixture
steps for proteins to be released from the cell during purification
step 1: disrupt the cell membranes of intact cells to forma homogenate
step 2: centrifuge the homogenate at low speed to yield a pellet consisting of heavy material and lighter supernatant•
step 3: centrifuge the supernatant at a higher centrifugal force to yield another pellet and supernatant
- This process of differential centrifugation is repeated many times to yield several fractions of decreasing density.
- One fraction will be enriched for the desired activity.
differential centrifugation
Procedure for separating cellular components according to their size and density by spinning a cell homogenate in a series of centrifuge runs. After each run, the supernatant is removed from the deposited material (pellet) and spun again at progressively higher speeds.
salting out
effect by which most proteins are less soluble at high salt concentrations
the salt concentration at which a protein precipitates differs from one protein to another.
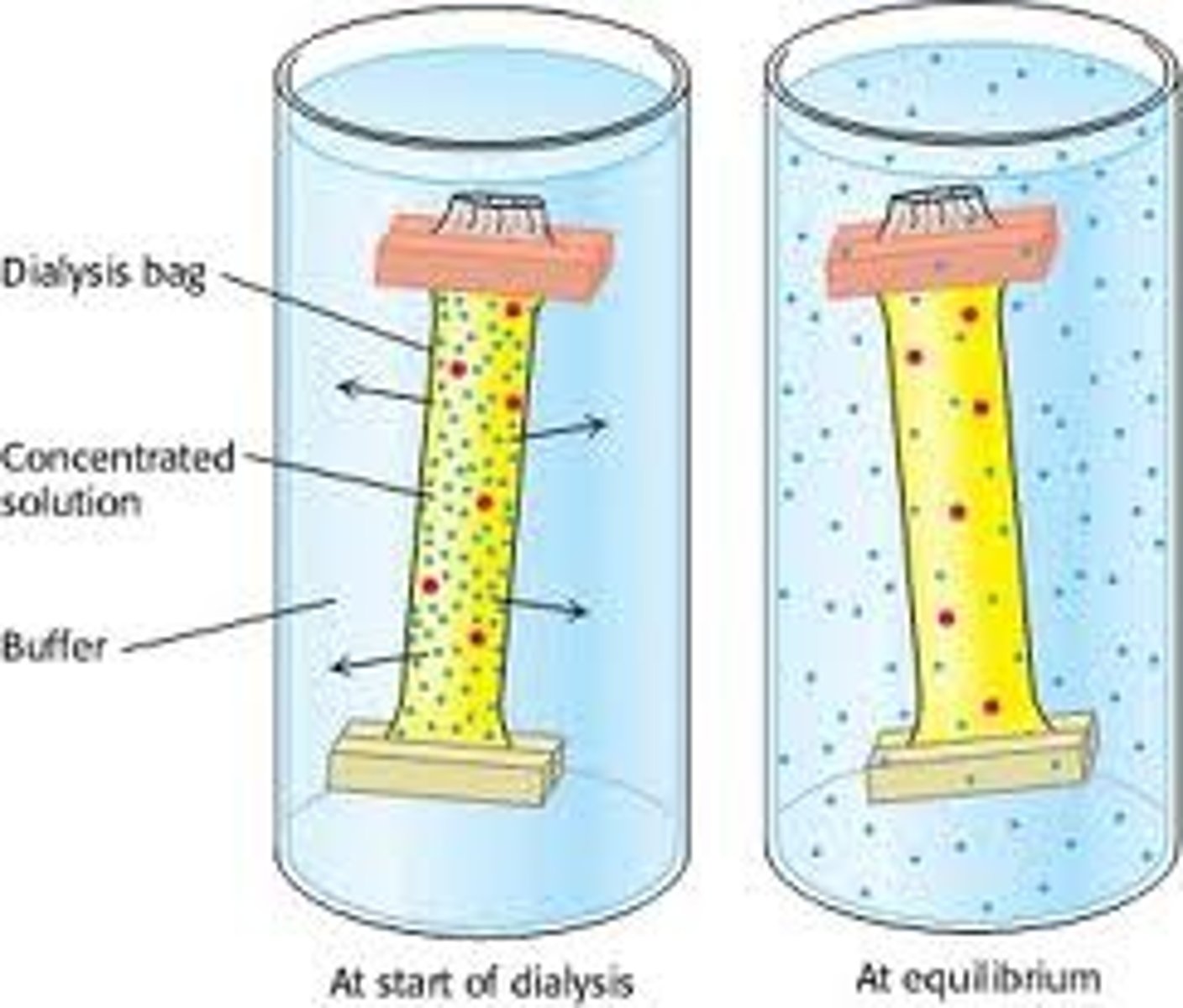
dialysis
Proteins can be separated from small molecules (e.g.,salt) by dialysis through a semipermeable membrane, such as a cellulose membrane with pores.
• Molecules larger than the pore diameter remain inside the dialysis bag.
• Smaller molecules and ions diffuse down their concentration gradients and emerge in the solution outside the bag
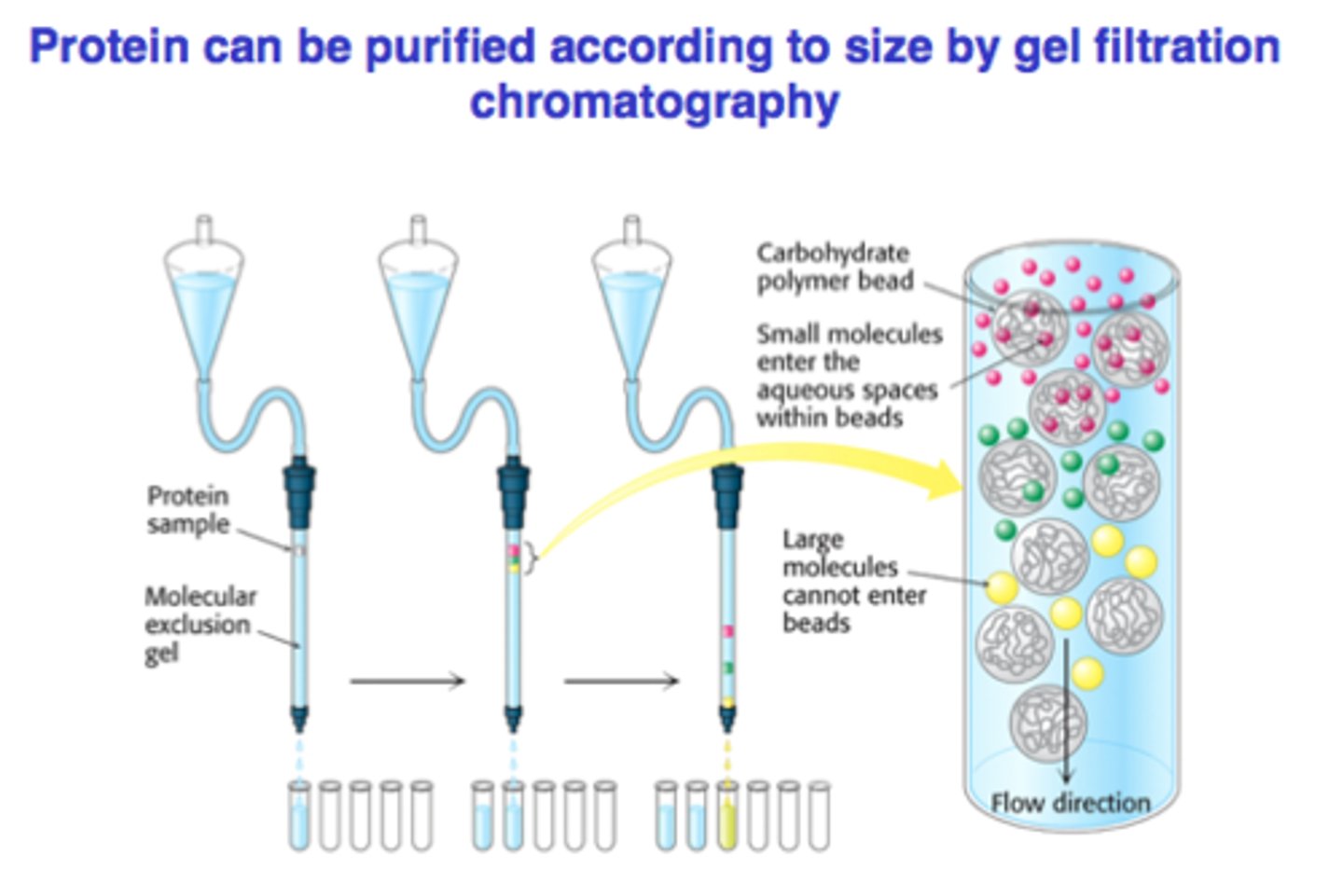
gel filtration chromatography
seperates proteins based on size
A column is filled with porous beads, and the sample is applied to the top of the column.•
When a protein solution is passed over the beads, large proteins cannot enter the beads and exit the column first.
• Small proteins enter the beads and exit the column last.
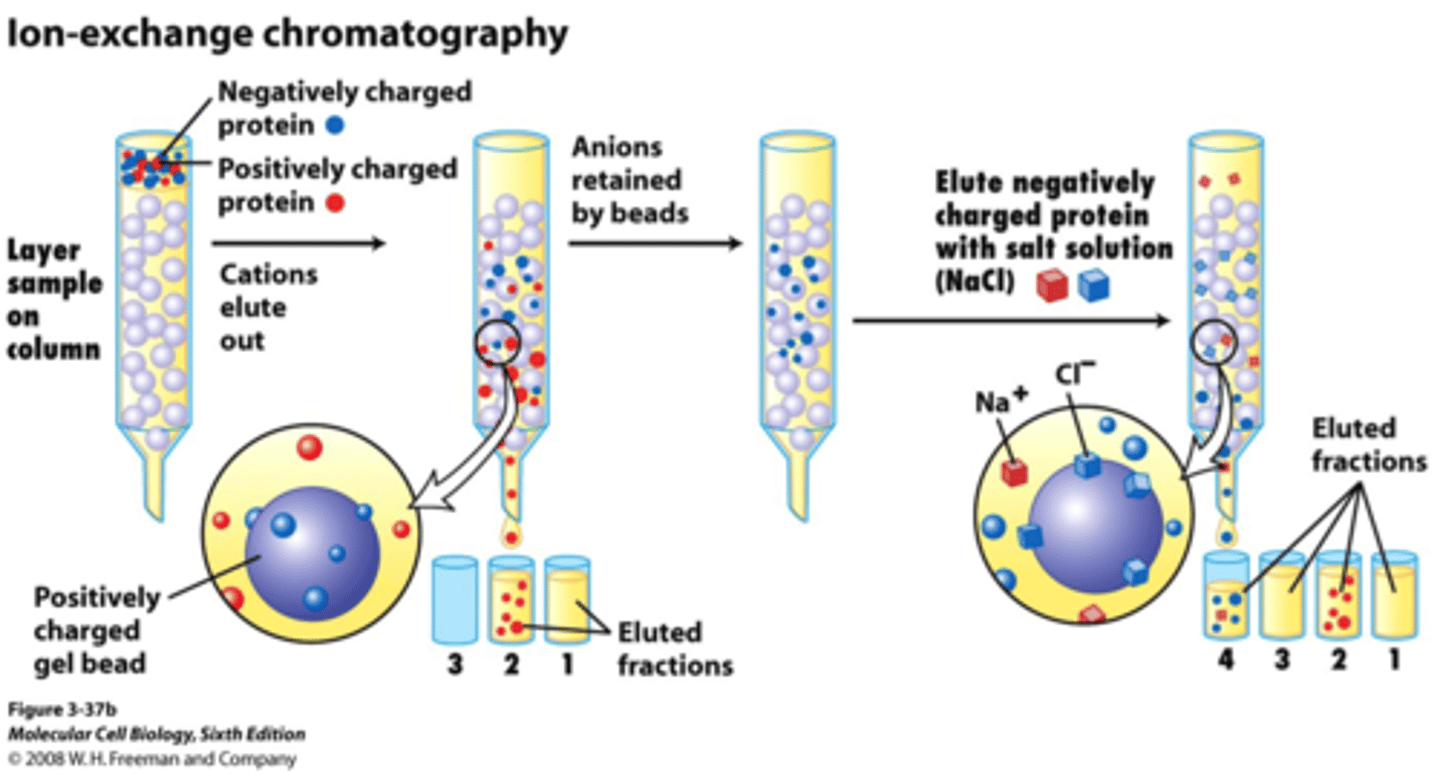
ion-exhange chromatography
separates proteins on the basis of charge
• A column is filled with charged beads, and the sample is applied to the top of the column.
• When a protein solution is passed over the beads, proteins with the same charge as that on the column will exit the column quickly.
• Proteins with the opposite charge will bind to the beads
– are ultimately released by increasing the salt concentration of the buffer that is passed through the column
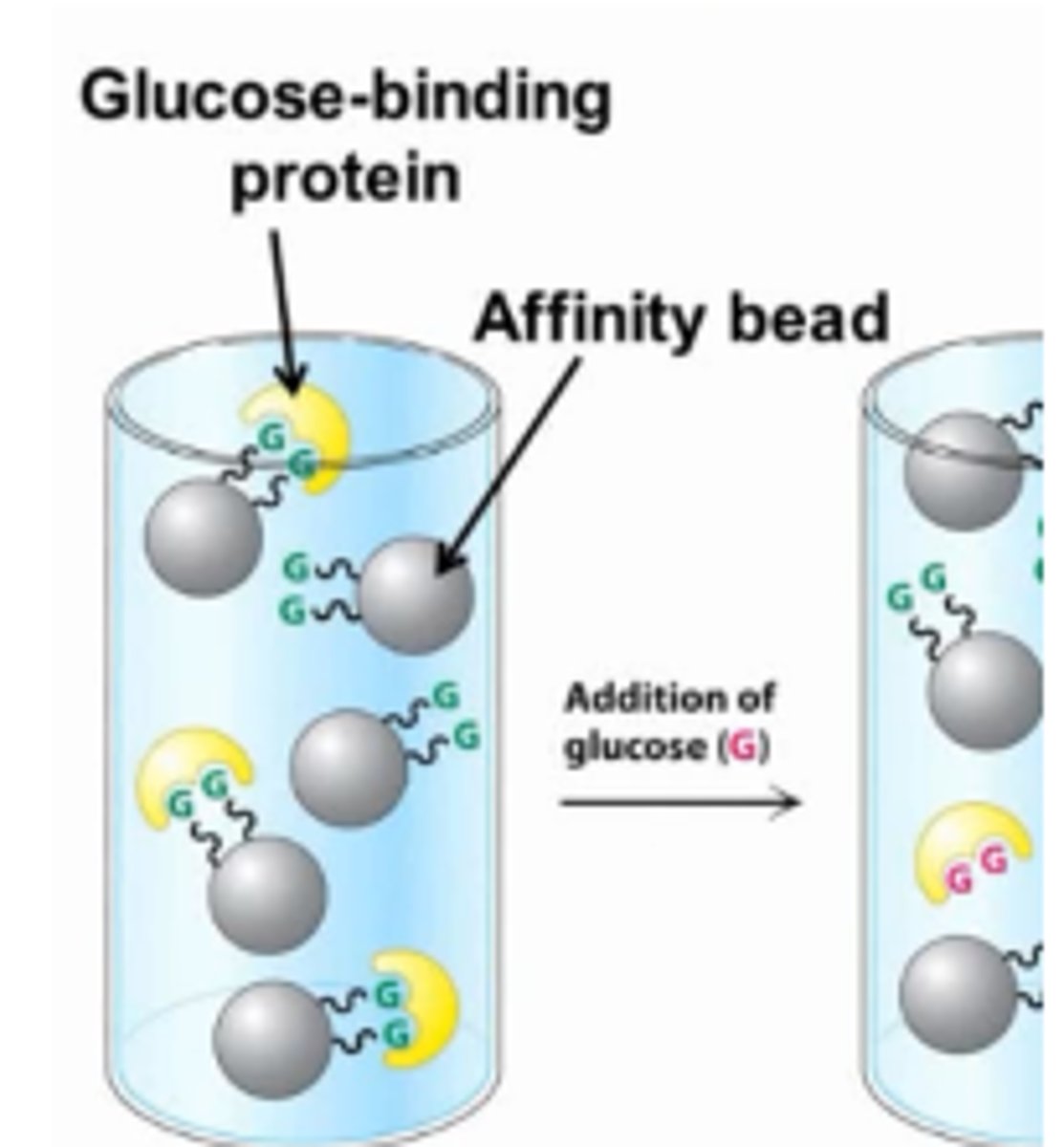
affinity chromatography
takes advantage of the fact that some proteins have a high affinity for specific molecules called ligands
• A column is filled with beads attached to the specific ligand.
• When a protein solution is passed over the beads,proteins with affinity for the attached group are retained.
• The bound protein is then released by passing a solution enriched in the ligand to which the protein is bound through the column.
High Performance Liquid Chromatography (HPLC)
Resolving Power: Depends on how many interaction sites exist between the protein and the column beads.
Fine Beads: More interaction sites → better separation (higher resolution), but slower flow.
HPLC (High-Performance Liquid Chromatography):
Uses very fine beads for high resolution.
Applies pressure to push liquid through quickly.
Results in sharper & faster protein separation.
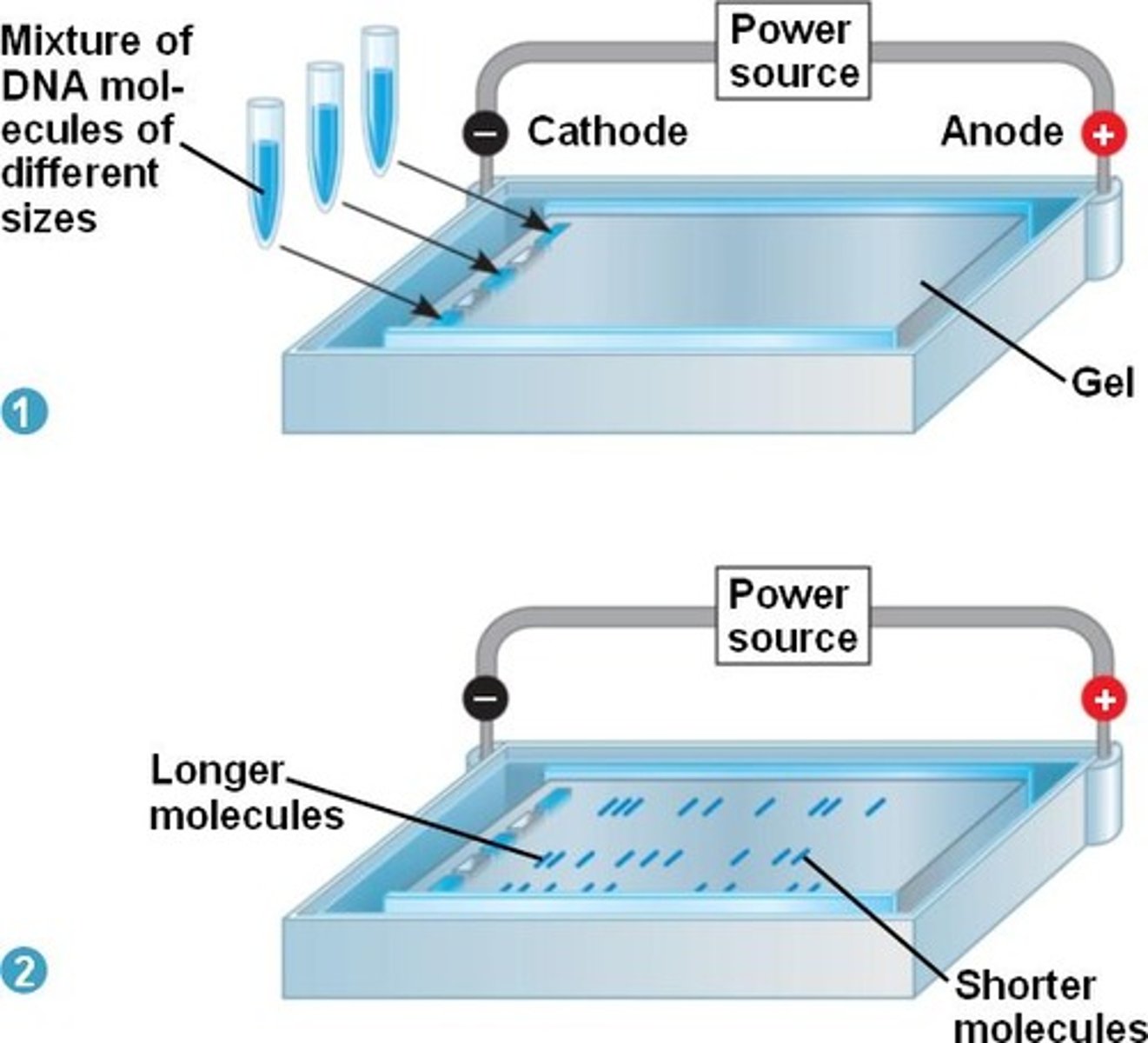
gel electrophoresis
separates mixtures of molecules with a net charge by applying an electric field
– used to separate proteins and nucleic acids
– carried out in gels (gel electrophoresis) which act as molecular sieves to enhance separation
– small molecules move quicker through the gel than larger molecules
Polyacrylamide Gel Electrophoresis (PAGE)
Polyacrylamide Gel Electrophoresis (PAGE)
Purpose: Separates proteins based on size and charge.
Gel: Made of polyacrylamide, acts like a molecular sieve.
Smaller proteins move faster through the gel.
Often used with SDS (a detergent) to:
Give proteins a uniform negative charge
Eliminate shape and charge differences → separates only by size
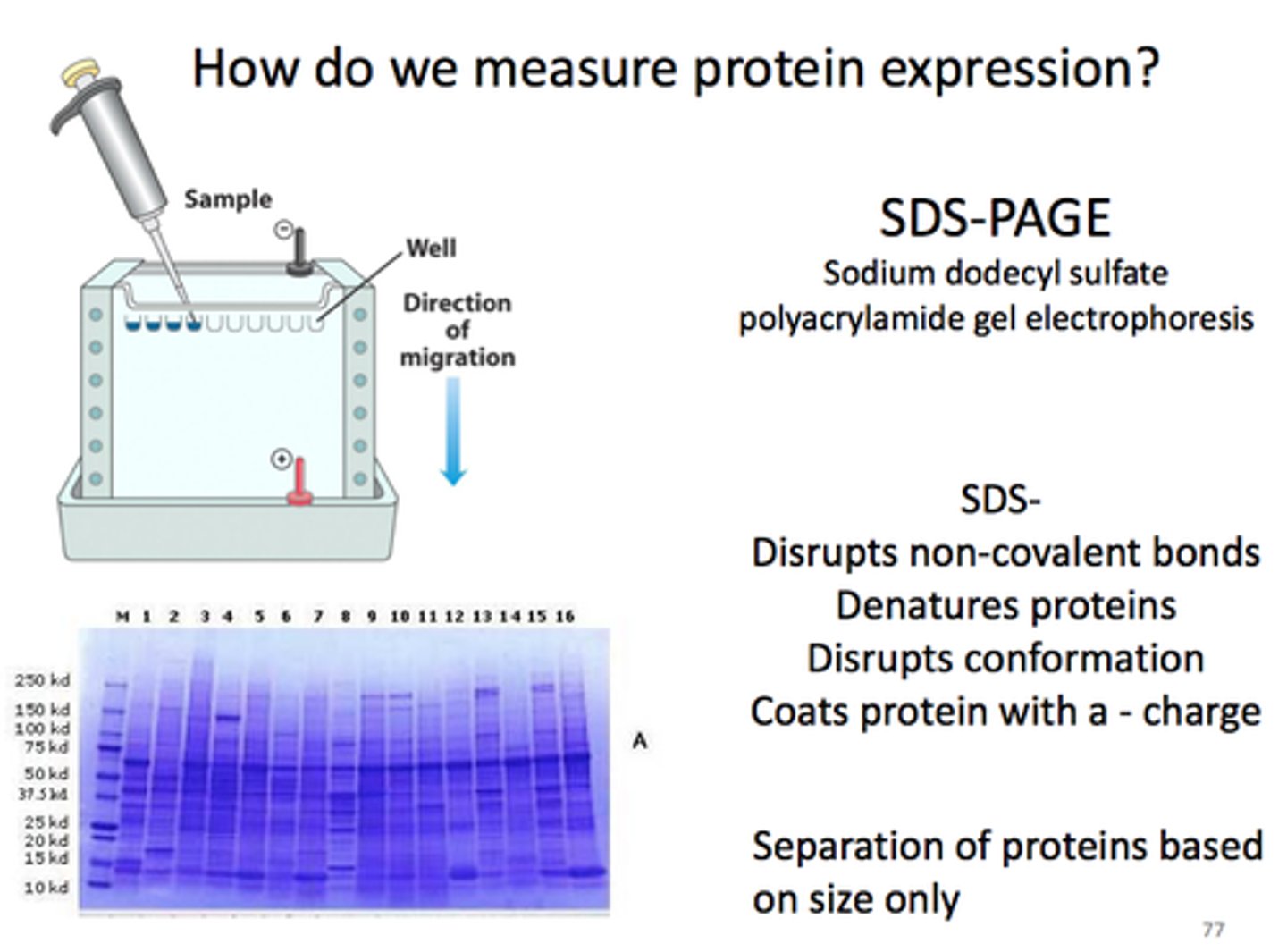
SDS-PAGE
-Used to measure protein size (mass) accurately.
-SDS is a detergent that:
Unfolds (denatures) proteins
Gives them a uniform negative charge
-SDS binds at a ratio of 1 SDS per 2 amino acids.
-Since all proteins have the same charge-to-mass ratio,→ they are separated only by size (not shape or charge).→ Smaller proteins move faster through the gel.
staining for proteins after electrophoresis
Proteins separated by SDS-PAGE are visualized by staining the gel with dyes such as Coomassie blue.
how can electrophoresis determine protein mass?
Electrophoretic mobility of many proteins inSDS-polyacrylamide gels is linearly proportional to the logarithm of their mass.
SDS-PAGE determines protein mass by separating proteins based on size. SDS denatures proteins and gives them a uniform negative charge. This removes shape and charge differences, so proteins move through the gel only based on size. Smaller proteins move faster. By comparing to a size standard, the protein's mass can be estimated.
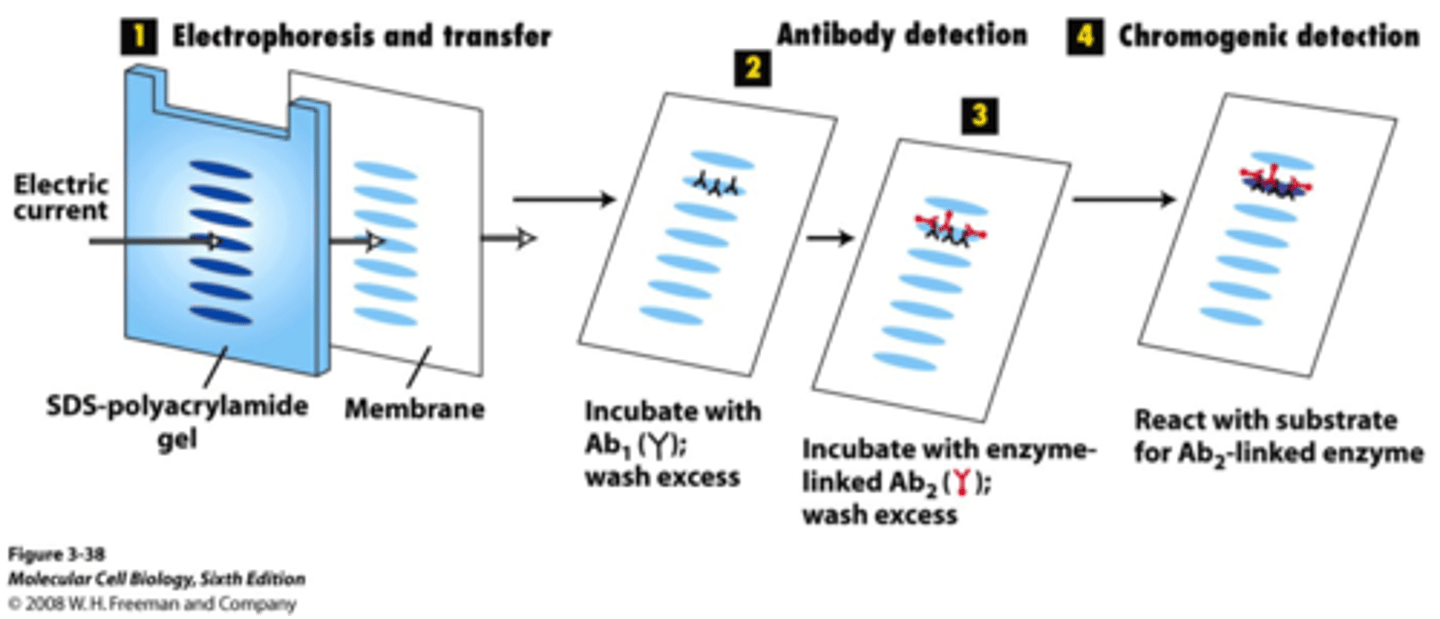
primary antibodies
an antibody specific for the protein
secondary antibody
an antibody specific for the primary antibody shapes
- attached to an enzyme that generates a chemiluminescent product or contains a fluorescent tag to enable identification and quantification
western blotting
proteins are separated in an SDS-PAGE gel, transferred to a polymer, stained with a primary antibody, stained with a secondary antibody, and quantified
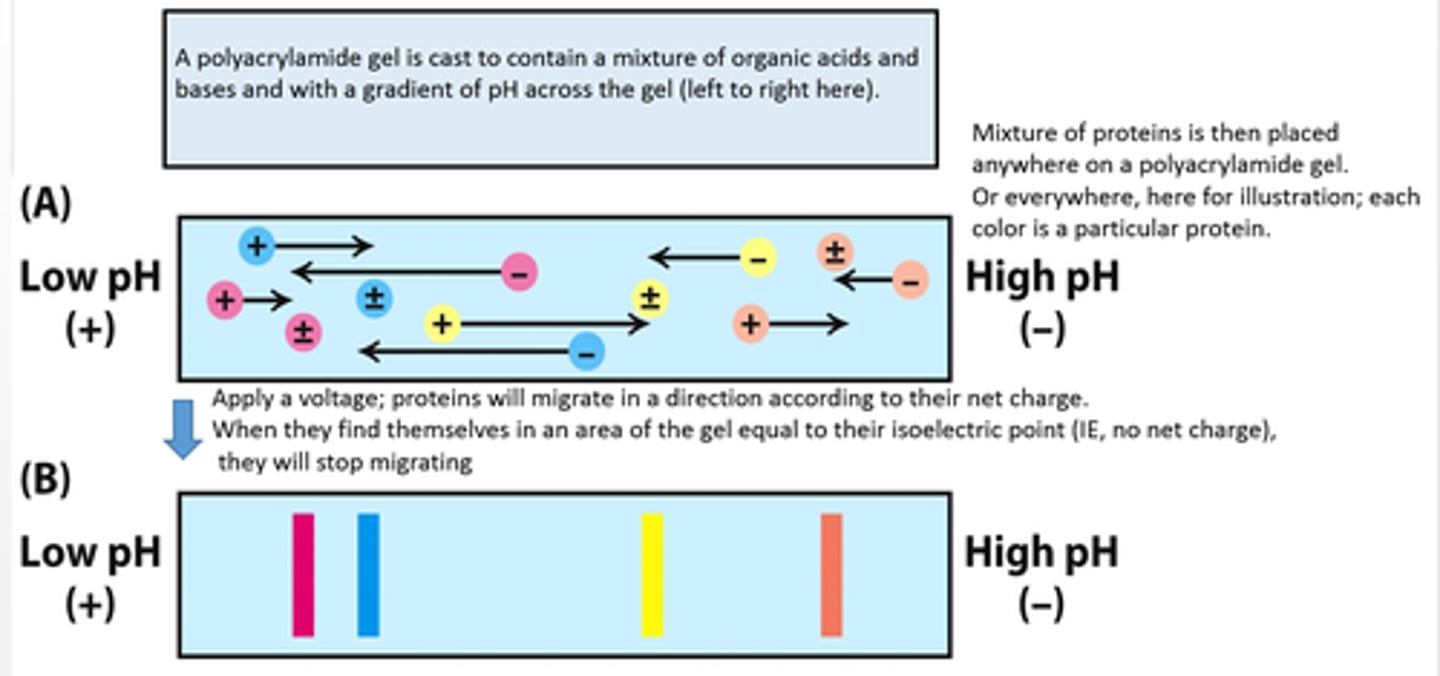
isoelectric point (pl)
pH at which a protein has no net charge
isoelectric focusing
separates proteins in a gel on the basis of pl
- If a mixture of proteins is placed in a gel with a pH gradient, each protein will migrate to its pI.
- At a protein's pI, its electrophoretic mobility is zero.
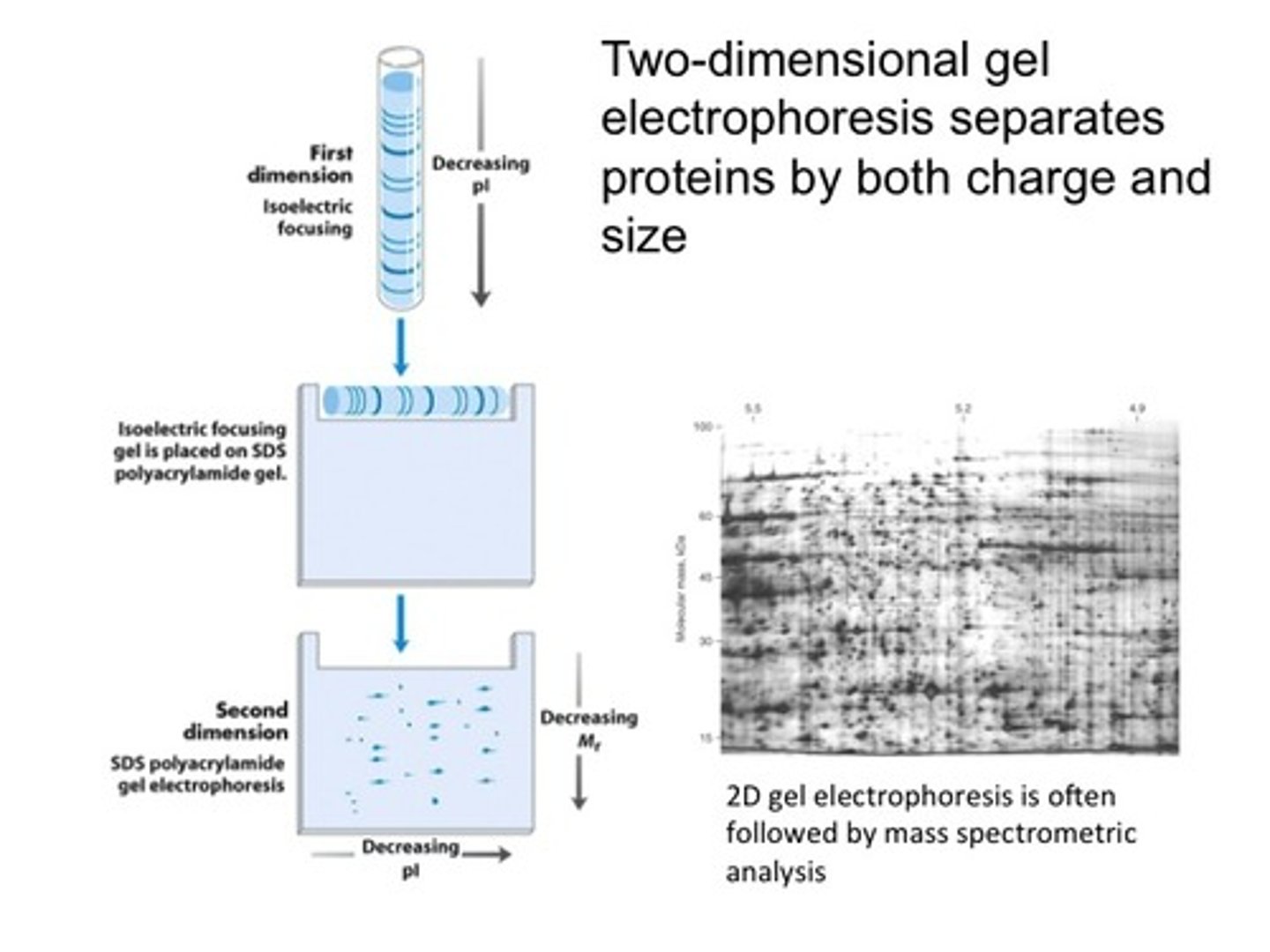
two dimensional electrophoresis
separates proteins in two directions.
– isoelectric focusing in a horizonal direction– SDS-PAGE in a perpendicular (vertical) direction
– yields a gel with proteins that have been separated based on pI in the horizontal direction and on mass in the vertical direction
-Two-dimensional electrophoresis can detect differences in protein expression under different physiological circumstances.
how is the effectiveness of purification scheme measured?
Effectiveness of a purification scheme is measured by calculating the specific activity after each separation technique.
specific activity
The ratio of enzyme activity to total protein concentration
- should increase with each step of the purification procedure since total protein is being removed while desired enzyme is being retained
Ultracentrifugation
used to analyze the physical properties of biomolecules, such as mass, density,shape, and interactions with other molecules
sedimentation coefficient
quantify the rate of movement when exposed to a centrifugal force
– expressed in Svedberg units (S)
– The smaller the S value, the more slowly a molecule moves in a centrifugal field.
– A more massive particle sediments more rapidly than doesa less-massive particle.
– Elongated particles sediment more slowly than sphericalones.
how can recombinant DNA technology used to make protein purification easier
Recombinant DNA technology affords several advantages in the production and purification of proteins:
– Proteins can be expressed in large quantities.
– Proteins can be modified with affinity tags that allow purification of the protein or visualization of the protein in the cell.
– Proteins with modified primary structure can be generated.
antibodies
proteins synthesized in response to thepresence of a foreign substance called an antigen
Antibodies to specific proteins can be generated
epitope (antigenic determinant)
specific group orcluster of amino acids on the target molecule that anantibody recognizes
polyclonal antibodies
heterogenous mixtures ofantibodies
- derived from multiple antibody-producing cell populations
- each antibody is specific forone of the various epitopes onan antigen
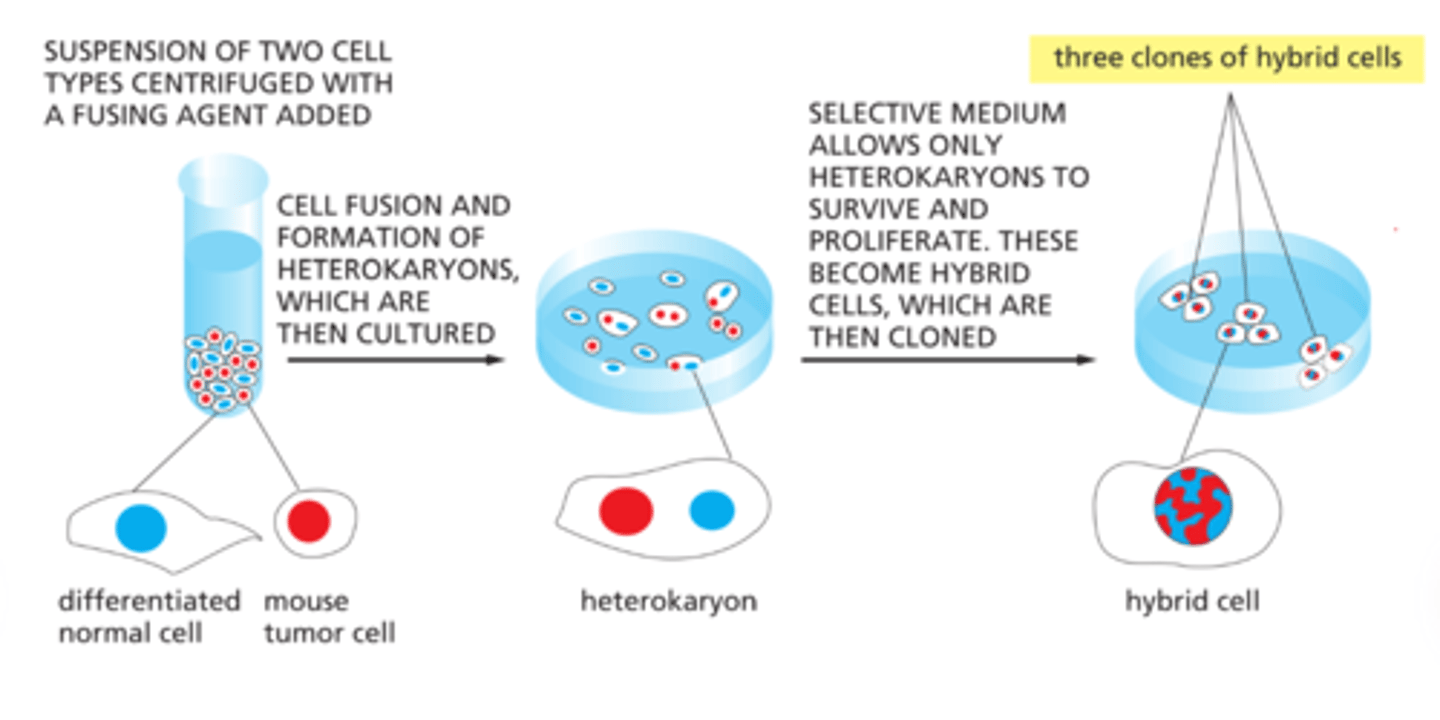
monoclonal antibodies
identical antibodies producedby clones of a single antibody-producing cell
Immortal cell lines produce monoclonal antibodies.
- generated by fusing normal, short-lived antibody
-producingcells with immortal cells from a type of cancer calledmultiple myeloma- results in hybrid cells called hybridoma cells
• A monoclonal cell line is isolated by screening for theantibody of interest
What does ELISA stand for?
A: Enzyme-Linked Immunosorbent Assay
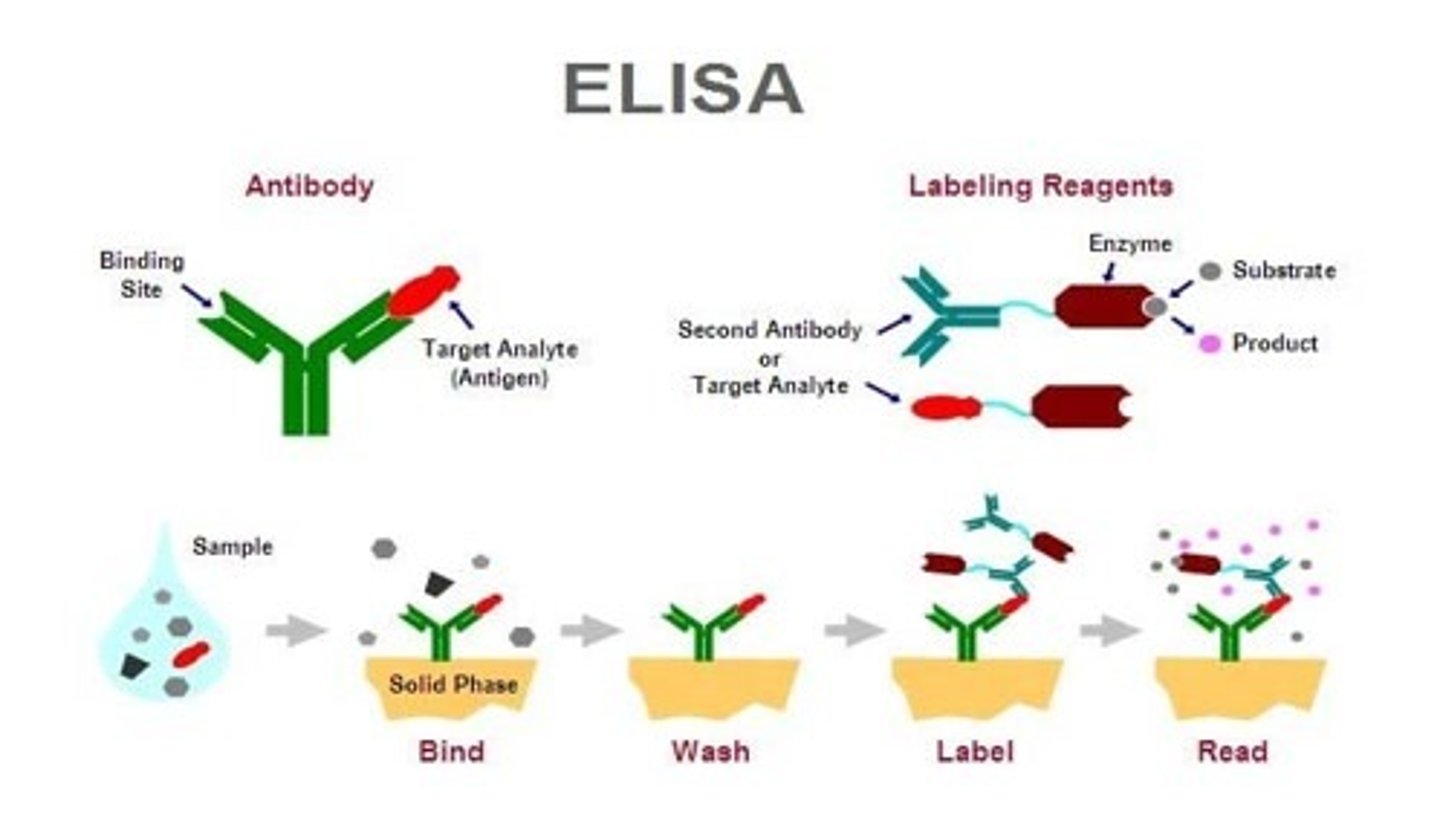
What is the primary purpose of ELISA?
A: To quantify the amount of a specific protein or antigen present in a sample.
Q: What type of molecule is used in ELISA to detect proteins?
A: Antibodies
Q: How do antibodies function in ELISA?
A: They bind specifically to the target protein (antigen).
Q: What is the antibody linked to in an ELISA?
enzyme
Name a common enzyme used in ELISA.
Horseradish peroxidase (HRP)
What does the enzyme in ELISA do?
A: It reacts with a substrate to produce a colored product.
Q: What does the color intensity in ELISA indicate?
A: The amount of protein or antigen present in the sample.
What is the role of the substrate in ELISA?
A: It produces a detectable signal (usually color) when cleaved by the enzyme.
Q: Why is ELISA considered quantitative?
A: Because the intensity of the color change correlates with the concentration of the target protein.
Q: What is the purpose of co-immunoprecipitation (Co-IP)?
A: To identify proteins that bind to a specific protein (binding partners).
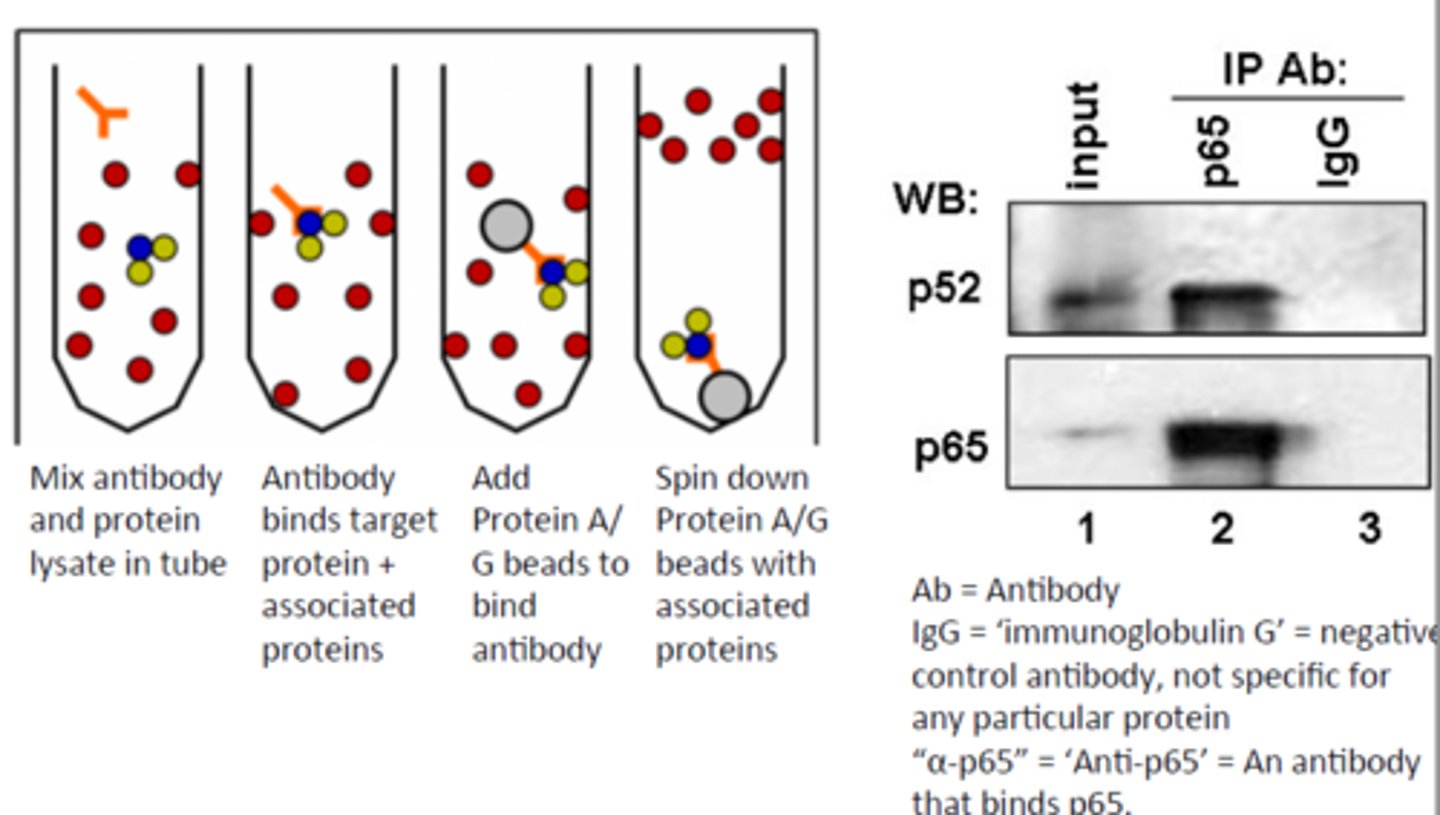
Q: What is the first step in Co-IP?
A: Incubate a cell extract with a monoclonal antibody against a specific protein.
Q: What is the second step in Co-IP?
A: Add agarose beads coated with an antibody-binding protein (e.g., Protein A).
Q: What is the third step in Co-IP?
A: Use centrifugation to separate the antibody-bound protein complex.
Q: What happens during centrifugation in Co-IP?
A: The antibody-protein complex (and any proteins bound to it) is pulled down as a pellet.
Q: What ensures that protein-binding partners co-precipitate?
A: Optimal buffer conditions that preserve protein-protein interactions.
Q: What is Protein A used for in Co-IP?
A: It binds to the Fc region of antibodies, helping attach them to the agarose beads.
Q: What does the term "precipitate" mean in the context of Co-IP?
A: To separate a complex from solution as a solid (pellet) using centrifugation.
Q: After Co-IP, how can other binding partners be identified?
A: Using methods such as Western blotting, mass spectrometry, or other biochemical assays.
Q: What do fluorescent markers allow scientists to do?
A: Visualize proteins within cells.
Q: How can cells be made to reveal specific proteins?
A: By staining them with fluorescence-labeled antibodies.
Q: What is the purpose of using fluorescence microscopy in protein studies?
A: To determine the cellular location of a protein.
Q: Why is knowing a protein’s location in the cell important?
A: It provides clues about the protein's function.
Q: How can fluorescent proteins be used in living cells?
A: To tag and track the movement of proteins within the cell.
Q: What is a common example of a fluorescent protein used to tag proteins?
A: Green Fluorescent Protein (GFP)
Q: What technique uses antibodies labeled with electron-dense metals?
A: Immunoelectron microscopy
Q: What is the purpose of immunoelectron microscopy?
A: To visualize proteins at high resolution using electron microscopy.
Q: What kind of label is used for immunoelectron microscopy?
A: Clusters of electron-dense metal on antibodies
Q: What advantage do fluorescent markers offer in cell biology?
A: They allow dynamic and spatial tracking of proteins in real time.
Q: What is mass spectrometry used for in protein analysis?
A: It allows precise and sensitive detection of the mass of an analyte (e.g., peptides/proteins).
Q: What does a mass spectrometer measure?
A: The mass-to-charge ratio (m/z) of ions.
Q: What are the three main components of a mass spectrometer?
A: An ion source, a mass analyzer, and a detector.
Q: What happens to the analyte in mass spectrometry?
A: It is converted into gas-phase ions.
Q: How does a mass spectrometer move and sort ions?
A: By applying electrostatic potentials to separate ions based on their m/z values.
Q: What does the "m/z" ratio represent?
A: Mass-to-charge ratio of an ion.
Q: What is MALDI?
A: Matrix-Assisted Laser Desorption/Ionization - a method to ionize analytes without fragmentation.
Q: What is MALDI commonly paired with in protein identification?
A: A time-of-flight (TOF) detector.
Q: How does a TOF detector work?
A: It measures the time it takes ions to reach the detector—lighter ions reach it faster.
Q: Why is mass spectrometry useful in proteomics?
A: It enables identification and characterization of peptides and proteins from complex mixtures.
Q: What is the purpose of ionization in mass spectrometry?
A: To convert analytes into gas-phase ions for detection.
Q: What does MALDI stand for?
A: Matrix-Assisted Laser Desorption/Ionization
Q: How does ionization occur in MALDI?
A: The analyte is evaporated with a light-absorbing matrix using a laser.
Q: What is the role of the matrix in MALDI?
A: It absorbs laser light and helps transfer energy to the analyte for ionization.
Q: What type of samples is MALDI commonly used for?
A: Large biomolecules like peptides and proteins.
Q: What does ESI stand for?
A: Electrospray Ionization
Q: How does ionization occur in ESI?
A: The analyte solution is passed through a charged nozzle, creating a fine spray of ions.
Q: What type of samples is ESI especially suited for?
A: Liquid samples of biomolecules and non-volatile compounds.
Q: What is a key difference between MALDI and ESI?
A: MALDI uses a laser and matrix; ESI uses an electric field and does not require a matrix.
Q: What do both MALDI and ESI have in common?
A: They produce gas-phase ions suitable for mass spectrometry analysis.
what are two ways to measure proteins?
size and isoelectric point
Q: What is a mass analyzer in mass spectrometry?
A: A component that distinguishes ions based on their mass-to-charge (m/z) ratios.
Q: How does a TOF mass analyzer work?
A: Ions are accelerated through a long chamber using a fixed electrostatic potential.
Q: What determines how fast an ion travels through the TOF chamber?
A: Its mass-to-charge ratio (m/z)
Q: What do lighter ions do in a TOF analyzer?
A: Reach the detector faster than heavier ions.
Q: How is the mass of an ion determined in TOF?
A: By measuring the time it takes for the ion to travel through the chamber.
Q: Why is TOF useful in protein identification?
A: It allows accurate measurement of peptide/protein mass for identification.
Q: What property must remain constant for TOF analysis to be accurate?
A: The electrostatic potential accelerating the ions.
Q: What does a TOF detector produce as output?
A: A spectrum showing ion intensity vs. time, which correlates with m/z.
Q: Why are proteins cleaved into smaller peptides before analysis?
A: To make sequencing and identification easier, especially for proteins longer than 50 amino acids.
Q: What is Edman degradation used for?
A: Sequencing short peptides by identifying amino acids one at a time from the N-terminus.
Q: What is a limitation of Edman degradation?
A: It is effective only for peptides with fewer than 50 residues.
Q: Why doesn’t Edman degradation work well on longer peptides?
A: Not all peptides release the amino acid derivative at each step, leading to signal loss.
Q: Why is mass spectrometry challenging for long peptides?
A: It produces complex spectra that are difficult to interpret.
Q: What must be done to sequence an entire protein?
A: Chemically or enzymatically cleave the protein into shorter peptides (under 50 amino acids).
Q: How are peptide sequences ordered after cleavage?
A: By using different cleavage procedures to produce overlapping peptides.
Q: What is the purpose of generating overlap peptides?
A: To reconstruct the full protein sequence by aligning overlapping regions.
Q: What enzymes are commonly used for protein cleavage?
A: Trypsin, chymotrypsin, and others that cut at specific amino acid residues.