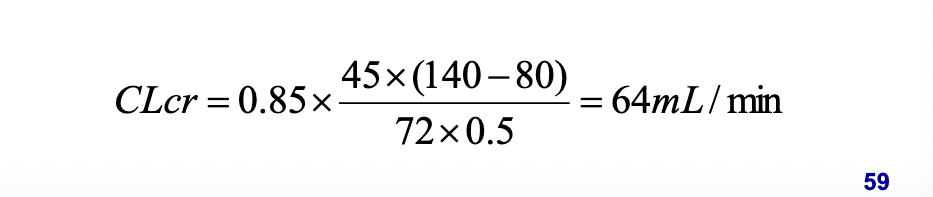lec 24 - precision med (guo)
1/47
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
48 Terms
what is precision med?
‘personalized' medicine’ = older term with a meaning similar to ‘precision medicine’
precision med = focus is on identifying which approaches will be effective for which patients based on genetic, environmental and lifestyle factors
many drugs can be ineffective in…
drug → non-response rate
hypertension drugs → 10-30%
heart failure drugs → 15-25%
anti depressants → 20-50%
cholesterol drugs → 30-70%
asthma drugs → 40-70%

some experience adverse effects
just in hospitals → 6.7% of patients experience serious adverse drug reactions
serious adverse drug reactions in even smaller percentages of treated populations have led to withdrawal of several drugs from the market
baycol
fen-phen
lotronex
propulside
tysabri
vioxx
are good drugs going to wrong people?
qualitative understanding of variability
assumption that all people are alike in PK should be challenged
differences among people do exist including their responsiveness to drugs (PD)
frequency need to tailor dosing regimen to individual patient
a failure to do so can lead to ineffective therapy, toxiciy or both
magnitude and relative contribution of PK/PD to variability in response to a given dosage within a pt population vary with the drug and to some extent, the condition being treated
quantitative understanding of variability
to minimize the confounding (inability to tell whats truly causing variability) from various doses and time in explaining variability, let’s express variability in terms of parameter values defining PK and PD
PK
bioavailibility
absorption rate constant
CL
V
t1/2
PD
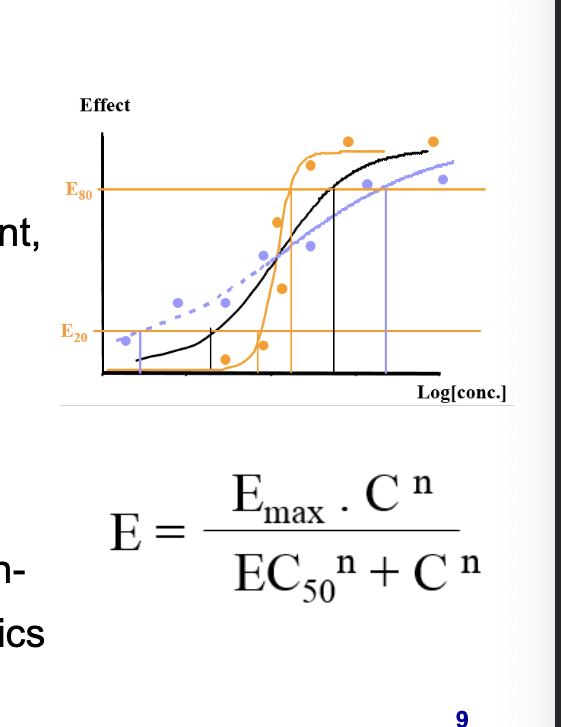
maximal response (Emax)
concentration to achieve 50% of max response (EC50)
factor defining the steepness of the concentration-response relationship for PD (n: Hill coeff)
equation in photo = hill equation
E = effect
variables (‘covariates’) affecting PD/PD variability
age
size (WT, BSA)
genderrace
genotype/phenotype
food effect
renal function status
hepatic function status
concomitant medications
enzyme inducers
enzyme inhibitors
compete renal CL
displace plasma protein binding
impair GI absorption
route of admin
enantiomeric drugs
disease states
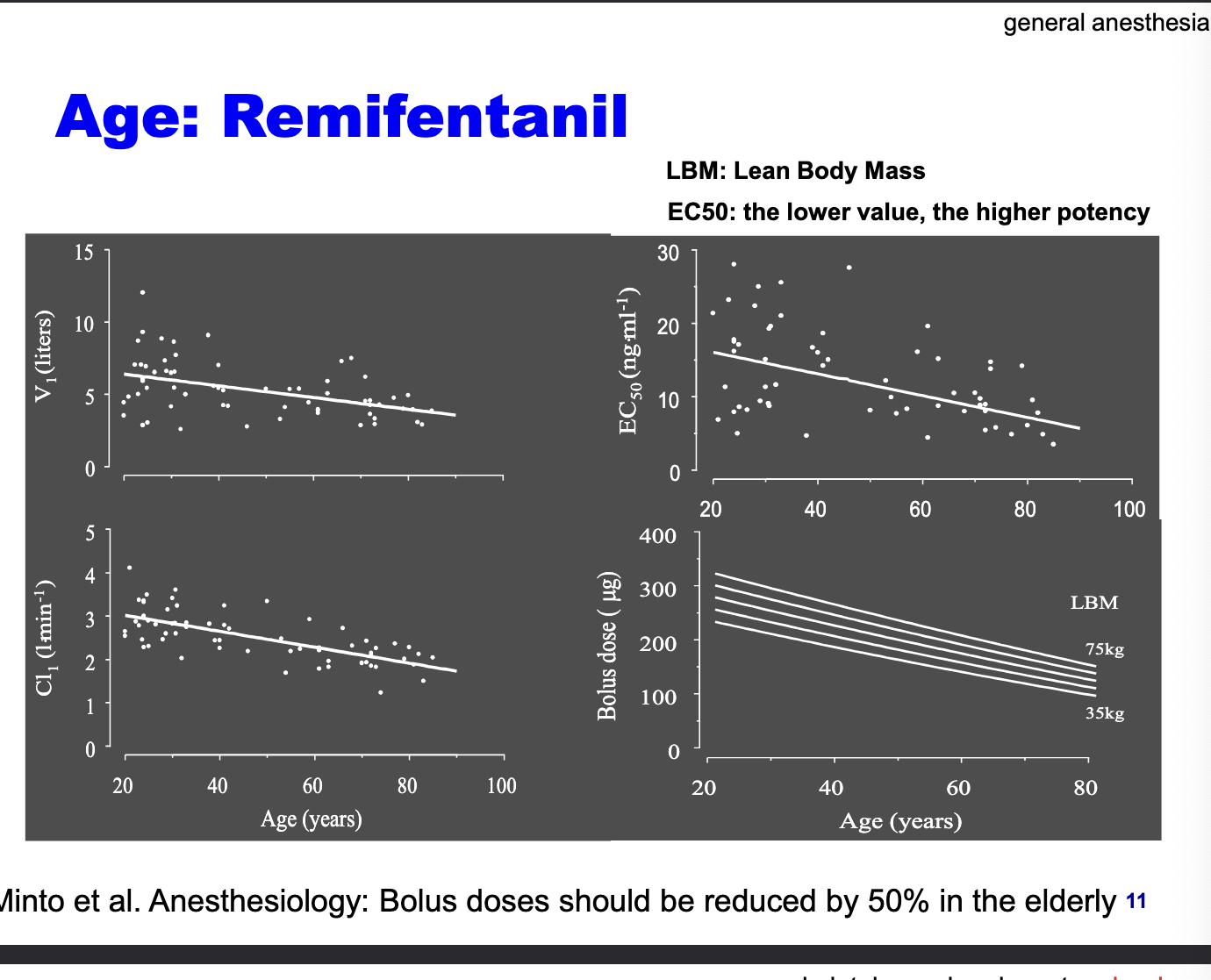
age: remifentanil
bolus doses should be reduced by 50% in the elderly
V1 decreases with age → older adults have less volume to distribute the drug
CL decreases with age
EC50 decreases with age → older adults require less drug to achieve the same effect → increased sensitivity
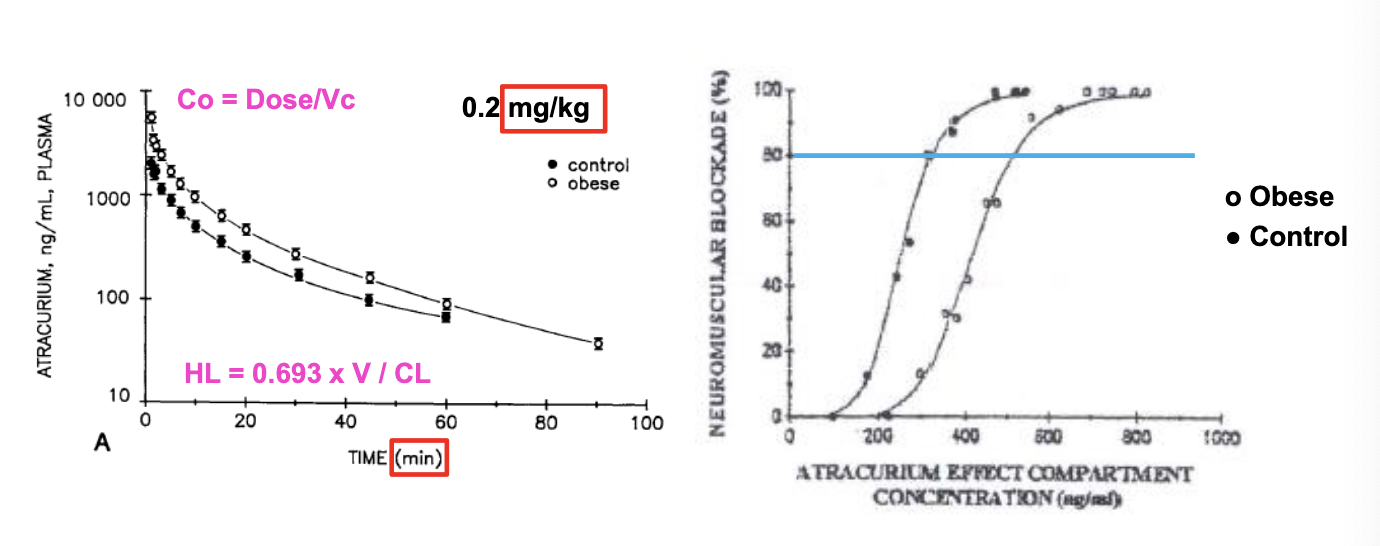
body weight: atracurium
obese subjects have higher exposure but as less sensitive to drug
Vss and CL = decreased but PK t1/2 is unchanged
CL decreased → more drug stays in plasma as time goes on but not as sensitive to drug so need higher concentrations to achieve same effect
NO difference in the time of recovery of neuromuscular blockade, so dosing based on total body weight (TBW)
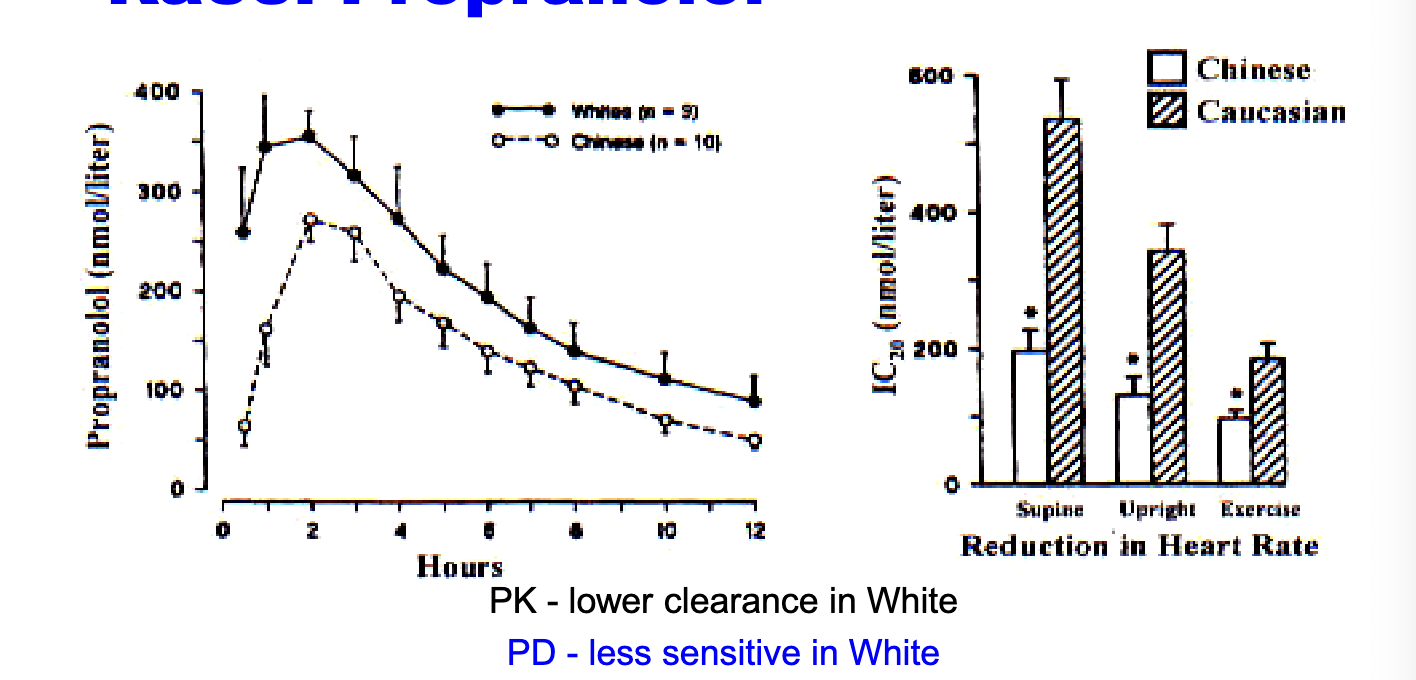
race: propranolol
PK → lower clearance in white
PD → less sensitive in white → higher doses
label → 45% higher free fraction (of propranolol due to NO protein binding) in chinese
to produce the same degree of beta-blockage, plasma propranolol concentrations had to be twice as high in whites v chinese
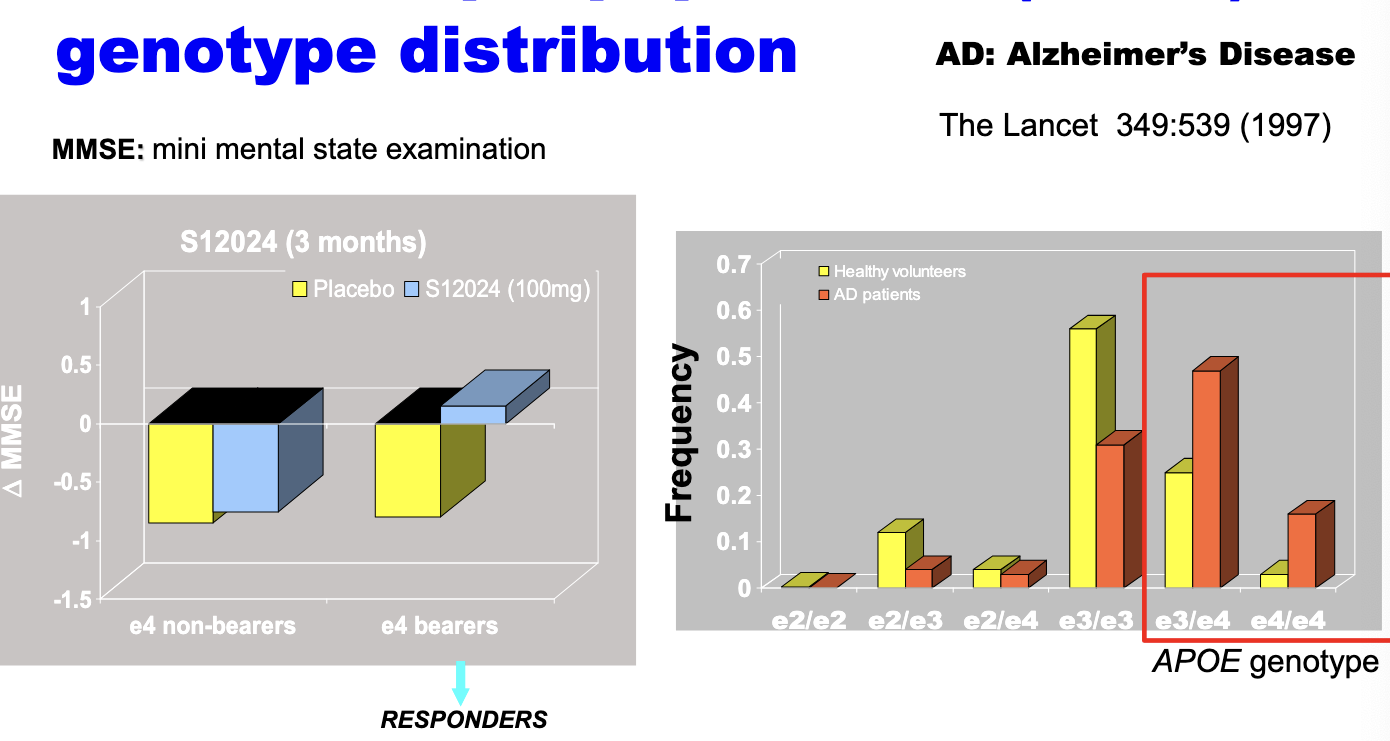
genetics: apolipoprotein E (APOE) genotype distribution
one of the variants of the APOE genes, e4, has been found to be associated with increased risk of Alzheimers disease
impact of NOT having genotype information in this case → may discontinue the development of this drug
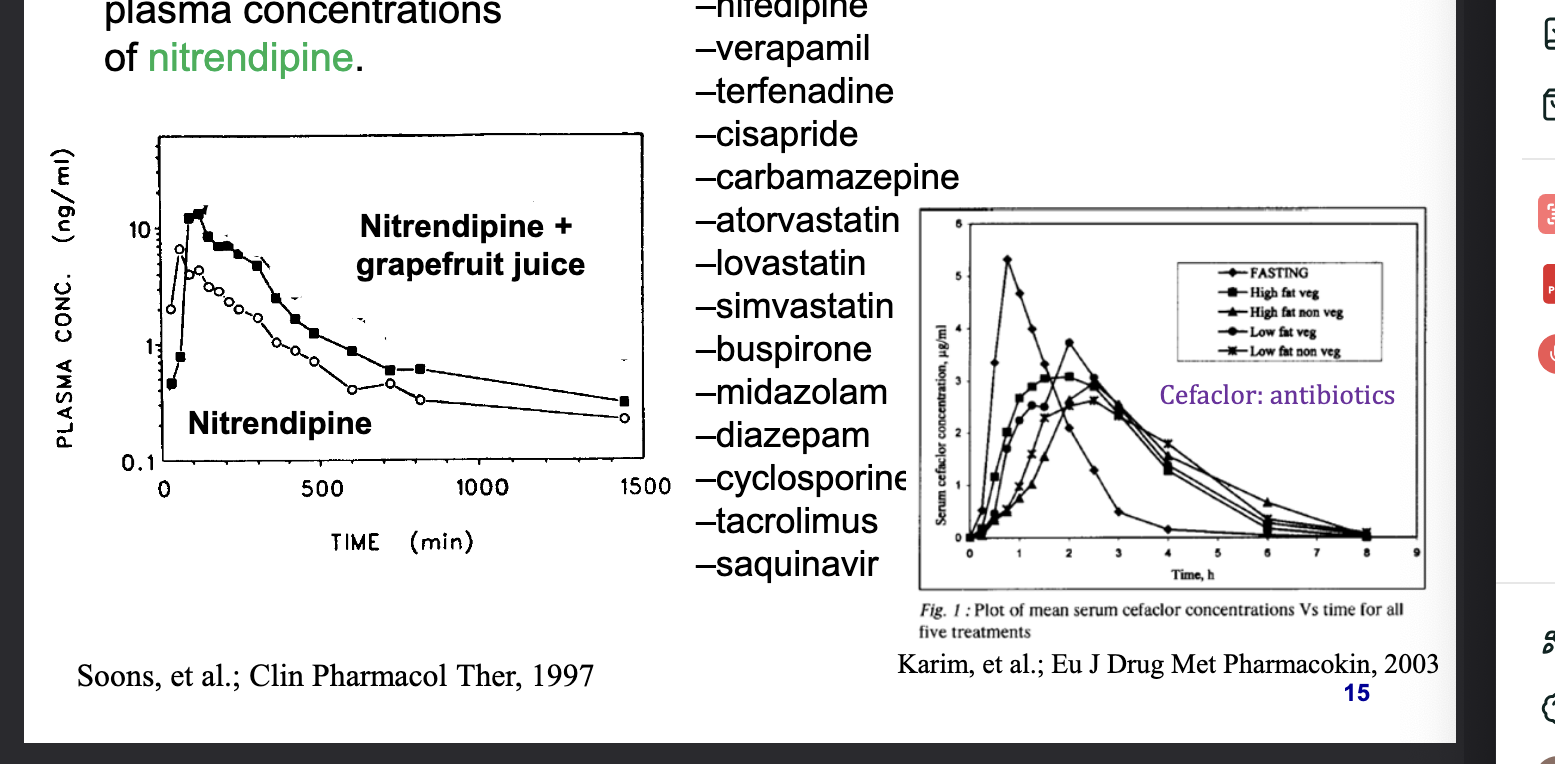
impact of food
chemicals in grapefruit juice inhibits CYP3A4 and increases the plasma concentrations of nitrendipine
other drugs that interact with grapefruit juice
felodipine
nifedipine
verapamil
terfenadine
cisapride
carbamazepine
atoravastatin
lovastatin
simvastatin
buspirone
midazolam
diazepam
cyclosporine
tacrolimus
saquinavir
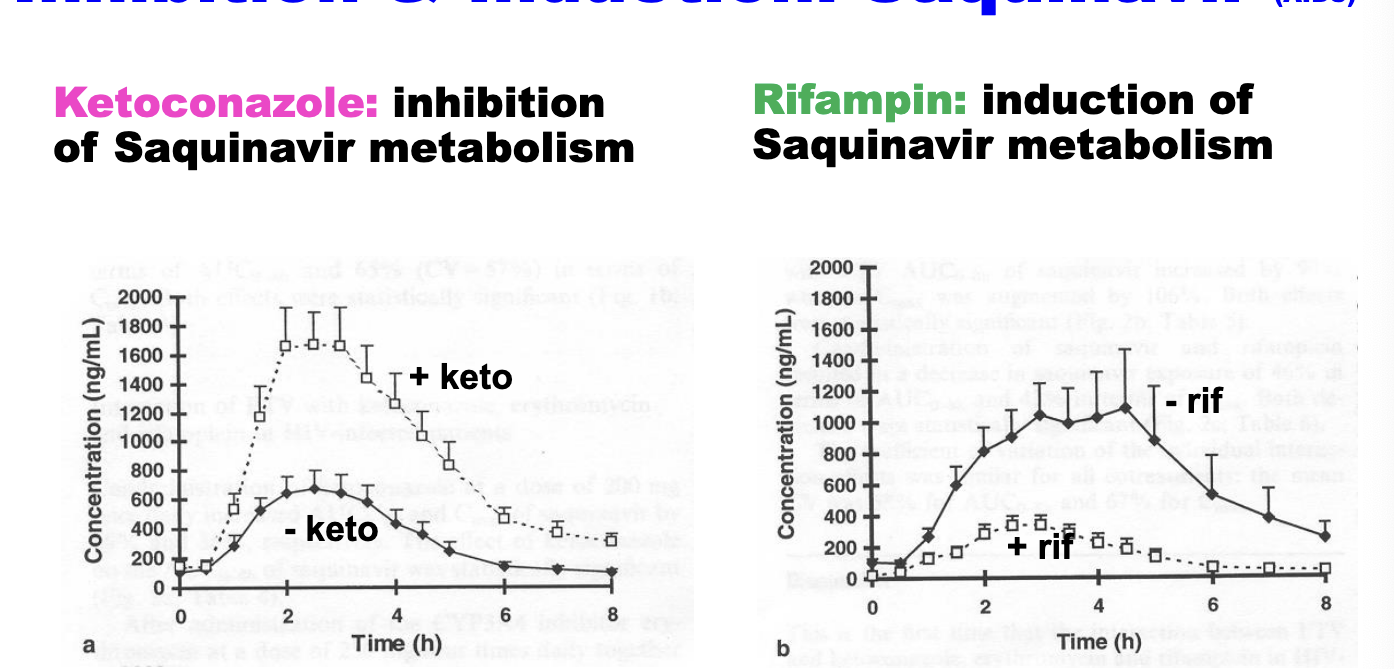
inhibition and induction: saquinavir (AIDS)
ketoconazole → inhibition of saquinavir metabolism
rifampin → induction of saquinavir metabolism
CYP3A4 plays major role in elimination of saquinavir
disease states
hepatic impairment
renal impairment
GI diseases
CV diseases
respiratory diseases
endocrine diseases
hepatic impairment
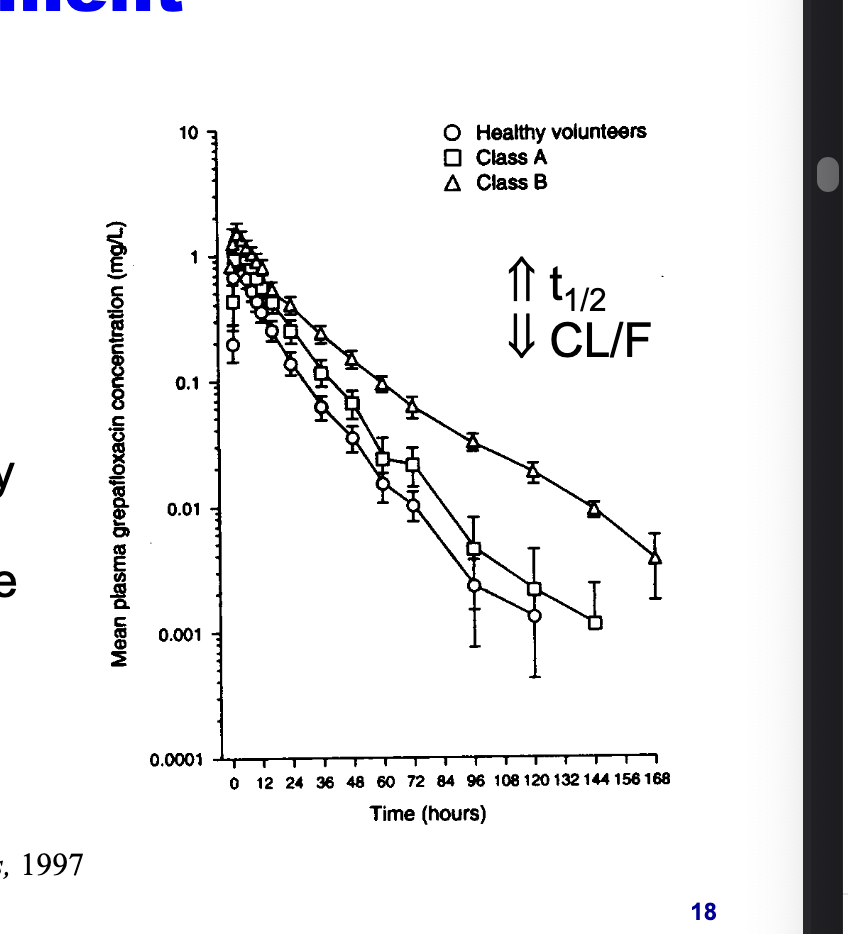
hepatic diseases can result in decrease in CYP3A4 activity
grepafloxacin, CYP3A4 substrate, eliminated more slowly in patients with advanced liver disease
increased half life
decreased CL/F since CL decreases
hepatic impairment cont.
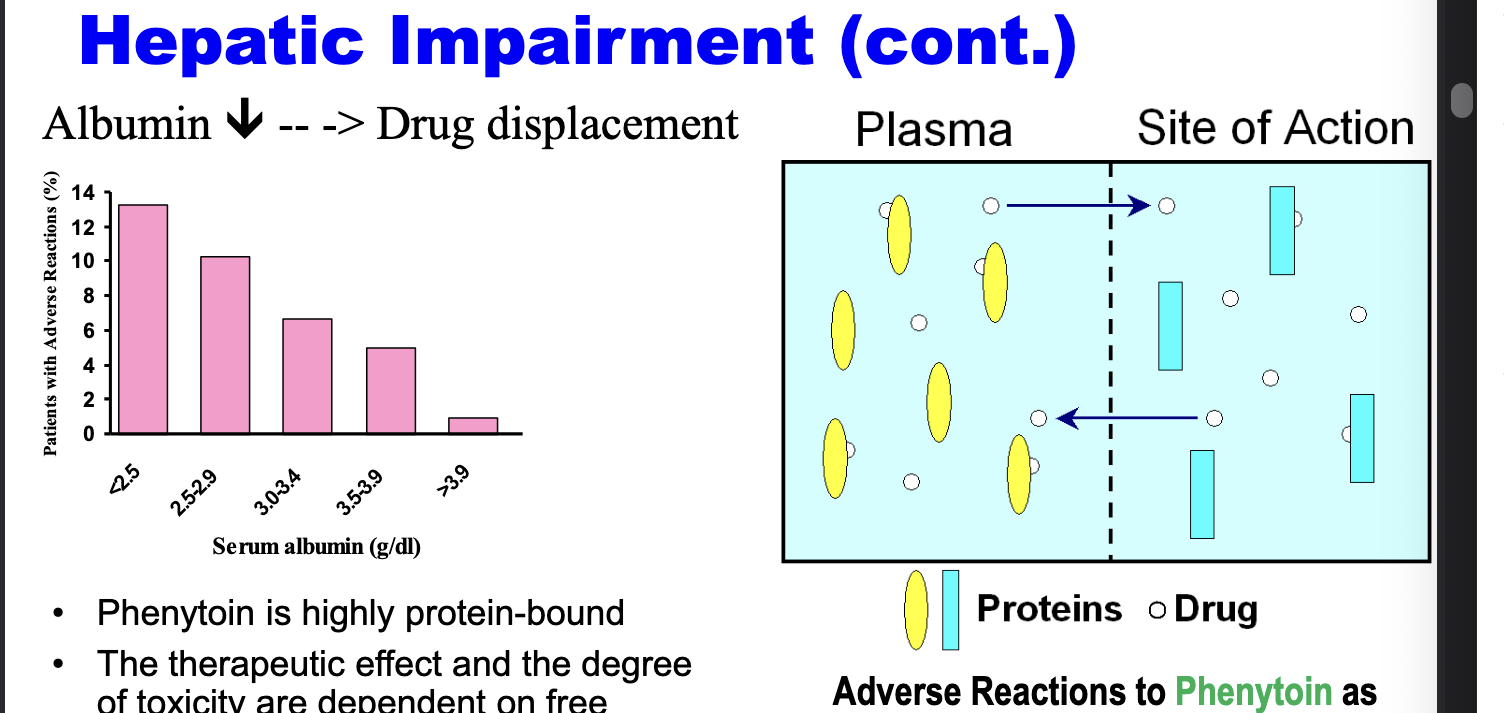
phenytoin is highly protein bound
therapeutic effect and the degree of toxicity are dependent on free phenytoin concentration which is turn depend on the albumin binding
less albumin → greater drug displacement → more adverse rxns
renal impairment
levofloxacin excretes primarily as unchanged drug into the urine
drug CL is dependent on the renal function status of the patients
CL of levofloxacin is substantially reduced in patients with impaired renal function → requires dosage adjustment to avoid accumulation
CYP450s involved in phase I drug metabolism
predominant phase I metabolizing enzymes
high inter-individual variation in expression
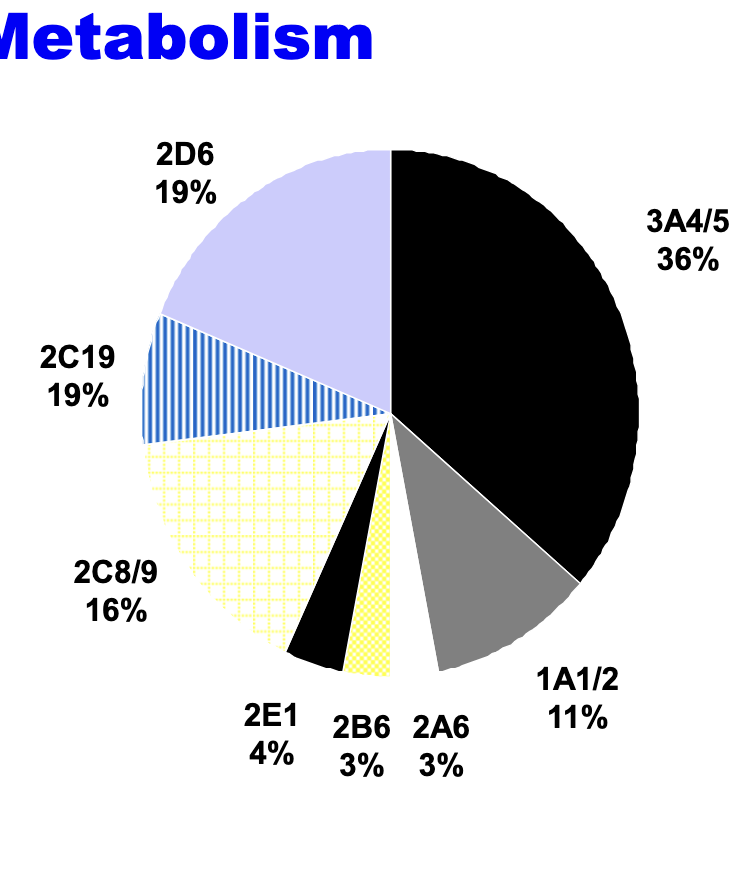
polymorphism in phase I metabolism
individuals are classified as poor (PM) or extensive (EM) metabolizers
mutation on the gene coding for a metabolizing enzyme = major cause of variation in drug metabolism → results in allelic variants producing enzymes with altered metabolizing activity
affect the efficacy and adverse effects of drugs
incidence of poor metabolizers
CYP2D6 (marker: dextromethorphan)
5-10% caucasians
3.8% blacks
0.9% asians
1% arabs
2C9 and 2C19 also have polymorphism
half life of dextromethorphan was significantly increased in PM compared with EM
dextrophan = active metabolite with anticonvulsant, sedative and antitussive properties → contributes to dextromethorphan abuse liability → PMs less likely to abuse dextromethorphan
phase II metabolism
drugs that are subject to this metabolism pathway have high water solubility and their metabolisms become usually biologically inactive
in most phase II reacitons
conjugation groups are activated by coenzyme
many endogenous processes utilize the same coenzymes
acetylation, methylation, sulfation, glucuronidation
polymorphism in phase II metabolism
acetylation activity is bimodally distributed in human populations
classified as either slow or rapid acetylators
marker = isoniazid (primarily metabolized by acetylation)
most asians are rapid acetylators
a little more of caucasians are slow but pretty even
more middle easterns are slow acetylators

drug-drug interactions
enzyme induction
nicotine induces CYP1A2
rifampin and st johns wort induce CYP3A4
enzyme inhibition
grapefruit juice and ritonavir inhibit CYP3A4
transporter drug-drug interaction
P-glycoprotein 1 (Pgp) also known as multidrug resistance protein 1 (MDR1) or ATP-binding cassette sub-family B member 1 (ABCB1) or cluster of differentiation 243 (CD243)
cell membrane protein that pumps many foreign substances out of cells; more formally = ATP-dependent efflux pump with broad substrate specificity
extensively distributed and expressed in the intestinal epithelium where it pumps xenobiotics (such as toxins or drugs) back into the intestinal lumen in liver cells where it pumps them into bile ducts in the cells of the proximal tubule of the kidney where it pumps them into urine-conducting ducts and in the capillary endothelial cells composing the BBB and blood-testis barrier where it pumps them back into the capillaries
some cancer cells express large amounts of PGP which ernders these cancers multi-drug resistant
drug development and drug interactions: table of substrates, inhibitors and inducers

physiology: a source of variability
age
children cannot be viewed simply as little adults
PK differences
altered PD responses
process of growth (body proportion) and development (organ functionality)
age classification
pre-term newborn infants
term new born infants → 0-27 days
infants and toddlers → 28 days - 23 months
children → 2-11 years
adolescents → 12 to 18-18-21 years (dependent on country)
elderly → >65 years
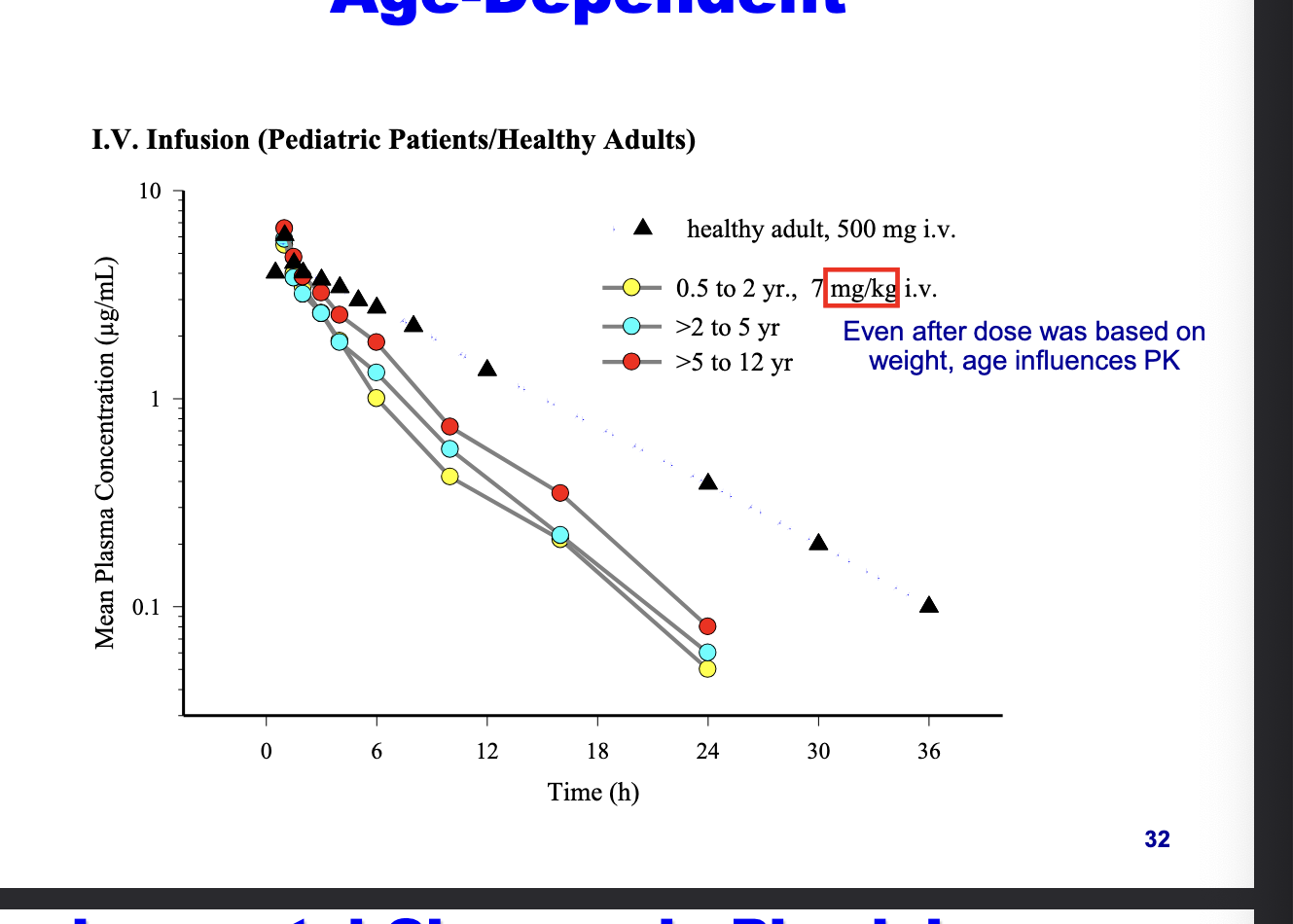
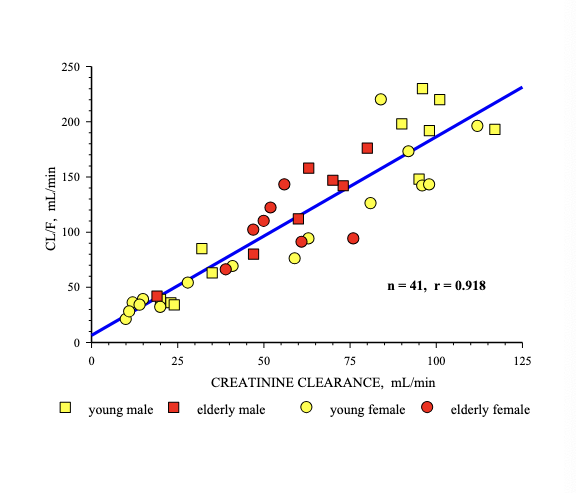
PK of levaquine is age-dependent
even after dose was based on weight, age influences PK
healthy adult = 500 mg
peds = 7 mg/kg
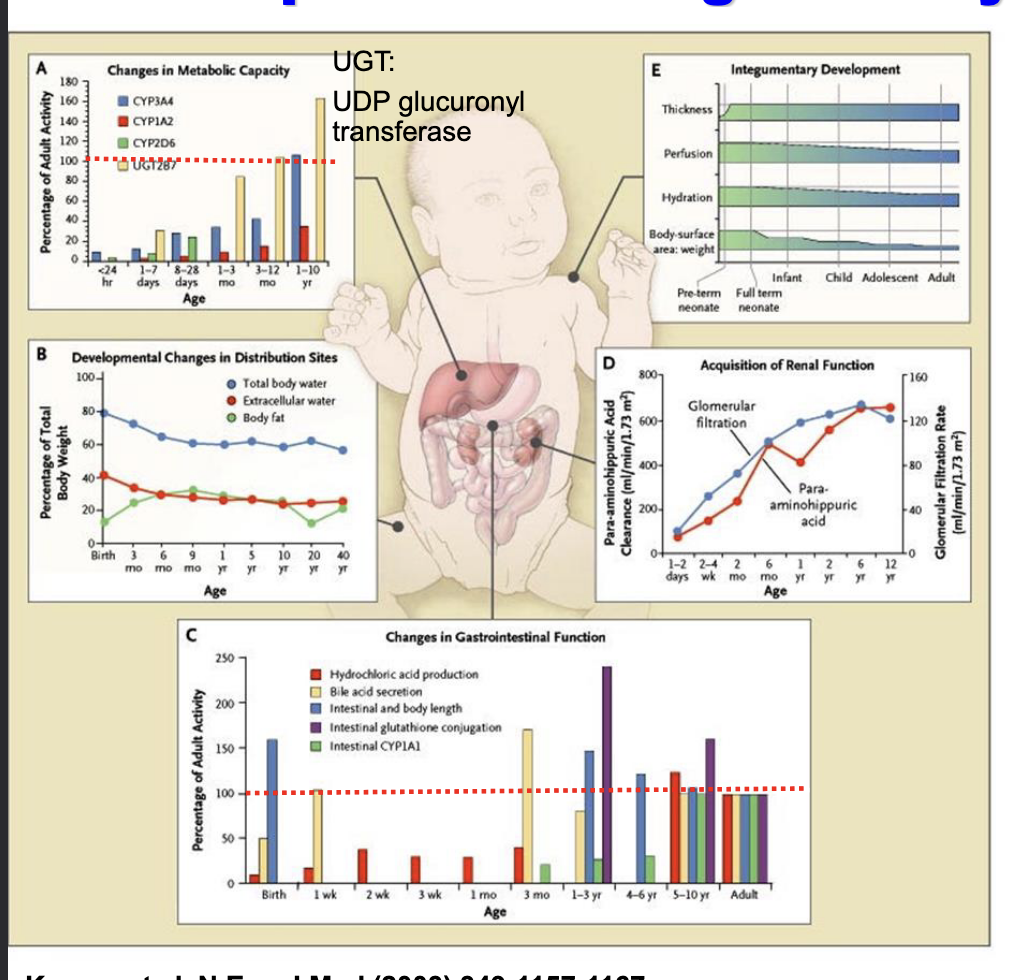
developmental changes in physiology
integumentary system = organ system that protects the body from various kinds of damage such as loss of water or abrasion from outside
comprises the skin and its appendages (including hair, scales, feathers, hooves, and nails)
aminohippuric acid or para-aminohippuric acid (PAH), a derivative of hippuric acid = diagnostic agent useful in medical tests involving the kidney used in the measurement of renal plasma flow
changes in metabolic capacity
CYP enzymes activity gradually increase with age
distribution changes
total body water is very high at birth and decreases with age
body fat increases later in infancy
changes in GI function
neonates have low hydrochloric acid, immature bile acid secretion, incomplete enzyme activity
renal function
GFR and tubular secretion = immature at birth and increase as you get older
integumentary development
preterm infants = thin, highly permeable skin and a high surface area-to-weight ratio
absorption factors
factors influencing GI absorption
gastric acidity
high pH in neonates, infants and young children
GI motility
low in neonates and young infants
high in older infants and children
enzymatic activity
low beta-glucuronidase and UDP-glucuronyl transferase in neonates
bile acids
low in neonates
mucosal membrane permeability
bacterial flora
dietary components
diarrheal episodes
distribution factors
body water composition
total body water
~75% of body weight in neonates and infants vs 55% in adults
high Vd
extracellular water (high in neonates, infants and children)
high Vd
intracellular water (similar % to adult)
albumin concentration & plasma protein binding
low in neonates and infants
body fat content
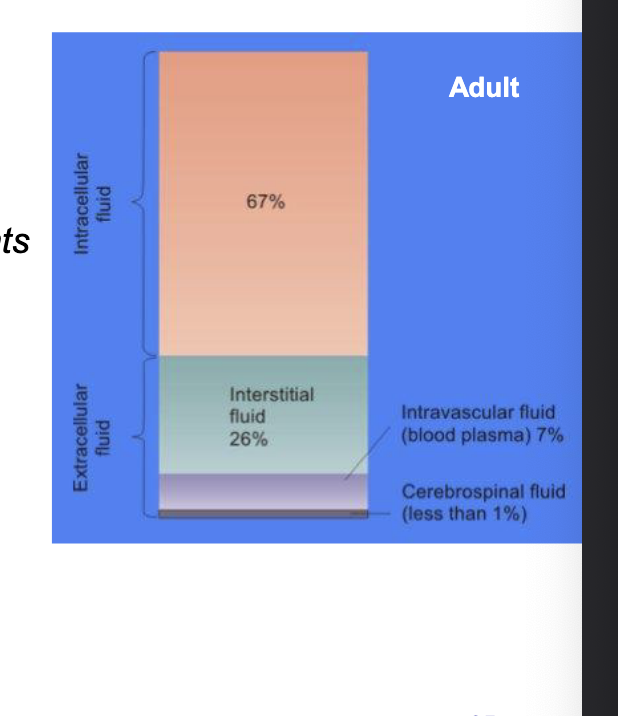
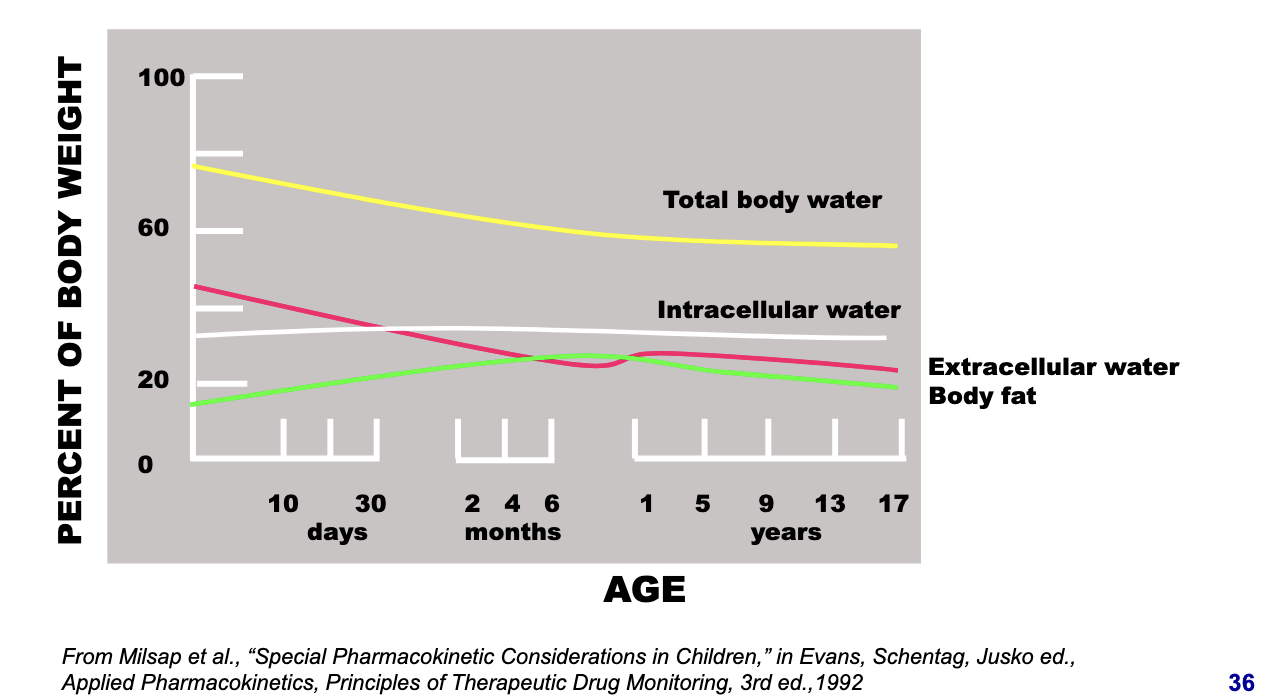
distribution factors: developmental changes in total, intra- & extracellular water & body fat
total body water
starts high in neonates and gradually decreases
higher total water = more distribution space for hydrophilic drugs → higher Vd
extracellular water
very high in neonates and drops with age
hydrophilic drugs tend to distribute in extracellular fluid → higher Vd in neonates
intracellular water
fairly stable across age groups
slightly lower in neonates, increases then plateaus
body fat
low in neonates
increases during infancy and childhood
lipophilic drugs will have lower Vd in neonates due to less fat to accumulate in
metabolism factors
drug metabolism occurs in
liver (majority)
GI tract
kidney
lung
skin
4 principal metabolism pathways
oxidation → phase I, mainly via CYP450
reduction → phase I
hydrolysis → phase I
conjugation → phase II
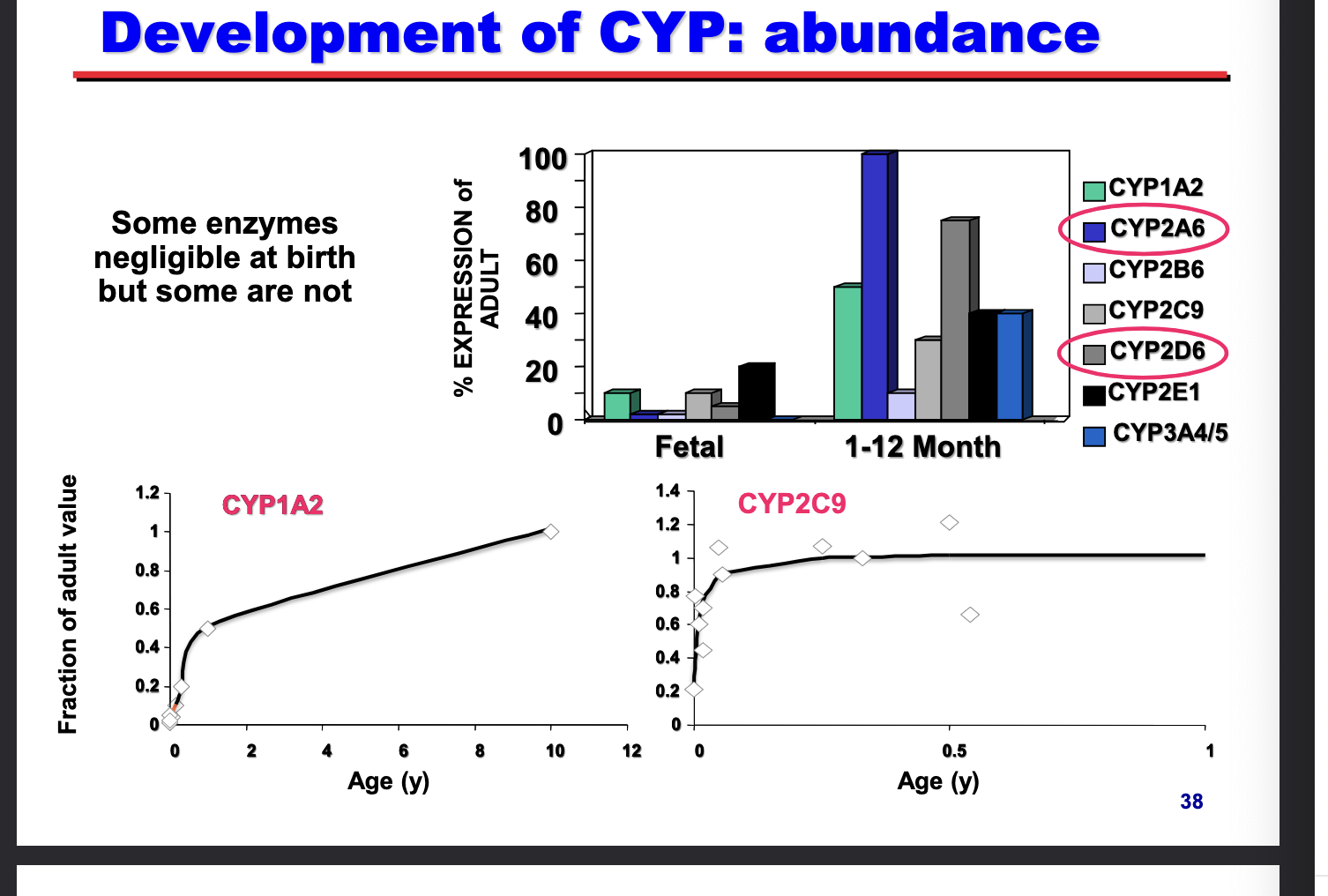
development of CYP: abundance
some enzymes negligible at birth but some are NOT
CYP2A6/CYP2D6 → faster postnatal maturation → most dominant enzymes
CYP1A2 → slow onset, does NOT reach adult levels until around 10 yrs
CYP2C9 → rapid onset, reaches near adult levels within months
metabolism factors
conjugation
glucuronide conjugation
UDP-glucuronyl transferase activity depressed at birth → reaches adult levels by 3 yo
chloramphenicol (antibacterial) accumulation → grey baby syndrome
glucuronidation = only pathway for detoxification
sulfate conjugation
phenosulfotransferase activity higher in neonates than adults
acteaminophen glucuronide conjugates lower but sulfate conjugates higher in neonates
compensatory mechanism
excretion factors
drug & metabolite excretion routes
renally into urine (majority)
biliary into gut
pulmonary thru lung
transfermal thru skin
renal excretion is controlled by:
renal blood flow
GFR
tubular secretion
tubular reabsorption
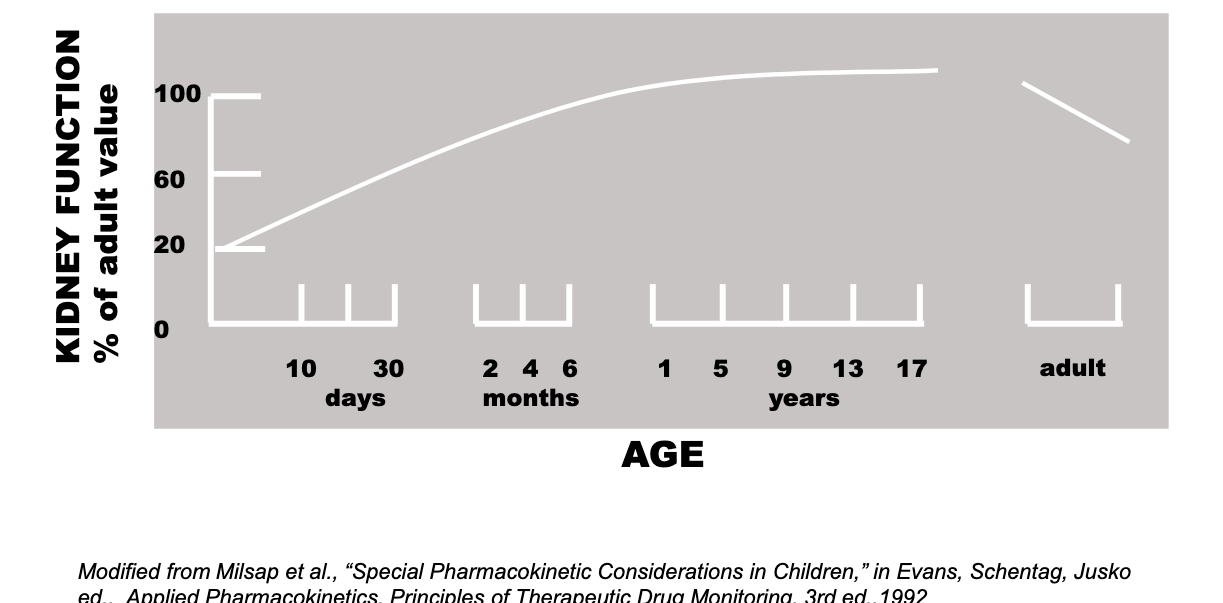
excretion factors: kidney function
developmental changes in kidney function
kidney function increasing till you reach adult levels around like 9??? and then elderly decreases
excretion factors: kidney cont
dosage adjustment based on maturation of kidney function
digoxin CL → lower in neonates
decreased GFR and tubular function
aminoglycoside → correlate with creatinine CL
controlled by GFR
excretion factors: renal function estimation (schwartz equation)
just look at photo bruh
basically a greater factor you multiple length by for children 1-12 years
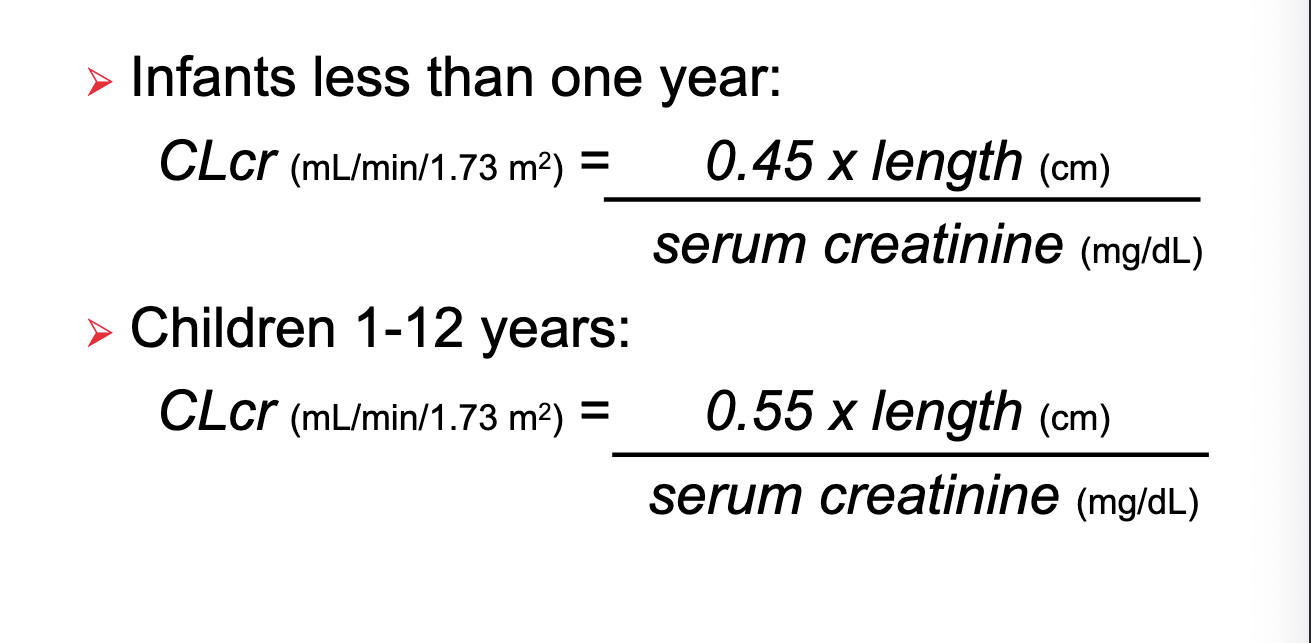
renal function estimation for adults
cockcroft and gault equation
serum creatinine should represent a steady state of renal function

closing remarks for everything before this
understanding the various factors that can alter the PK of a drug in targeted patient population = crucial proper for proper dosage regimen recommendation
integration of ADME principles, population PK analysis approach and simulation technique maximizes the quality/utility of the info generated
data analysis methodology to explain variability
descriptive → population modeling analysis
describes observed data by identifying how pt specific factors affect drug exposure and response
predictive → clinical trial simulation
uses the descriptive model to simulate future clinical trials
population PK/PD modeling
advantage
study broader spectrum of patients
can analyze data pooled from multiple studies
screen for drug-drug interactions (co-medication)
identify important co-variates
may also provide some understanding of drug exposure-response relationships for efficacy and toxicity
limited expert available (who know physiology, pharmacology, pharmaceutics and statics) → teamwork
population PK/PD modeling: fixed and random effects
fixed effect → estimate the population mean PK/PD parameters and their relationship with patient specific co-variate (age, weight…) in order to explain the observed inter-variability in response
random effect - true biological variability → estimate the probability distribution of inter-individual random effect that is NOT explicable by patient specific covariate
random effect — background noise → estimate the residual variability due to measurement error and intra-individual variability
what is clinical trial simulation
generation of virtual patient data by approximating human, disease and drug behaviors with proposed trial designs using mathematical models and numerical methods
human factors
account for variability in:
trial execution characteristics (patient compliance, missing records, demography distribution)
disease model
disease progress models
drug
drug action models (PK, PD, placebo)
clinical trial simulation
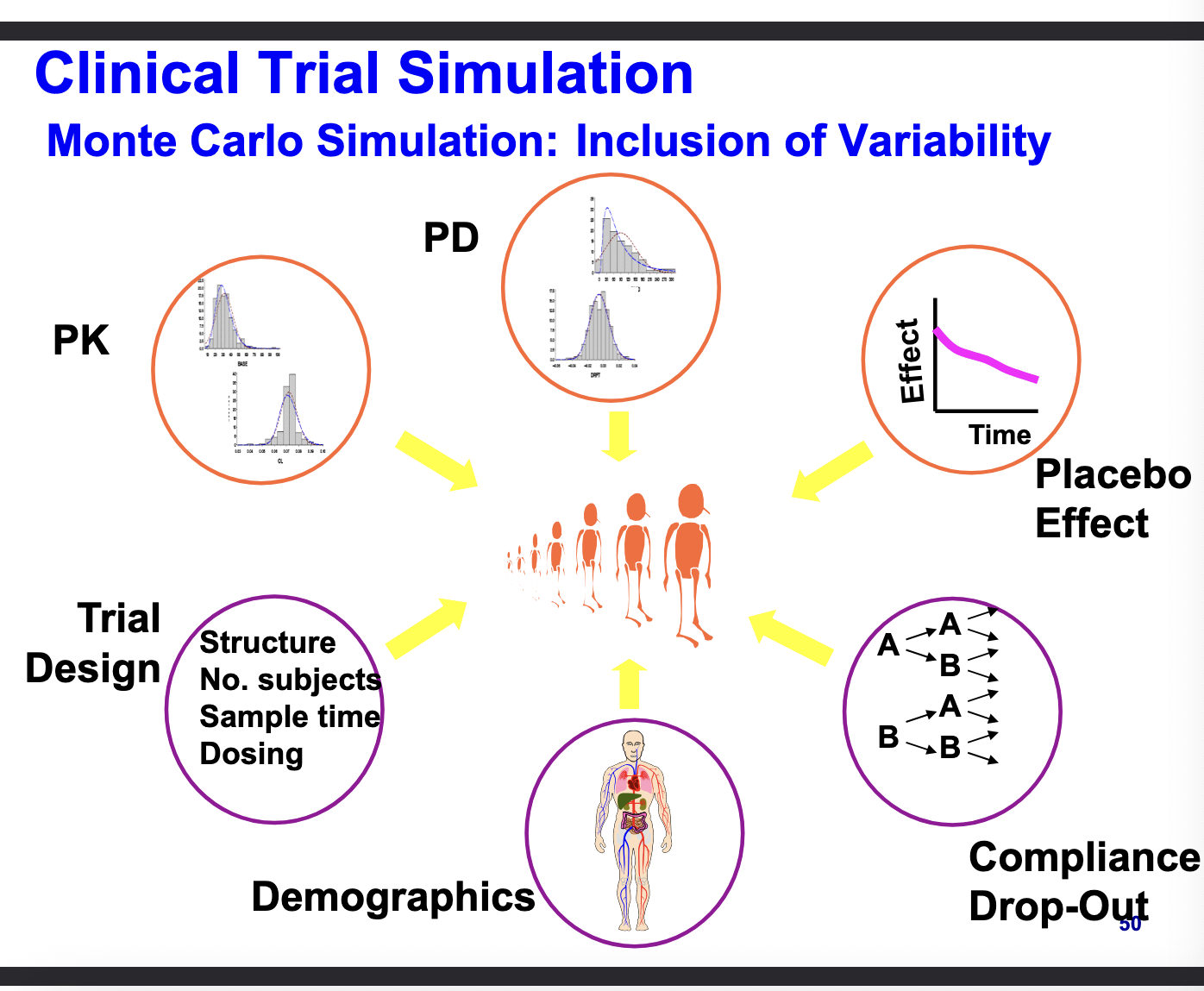
monte carlo simulation: inclusion of variability
computational technique that stimulates thousands of ‘virtual’ patients
randomly samples from distribution of paramters to capture real-world variability
PK
PD
placebo effect
compliance & drop outs
demographics
trial design
designing an optimal trial
objectives → overall goals of an optimal trial
recommend optimal trial designs
to achieve reasonable statistical criteria
assess precision/accuracy of model parameters
specific aims → actionable items that guide trial implementation so things you decide for an optimal trial
provide
dosage regimen
# of subjects
sampling times
patient inclusion/exclusion critera
study period
package insert of the drug

dosage adjustment information from package insert of X


case for dosage recommendation
consider the following scenario and questions
X can be taken with food as X has Large Vd (100L) which indicates that majority of X is NOT in the plasma compartment (or systemic circulation). under such circumstance a slight decrease in absorption rate but NOT in extent (Tmax shifts one hour) will have minimal effect in altering the plasma concentration-time profile hence the resulting treatment effect
food impacts absorption rate by delaying gastric emptying meaning it takes longer for drug to move from stomach to small intestine where most drug absorption occurs
ok so its saying that b/c of food you’re going to have drug being absorbed at a slightly slower rate but since Vd is high it doesn’t matter b/c most of the drug will still enter the tissue aka the extent will be the same itll just be slower
X should NOT be given with milk to avoid chelation with metal ions in milk
X is slightly bound to serum albumin only so it is unlikely for X to displace warfarin from binding site. X is hardly metabolized so it will NOT influence the metabolism of warfarin. however, warfarin has very narrow TI needs to be monitored closely.
X is minimally metabolized and slightly bound to plasma albumin so NO (liver does NOT need to metabolize it so don’t matter)
influence of hepatic impairment on PK of X has NOT been evaluated. because greater than 90% of the dose is excreted in the urine as unchanged drug, hepatic impairment would NOT be expected to have a significant effect on X elimination
look at image for calculation. NO, as calculated from equation, renal function of the patient is 64 mL/min which is only moderately impaired and NO dosage adjustment is recommended under such circumstance according to package insert