Bio98 Lecture 17 Regulation of fatty acids metabolism & degradation of amino acids
1/29
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
30 Terms
Where are the building blocks coming from?
-Malonyl-CoA: build from acetyl-CoA
-Both Malonyl-CoA & Acetyl-CoA are used in FA biosynthesis
-can break down FAs to make Acetyl-CoA; FUTILE CYCLE bc it’d waste all energy generated to rebuild them
-TCA Cycle is also a hub of biosynthesis (fats are createed and stored from other foods like sugar bc of the inhibition of siocitrate dehydrogenase)
citrate → acetyl-CoA → FAs → Lipids
Understand the ways that FA metabolism is regulated
1) FA biosynthesis and oxidation are OPPOSING ACTIVITIES:
when one is activated the other is inhibited (no futile cycle)
2) FA biosynthesis STORES energy, oxidation PRODUCES energy
3) Regulated by citrate and energy state (high/low ATP)
4) Regulated by transporters/compartmentalization
5) Regulated by hormones like insulin and glucagon
6) Regulated by enzyme phosphorylation state
Activation of FA biosynthesis
1) Phosphoprotein phosphatase 2A (PP2A) dephosphorylates the enzyme (citrate) → returns to active state
stimulated by insulin signaling pathway (blood glucose = high)
2) Phosphorylation of ACC I by AMP kinase (AMPK) or protein kinase A (PKA) → ACC adopts less active state.
PKA is stimulated by epinephrine and glucagon (blood glucose = low)
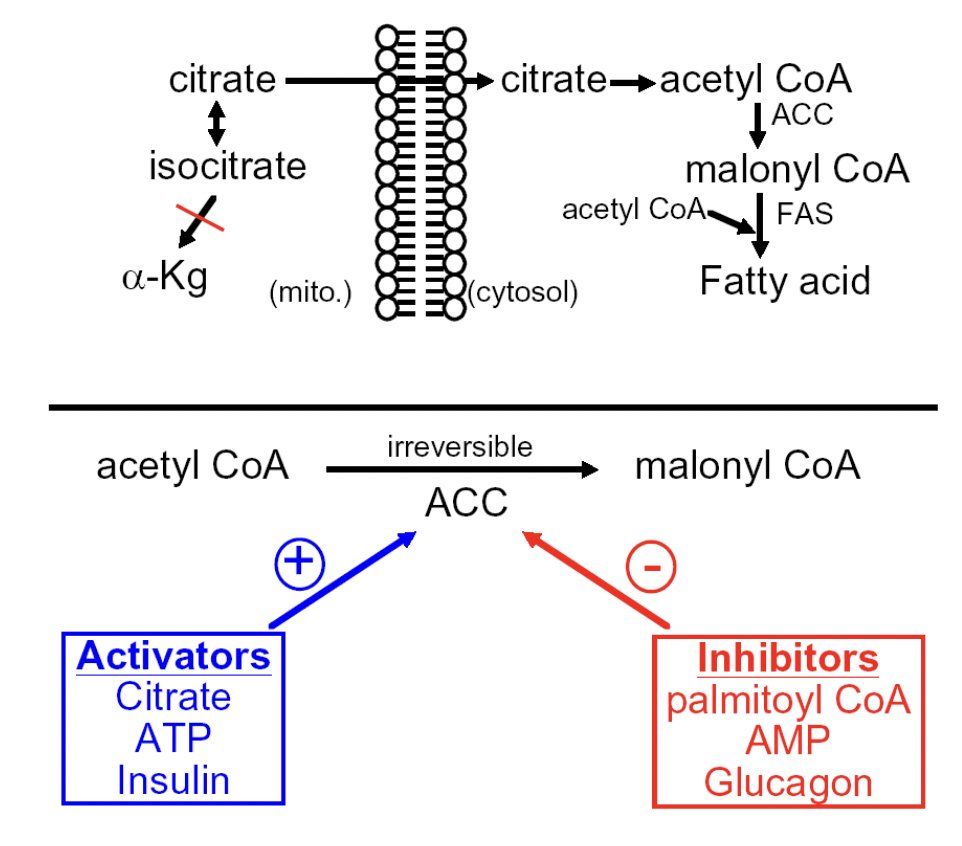
^^ Regulation of ACC (Acetyl-CoA carboxylase)
-activators: citrate, ATP, insulin
-inhibitors: palmitoyl CoA, AMP, glucagon
Regulation of FA oxidation via ?
transport control
FA - CoA in cytoplasm
FA - CoA in mitochondrion : degradation of FATTY ACYL-CoA
—> CARNITINE SHUTTLE involved
- INHIBITOR: malonyl-CoA
—> commits BIOSYNTHESIS not OXIDATION
- EXERCISE —> ACTIVATES oxidation
—> inhibits ACC from producing MALONYL-CoA
—> no MALONYL-CoA is there to inhibit FA transport for fat burning
Regulation of Lipase
- pancreatic enzyme that breaks down FATS into FATTY ACIDS + GLYCEROLS
- HORMONE SENSITIVE enzyme (lipase)
—> ACTIVATOR: glucagon
—> INHIBITOR: insulin
- EXERCISE also ACTIVATES this enzyme (lipase)
Understand different impacts of anerobic vs aerobic exercise
-Anaerobic: breakdown sugar (glycolysis), accumulate lactate.
Training: reduce fatigue and increase power burst (think back to the alligator)
-Aerobic: mostly burn fat, minor breakdown of sugar, produce CO2.
Training: reduce fatigue and increase endurance
Aerobic exercise training causes increases and decreases in?:
1) increase in hexokinase rate
2) Increase gluconeogenesis ability
3) Decrease in total LDH activity
Purpose: to decrease lactate (causing soreness) formation
4) Increase the # and size og mitochondria; enhances:
pyruvate dehydrogenase enzyme rate
Krebs cycle enzyme rate
Fatty acid Beta-oxidation enzyme rate
Electron transport chain
5) Fatty acid availability for oxidation due to increased:
lipase activity: increased uptake of fatty acids by skeletal muscle
Acyl-CoA synthetase activity: increases activation of fatty acids for transport into mitochondria
Carnitine transporter activity
Anaerobic training: it increases what and has little effect on what?
1) Increases anaerobic capacity
more muscle cells to use more ATP
increased glycolysis rate (raise PFK rate)
increased gluconeogensis rate
increase lactate tolerance in blood and muscle
• But has little effect on:
• Oxidative capacity (e.g. mitochondria number, burning fat, TCA cycle or ET)
• Cardiovascular adaptation (heart pumping rate)
Digestion: Stomach
-Enzymes are released as (1).
-Pepsinogen is activated by (2).
1) zymogens: an inactive form meant to prevent digestion of proteins within the cells making them
2) the low pH of the stomach to the active enzyme pepsin.
Digestion: secondary enzyme
-Other zymogens are activated by other enzymes.
ex. chymotrypsin is activated by trypsin
Methods of Amino Acid Digestion
1) Digested amino acids enter the blood stream in the intestine
—> can import SHORT POLYPEPTIDES or individual AAs into the intestinal wall
—> PEPTIDASE can break down the polypeptides into SINGLE AAs
2) Proteins can also be broken down inside cells + transported to blood
—> LYSOSOMES can digest proteins —> released into blood as SINGLE AAs
—> ENDOSOMES can ingest proteins —> released into blood as INTACT PROTEIN
targeted protein degradation
- need to RECYCLE proteins after they fulfilled their purpose
- need to DESTROY proteins if they are accumulating or aggregating or causing issues
ubiquitin
- small protein "tag" that COVALENTLY LINKS to a targeted protein that shows that PROTEIN NEEDS to be DEGRADED by a PROTEASOME
- gets popped off and REUSED
- repeated cycles lead to attachment of ADDITIONAL ones
—> POLYUBIQUITINYLATION (regulates protein levels)
proteasome
- protein DEGRADATION machine
- degrades any protein that has an ubiquitin "tag"
- MUTATION of this enzyme can cause: PARKINSON'S DISEASE, DEMENTIA, MISCARRIAGE
non-protein degradation tangent
- other tags that are similar to ubiquitin
- SUMO (small ubiquitin-like modifier)
—> regulates enzyme activity rather than destroying them
—> targeted proteins have ALTERED FUNCTIONS
- ISG15 (interferon stimulated gene of 15 kDa) is ANTIVIRAL
—> looks like 2 fused ubiquitin domains in 1 polypeptide chain
dietary amino acids
- can be used to MAKE proteins + be used to STORE energy for later
- organisms DO NOT have a way to store AAs for energy
—> use CARBOHYDRATES (glycogen) + LIPIDS (triaglycerols)
- AAs can be converted to things like α-keto acids BUT there is a BUILDUP of AMMONIA
—> amine can be lost as ammonia
α-keto acids
- AAs can be converted to this version by the enzyme TRANSAMINASE
- AMINE that is released can be converted to AMMONIA
- undergoes TCA cycle after NH4+ is released so that they can be further metabolized to produce ATP
glucose-alanine shuttle
- nitrogenous wastes generated in MUSCLE are transferred to PYRUVATE —> generates ALANINE
—> transamination of ANIMO ACID —> α-KETO ACID can occur between pyruvate and alanine
- ALANINE is transported from MUSCLE to LIVER through BLOOD
- alanine can undergo transamination + oxidative deanimation (cycle between α-ketoglutarate + glutamate)
—> AMMONIA (NH4+) is generated and is released as UREA
- pyruvate in the LIVER then undergoes GLUCONEOGENESIS
Urea cycle and how it is linked to the TCA cycle
- process in which excess ammonia (NH4+) that is released from dietary AAs is made into CARBONYL PHOSPHATE (high energy molecule) w/ carbamoyl phosphate synthetase; undergoes this cycle to make it into UREA (excreted)
- uses ATP
- primarily occurs in the LIVER CELLS
Urea Cycle Step 0. To produce carbamoyl phosphate, how many ATPs need to be spent?
2 ATP
Urea Cycle Step 1
- CARBAMOYL PHOSPHATE reacts with ORNITHINE (2 Ns; 21st AA) to form CITRULLINE (3 Ns)
- enzyme involved is ORNITHINE TRANSCARBAMOYLASE (OTC)
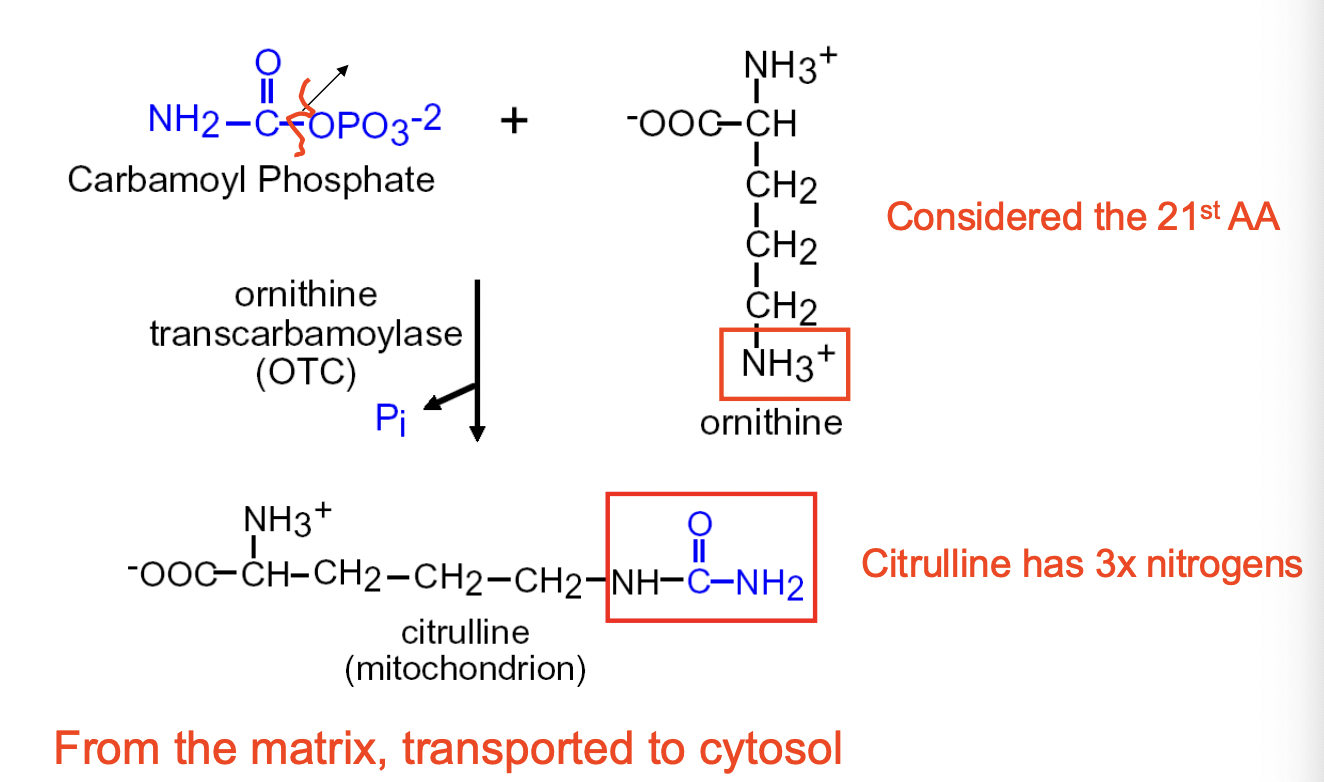
- happens in the MITOCHONDRIAL MATRIX —> transported to CYTOSOL
Urea Cycle Step 2
- CITRULLINE (in cytoplasm) reacts with ASPARATE to form ARGINOSUCCINATE (4 Ns)
- enzyme involved is ARGINOSUCCINATE SYNTHETASE
- ATP is needed (AMP + 2 Pi released)
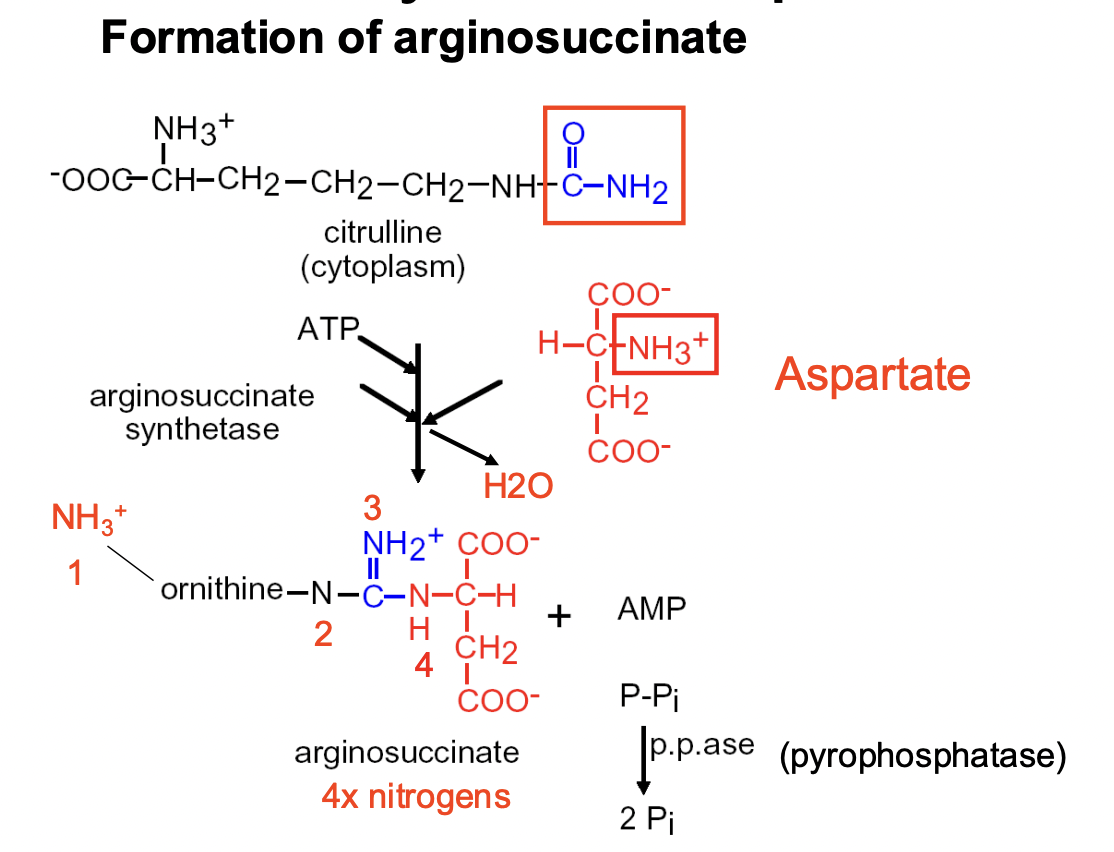
- happens in the CYTOPLASM/CYTOSOL
Urea Cycle Step 3
FORMATION OF ARGININE
- ARGINOSUCCINATE turns into ARGININE (4 Ns)+ FUMARATE (side-product)
- enzyme involved is ARGINOSUCCINASE
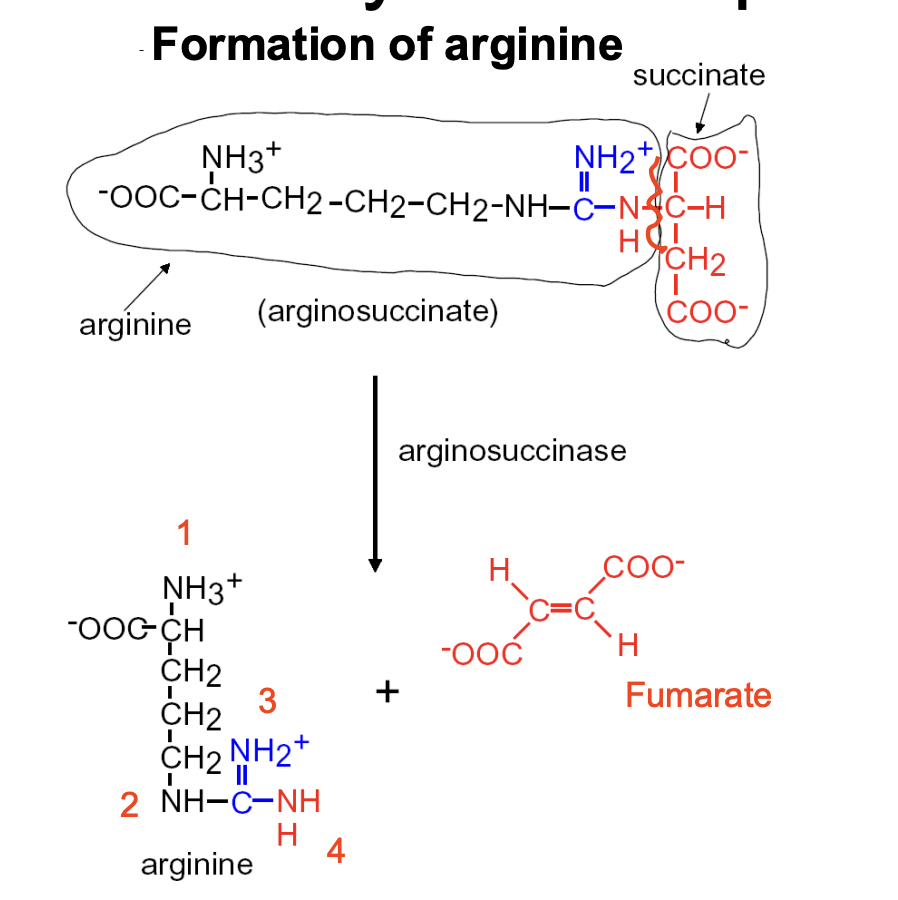
- happens in the CYTOSOL
Urea Cycle Step 4
FINAL STEP: formatioln of urea & ornithine
- ARGININE turns into ORNITHINE + UREA (cycle completed)
—> urea goes through the blood into the kidneys —> released in URINE
—> ornithine feeds back into the 1st STEP
- enzyme involved is ARGINASE (+ H2O)
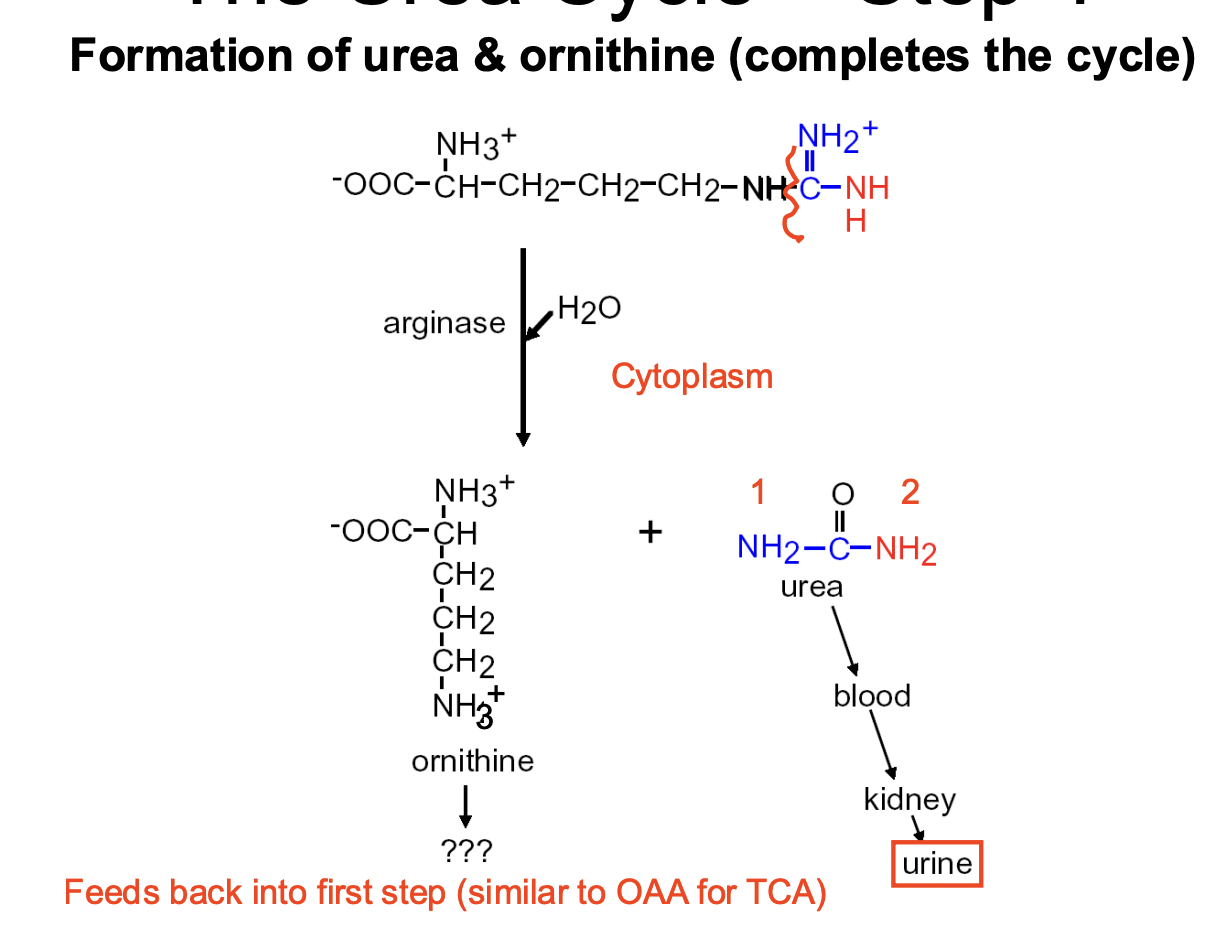
- happens in the CYTOSOL
How does the urea and TCA cycle connect?
Asp-Arginosuccinate shunt connect urea and TCA cycles
—> "communicates" between the two cycles
- transports malate (metabolite) into mitochondria
- TCA cycle in the mitochondrial matrix
- urea cycle in the cytosol
Example of TCA → AA degradation → Urea
Step #1: GLUTAMATE carries 1 NITROGEN
—> glutamate buildup can be TOXIC (ex. MSG) + needs to be broken down
Step #2: GLUTAMINE carries 2 NITROGENS
—> glutamate + ATP + NH4+ = GLUTAMINE
—> carried out by GLUTAMINE SYNTHETASE
—> NOT TOXIC but needs to be broken down
- GLUTAMINE ==> URINE
—> GLUTAMINE is transported through blood into the LIVER
—> GLUTAMINE is converted into GLUTAMATE + NH4+
—> carried out by GLUTAMINASE (liver and kidneys)
—> GLUTAMATE turns into α-KETOGLUTARATE + another NH4+ released
—> carried out by GLUTAMATE DEHYDROGENASE
—> NH4+ undergoes UREA CYCLE —> released in URINE
glucose-alanine cycle
- PYRUVATE is converted into ALANINE via ALANINE AMINOTRANSFERASE
—> GLUTAMATE transforms into α-KETOGLUTARATE
- ALANINE is then transported from MUSCLES to LIVER through the BLOOD
- ALANINE is converted back to PYRUVATE via ALANINE
—> α-KETOGLUTARATE is transformed into GLUTAMATE
- any EXCESS AMMONIA (NH4+) undergoes the UREA CYCLE
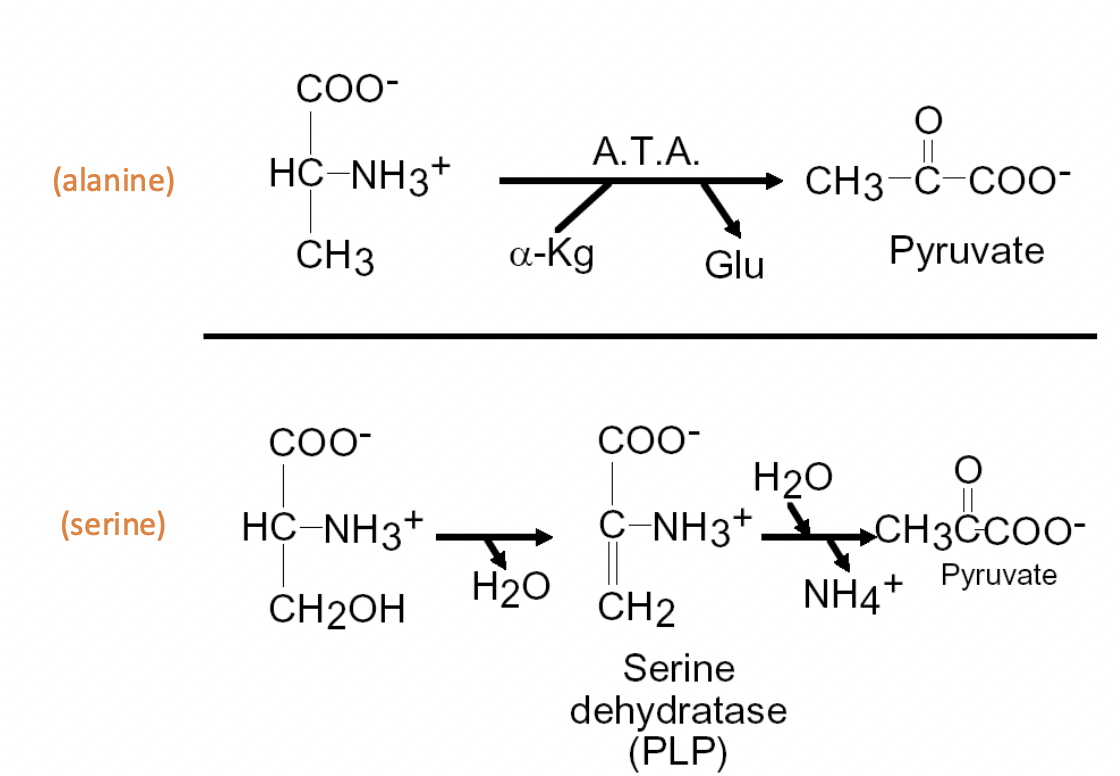
Oxidation of Selected Amino Acids: Same number of C as TCA
Alanine: ATA equals alpha-Kg & Glu = pyruvate
Serine: loss of H2O + serine dehydratase (PLP) + addition of H2O, out NH4+ = pyruvate
Defective A.A. oxidation leads to issues
Phenylketonuria (PKU): Lack ability to degrade phenylalanine
Albinism: Tyrosinase defect causes inability for melanin to be produced
Maple-syrup urine disease (MSUD): BCKDH defect; prevents B.C α-keto acid from undergoing TCA cycle
In total. To produce one Urea. How many ATPs need to be spent?
3