503 Clin Med - Hypothalamus/Pituitary/Adrenal Disorders
1/103
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
104 Terms
Causes of hypopituitarism? What is most common?
most common = pituitary infarction
tumor or surgical removal
damaged pituitary stalk
decreased hypothalamic releasing hormones
inability to produce hormones (synthesis)
Most common hormonally active pituitary tumor?
prolactinoma
Dwarfism: Etiology (2)
1) Congenital: GH deficiency (Autosomal Recessive)
2) Acquired: GH deficiency due to radiation, compression, or trauma
Dwarfism: Pathophysiology
Congenital
Acquired
Congenital: Mutation in gene that codes for GH receptor -> No GH release to tissues
Acquired: Injury caused by radiation, compression, trauma to pituitary cells --> low GH release
Dwarfism: Epidemiology (2)
Congenital: Newborns
Acquired: 40s-50s (pituitary tumor)
Dwarfism: Clinical Presentation (Congenital)
Metabolic
Skin
Appendages
Body Structure/Bones
Developmental
Newborn:
- Hypoglycemia
- Jaundice
- Small penis
- Reduced birth length
Childhood:
- Short stature
- Delayed bone age
- Delayed puberty
Dwarfism: Clinical Presentation (Acquired)
Body Habitus
Neurological
Systemic
NO HEIGHT INVOLVEMENT
- Central obesity
- Impaired concentration, memory, and depression
- Reduced bone/muscle mass
What would be indicative of a mass or lesion causing dwarfism?
Headache or visual field defects (bitemporal hemaniopia)
Before specific diagnostics tests are performed, how should children with suspected dwarfism be worked up?
growth charts, bone age, CBC (looking for anemia), malnutrition or malabsorption causes
Dwarfism: Dx
IGF-1
GH
MRI
IGF-1: Low
GH: Low
MRI: if suspected hypothalamic or pituitary mass
What is the disadvantage of IGF-1 for diagnosing dwarfism? What test can you do instead?
only kids >5 who are adequately nourish
IGFBP-3 is not affected by nutritional status
Dwarfism: Tx (3)
- SQ recombinant GH immediately post-diagnosis 3x/wk
- Stop all estrogen
- Refer to cardiology (increased cardiovascular morbidity)
Laron Syndrome (Congenital Dwarfism): Etiology/Pathophysiology
Etiology:
Autosomal recessive
Pathophysiology:
Mutation in GH receptor -> GH resistance + IGF deficiency
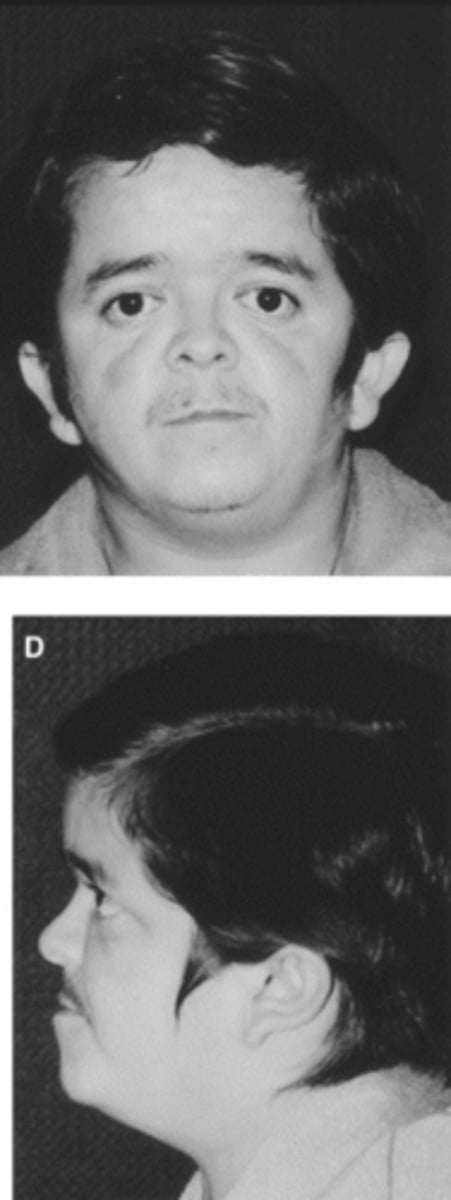
Laron Syndrome (Dwarfism): Clinical Presentation
Body Structure
Body Habitus
Head
Neurological
- Short stature
- Central obesity
- Large forehead, depressed nasal bridge, small jaw
- Hypoglycemic seizures
Laron Syndrome (Dwarfism): Dx
IGF-1
GH
IGF-1: Low
GH: High
Laron Syndrome (Dwarfism): Tx
Mecasermin (resistant to normal recombinant GH).
Diabetes Insipidus: Etiology
Insufficient ADH release from posterior pituitary -> no water retention -> polyuria
Diabetes Insipidus: Pathophysiology (3)
1) Central/Neurogenic: decreased ADH release
- Primary central: genetic, autoimmunity, induced by meds
- Secondary central: lesion of hypothalamus or pituitary
2) Nephrogenic:
Renal collecting tubules insensitive to ADH due to abnormal V2 receptors (lithium)
3) Dipsogenic:
Excessive fluid intake lowers plasma osmolality below threshold of ADH secretion
Diabetes Insipidus: Clinical Presentation
- Polyuria
- Polydipsia
- Nocturia
- Ice water craving
Diabetes Insipidus: Dx
24hr Urine
Urine Osmolality
Urine Specific Gravity
Serum Sodium
24hr Urine: >2L in 24 hours (should be a lot)
Osmolality: Low (<300)
Specific Gravity: Low (<1.006)
Sodium: Hypernatremia (high plasma osmolality)
Can you rule out diabetes insipidus if there is less than 2L of urine in 24 hours?
Yes.
How do you differentiate central/neurogenic diabetes insipidus from nephrogenic?
Plasma Vasopressin
Neurogenic: Low (<1)
Nephrogenic: Normal/high (>2.5)
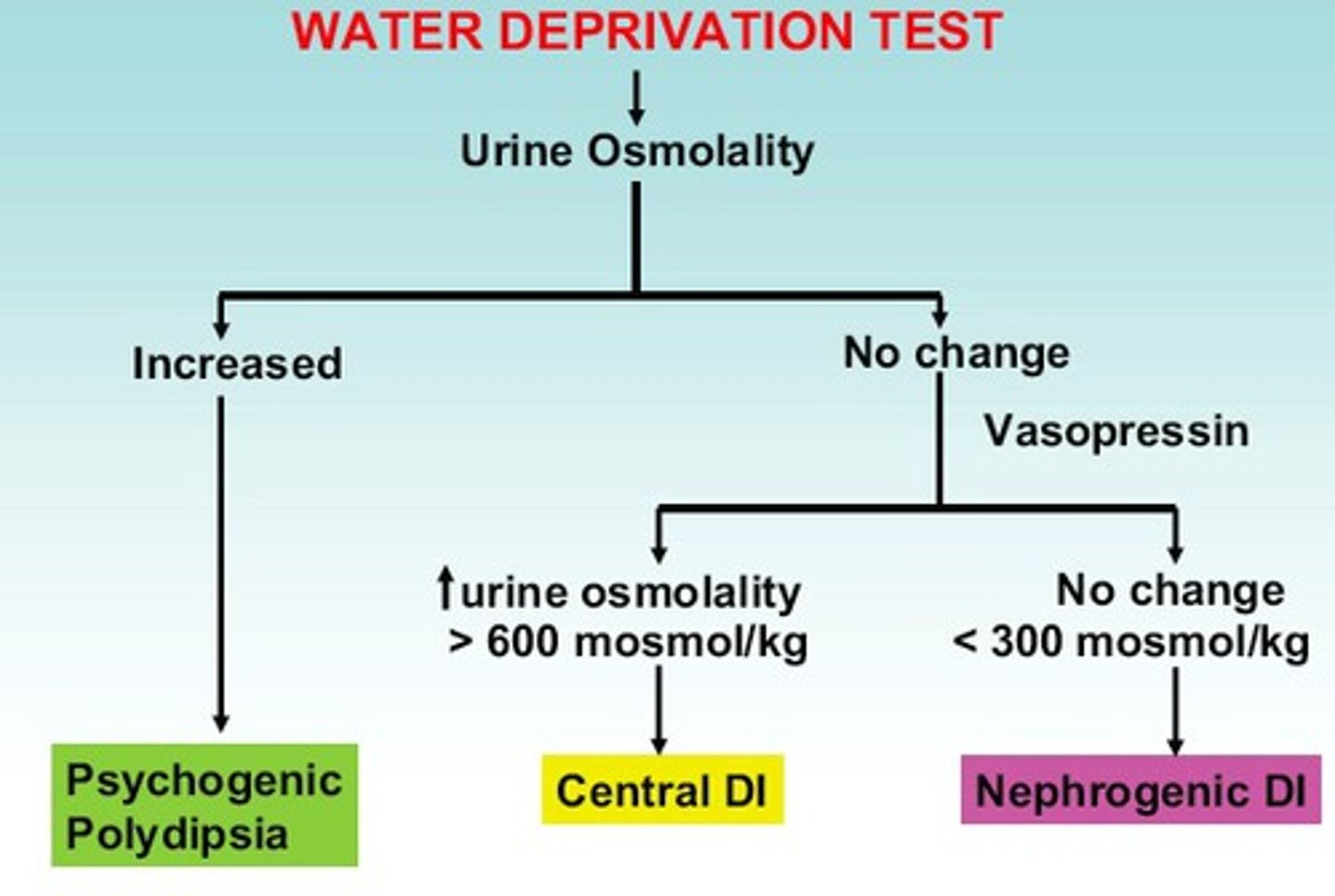
How is a water deprivation test done?
- Measure AM weight, serum electrolyte, and urine osmolality
- Collect urine hourly
- Allow dehydration (no PO intake)
- Recheck serum and urine osmolality
- Administer vasopressin
- Re-check serum and urine osmolality after 1 hour
What are the expected results for normal, central/neurogenic DI, and nephrogenic DI in the water deprivation test?
Normal:
- urine osmolality after dehydration > plasma osmolality
- after DDAVP, osmolality does not increase >5%
Neurogenic:
- unable to concentrate urine when dehydrated, remains dilute
- after DDAVP --> urine osmolarity increases
Nephrogenic:
- no response to DDAVP --> urine remains dilute
Diabetes Insipidus: Dx (MRI)
Neurogenic (primary central, not secondary)
Nephrogenic
Dipsogenic
What is normal?
Neurogenic: Absent posterior bright spot
Nephrogenic: Decreased posterior bright spot
Dipsogenic: Posterior pituitary bright spot present (normal)
Diabetes Insipidus: Tx
Mild Cases
Severe Cases (first and second line)
Mild: No treatment, maintain adequate fluid intake
Severe: Desmopressin (HCTZ if adverse rxn)
SIADH: Etiology
Ectopic Secretion of ADH:
Surgery, cancer (SCLC), or medications (desmopressin, anti-depressants)
SIADH: Pathophysiology (3-steps)
High ADH -> collecting ducts more permeable -> high water retention
SIADH is ____volemic, _____tonic, _______natremic condition.
euvolemic, hypotonic, hyponatremic
SIADH: Clinical Presentation
Mild
Moderate
Severe
Based on degree of hyponatremia:
Mild: Asymptomatic
Moderate: Neurological Issues (personality change, weak tendon reflexes, muscle weakness)
Severe: Coma, seizures, hypothermia, cranial nerve palsies
SIADH: Dx
Serum Sodium (mild, moderate, severe)
Mild: >120
Moderate: 105-120
Severe: <105
SIADH: Dx
Plasma Osmolality
Urine Osmolality
Plasma Sodium
Urine Sodium
Plasma Osmolality: Low
Urine Osmolality: High
Urine Sodium: >20
Plasma Sodium: Low
What do furosemide and urea do?
Diuresis.
SIADH: Tx
Mild (1)
Moderate (1)
Severe (2)
Mild: Restrict fluids (800-1000mL/day)
Moderate: Further restrict fluids (500mL/day)
Severe: Hypertonic saline and furosemide/urea (ICU admit)
At what point do know you have corrected severe SIADH and can slow down or stop treatment?
Sodium (125) and CNS involvement ceases.
Why is serum sodium normally corrected no more than 10-12 in 24 hours?
Central pontine demyelination:
- Neurologic injury/paralysis
Acromegaly: Etiology
Pituitary adenoma --> high GH secretion
(rarely ectopic GH or GnRH secretion)
Acromegaly: Pathophysiology
High GH secretion -> high IGF-1 -> hyperglycemia, connective tissue proliferation, bony proliferation, and hyperphosphatemia
Acromegaly: Epidemiology
Adults (sporadic, not familial) - growth plates have closed
Acromegaly: Clinical Presentation
Extremities
Head
Jaw
Voice
Extremities: Enlarged with carpal tunnel and large feet
Head: Enlarged skull/facial features, macroglossia
Jaw: Prominent jaw with enlarged tongue
Voice: Coarse
Acromegaly/Gigantism: Dx
IGF-1
MRI
X-Ray
IGF-1: VERY HIGH (if normal, rule out)
MRI: Likely will show pituitary tumor
X-Ray: Tufting of phalanges, thick heel pad
How would you use a glucose suppression test to rule out acromegaly?
- Give oral glucose syrup and measure GH after 60 minutes
- If GH is suppressed (< 0.4), then acromegaly is excluded
- Diagnose acromegaly if glucose fails to suppress GH
Acromegaly/Gigantism: Tx
Surgical
Transphenoidal Pituitary Surgery:
- Remove adenoma, preserve function
- Give corticosteroids over 1 week
What follows pituitary surgery if complete remission is not achieved? What must be done for life following this surgery?
Stereotactic Radiosurgery:
- Lifelong ASA because of small vessel strokes
Acromegaly/Gigantism: Tx
Pharmacological (3)
For incomplete biochemical remission post-operatively:
- Cabergoline (dopamine agonist)
- Octreotide/lanreotide (somatostatin analog)
- Pegvisomant (GH receptor antagonist)
Gigantism: Etiology
Pituitary macroadenoma -> high GH secretion
Gigantism: Pathophysiology
High GH -> INCREASED LONG BONE LENGTH
Gigantism: Epidemiology
CHILDREN:
- Epiphyseal plates have NOT closed
Gigantism: Clinical Presentation
Body Structure
Extremities
Development
- Very tall stature
- Large hands and feet
- Hypogonadism, delayed puberty
Medical complications of acromegaly?
HTN, cardiomegaly (increased mortality from CVD), glucose intolerance/DM, hypopituitarism (after surgery), obstructive sleep apnea
Pituitary Adenoma: Etiology/Pathophysiology
Etiology:
Benign neoplasm on gland
Pathophysiology:
Autonomous and excess hormone secretion without signaling from the hypothalamus -> no negative feedback
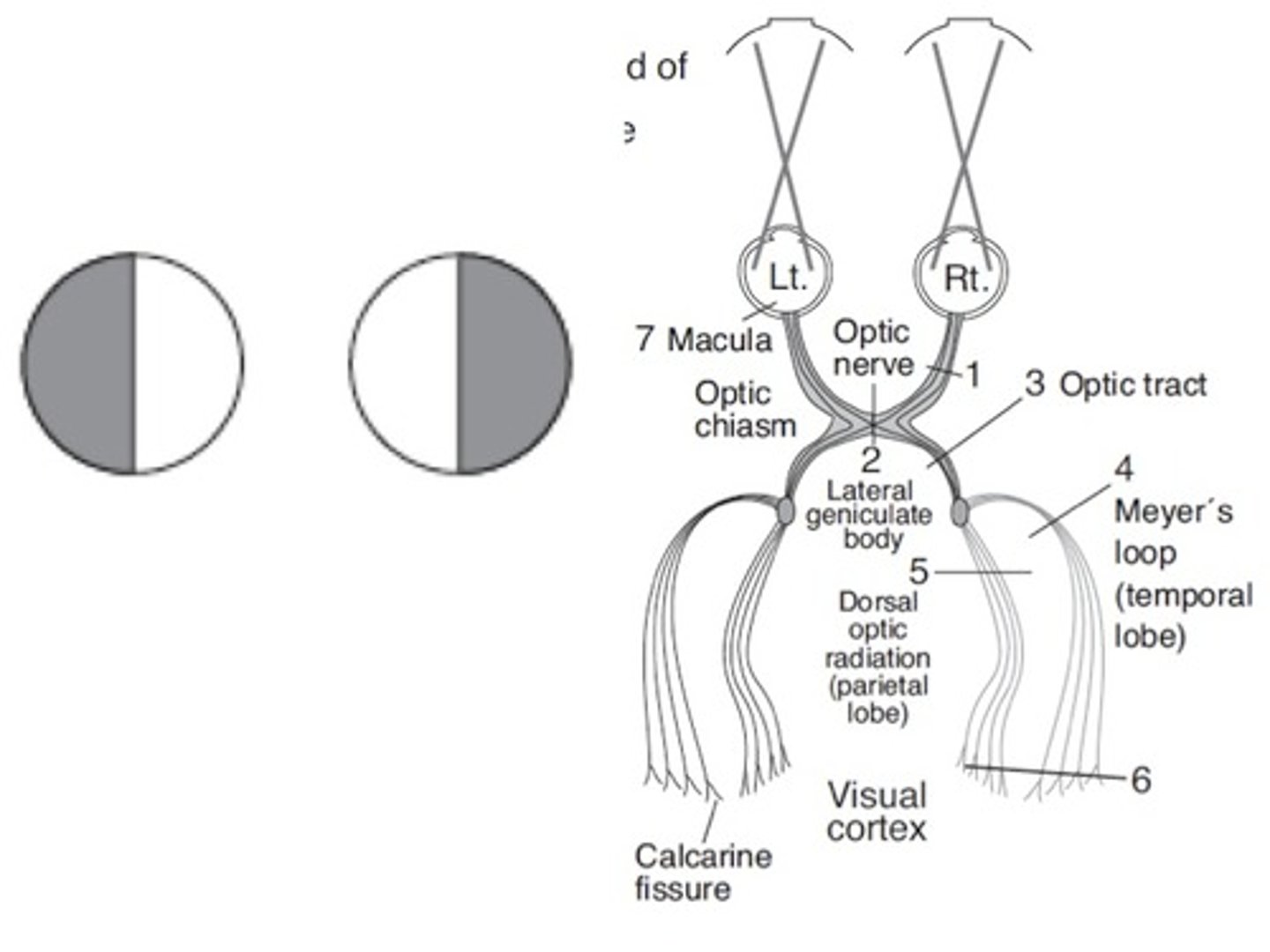
Pituitary Adenoma: Clinical Presentation (Mass Effects)
- Headache
- Vision loss (bitemporal hemianopia d/t compression)
- Diplopia
- Ptosis
- Ophthalmoplegia
- Decreased facial sensation (d/t CN compression)
Pituitary Adenoma: Clinical Presentation (Prolactinoma)
- Amenorrhea
- Galactorrhea
- Infertility
- Decreased libido
Pituitary Adenoma: Dx (by tumor type)
Prolactinoma
TSH
ACTH
Prolactinoma: High prolactin (check in morning)
TSH: elevated T4 and TSH
ACTH: 24 hour urine free cortisol, dexamethasone suppression test, plasma ACTH levels
Pituitary Adenoma: Dx
Imaging
MRI: With gadolinium before/after and specific cuts
CT: No contrast, coronal plane
Pituitary Adenoma: Tx
Surgical
Transsphenoidal resection (w/ radiation as an adjunct)
- Follow with medical hormone replacements if needed
Pituitary Adenoma: Tx (prolactinoma)
Dopamine Agonist:
- Cabergoline
- Bromocriptine
Addison's Disease: Etiology (MCC)
Autoimmune destruction of adrenal cortex.
Addison's Disease: Etiology (5 Less Common)
1) Infection (TB)
2) Adrenal hemorrhage (surgery or trauma)
3) Drug-Induced (mitotane/abiraterone)
4) Adrenoleukodystrophy (X-linked, males mainly)
5) Congenital (autosomal recessive, 21-hydroxylase deficiency)
Addison's Disease: Pathophysiology
Low corticosteroid and mineralocorticoid synthesis -> low cortisol
Will ACTH be elevated or decreased in Addison's disease? Why?
Is Addison's primary or secondary adrenal insufficiency?
ELEVATED
- Hypothalamus and pituitary work, but adrenal gland does not
- Primary insufficiency
Secondary Adrenal Insufficiency:
Etiology
MCC
What hormones are effected?
Etiology: anterior pituitary gland not making ACTH
MCC: exogenous steroid use (negative feedback on ACTH)
Hormone: only cortisol, adrenal still functioning and making mineralocorticoids
Addison's Disease: Clinical Presentation (4 main, 2 general)
Gradual onset over years:
- (Orthostatic) Hypotension
- Anorexia
- Skin hyperpigmentation
- Salt craving
- N/V/D, abd pain
- Cerebral edema --> gait disturbances
Addison's Disease: Dx
ACTH
Plasma Cortisol
Cosyntropin Stimulation
Imaging
BMP
ACTH: High
Plasma Cortisol: Low (especially morning)
Cosyntropin Stimulation: No rise in cortisol - diagnostic
Imaging: CT scan (if not autoimmune)
BMP: hyponatremia, hyperkalemia
How would the CT scan be different if Addison's disease was caused by TB instead of autoimmune? When is imaging more useful?
Autoimmune = small, non-calcified glands --> appear normal on CT (not useful)
TB or granulomatous = enlarged and calcified --> more useful diagnostic
Addison's Disease: Tx (2)
Corticosteroid Replacement:
- Hydrocortisone or prednisone
Mineralcorticoid Replacement:
- Fludrocortisone acetate
When should steroid replacement doses be increased in patient with Addison's?
sick, surgery, infection, trauma
summer time or extreme exercise (lose salt in sweat)
Congenital Adrenal Hyperplasia: Etiology/Pathophysiology
Etiology:
Autosomal recessive disorder -> no enzyme for cortisol synthesis (21-hydroxylase)
Pathophysiology:
Low cortisol + high ACTH = adrenal hyperplasia --> overproduction of other adrenal cortex products: androgens
Adrenal hyperplasia results in the overproduction of what?
Mineralcorticoids or androgens.
Congenital Adrenal Hyperplasia: Clinical Presentation
Female
Male/Female
Female: Virilization (due to high androgens)
Male/Female: Salt wasting
Congenital Adrenal Hyperplasia: Dx
ACTH
Serum Cortisol
ACTH Stimulation
17-Ketosteroid
ACTH: High
Serum Cortisol: Low
ACTH Stimulation: Unresponsive (cannot make cortisol)
17-Ketosteroid: High
Congenital Adrenal Hyperplasia: Tx
None.
Adrenal Crisis: Etiology (3)
Medical Emergency:
- Stressors (infection, surgery, stress)
- Medications that impair adrenal hormone synthesis (ketoconazole, mitotane)
- Steroid withdrawal
Adrenal Crisis: Pathophysiology
High corticosteroid demand with no increased production.
What specific history will you look for if an adrenal crisis is occurring in patient?
1) Addison's Disease
2) Adrenal Hyperplasia
Adrenal Crisis: Clinical Presentation
- Refractory hypotension (still hypotensive after fluids and pressors)
- Severe abdominal pain
- Shock
- Disorientation
- Dehydration
Adrenal Crisis: Dx
IDENTIFY STRESSOR/INFECTION/CAUSE:
- Blood
- Sputum
- Urine/blood cultures
Adrenal Crisis: Tx (2 main, 1 other)
- Hypotension: IV fluids (electrolytes and dextrose)
- Steroid Replacement: IV hydrocortisone
- Antibiotics
Cushing's Syndrome: Etiology
ACTH-Independent (2)
ACTH-Dependent (2)
Excess cortisol production:
- Adrenal Cortex Tumor: (primary, ACTH-independent)
- Iatrogenic, ectopic use of corticosteroids (ACTH-independent)
- Pituitary Adenoma: Cushing Disease (secondary, ACTH-dependent)
- Ectopic Tumor (secondary, ACTH-dependent)
Cushing's Syndrome: Pathophysiology (4-steps)
Catabolic Effects of Excess Cortisol:
- Muscle wasting
- Increased serum calcium (bone resorption)
- Loss of collagen/thinning of skin
- Melanocyte stimulation
Cushing's Syndrome: Clinical Presentation
- Purple Striae: Abdomen, thighs, breasts
- Buffalo hump
- Central obesity
- Moon face (with redness)
- HTN
- Osteoporosis
- ED, ameorrhea
How do you differentiate an ACTH-independant and ACTH-dependent cause of Cushing's disease in terms of ACTH and cortisol? Which is primary/secondary?
ACTH-Independant: ACTH low, Cortisol high (primary)
ACTH-Dependant: ACTH high, Cortisol high (secondary)
Cushing's Syndrome: Dx
Serum cortisol
Dexamethasone Suppression
24 Urine Free Cortisol
Cortisol: High (elevated @ midnight = diagnostic, loss of diurnal variation)
Dexamethosone Suppression: abnormal, cortisol >1.8
Urinary Free Cortisol: >100
Cushing's Disease: Dx (Imaging)
ACTH-Independant (primary)
ACTH-Dependant (secondary)
ACTH-Independant: CT of adrenals --> identify masses/lesion
ACTH-Dependant: MRI of pituitary
What should you do if MRI of pituitary shows normal or tiny irregularity in ACTH-dependent Cushing's?
First: selective catheterization of interior petrosal sinus veins draining pituitary --> measure ACTH levels compared to peripheral vasculature
Then: if sinus ACTH levels > 2x peripheral levels, indicative of pituitary Cushing's
If not: search for ectopic source of ACTH with CT scan of chest, abdomen, thymus, pancreas, adrenals
Cushing's Disease: Tx (3 surgical)
ACTH-Dependant (Pituitary):
- Transphenoidal resection of adenoma
ACTH-Dependant (Ectopic):
- Resect tumor or do laparoscopic bilateral adrenalectomy if it cannot be located
ACTH-Independant:
- Laparoscopic bilateral adrenalectomy
Cushing's syndrome: Tx
Pharmacologic (post-adrenal resection)
Pharmacologic (post-pituitary resection)
Adrenal resection: mitotane if suspected mets, hydrocortisone replacement if cortisol withdrawal
Pituitary: replace glucocorticoids
Cushing's Syndrome: Tx (no surgery)
Pasireotide, ketoconazole
Cushing's Syndrome: prognosis and complications
cognitive or psychiatric impairment
compression fractures from osteoporosis
HTN
**serious morbidity if untreated
Adrenal Cortex Neoplasm: Etiology
Somatic mutation in TP53 gene (tumor supressor).
Adrenal Cortex Neoplasm: Clinical Presentation
- Likely will not have S/S of excess corticosteroids
- Liver/lung metasteses common
Adrenal Cortex Neoplasm: Dx
Biopsies
Imaging (3)
Core Biopsy: Staging (fine needle = not good)
CT: C/A/P for metasteses
MRI: Offers better look
PET: Identify malignancy using FDG
Adrenal Cortex Neoplasm: Tx and prognosis
Surgery with chemotherapy
Highly malignant, median 15 month survival with treatment
Pheochromocytoma: Etiology
MEN2 mutation (autosomal dominant) -> tumor of adrenal medulla (Chromaffin cells)
Pheochromocytoma: Pathophysiology
Adrenal medulla hyperfunction -> excess catecolamine secretion
Pheochromocytoma: Clinical Presentation
- Episodic hypertension
- Palpitations, headache, perfuse sweating (triad)
If there is an episodic release of catecholamines in a patient with a pheochromocytoma, what will you likely see in the clinical presentation? What 4 things would cause these episodes?
Surgery/Position Change/Pregnancy/Medication:
- Anxiousness
- Pallor
- Palpitations
- Tachycardia
Pheochromocytoma: Dx (blood/urine)
Plasma/Urine Catecholamines/Metanephrines
Plasma fractioned free metanephrines
24hr urine fractioned free metanephrines
All high (plasma free, fractioned metanephrines are most sensitive).
Pheochromocytoma: Dx
Imaging (3)
CT: Non-contrast of abdomen to localize tumor --> add contrast if mass found
MRI: No gadolinium (preferred for pregnancy/childhood)
PET: Complements CT or MRI
Pheochromocytoma: Tx
Pharmacologic
- Alpha Blockers: 1st line
- Ca Channel Blocker: Added to alpha blockers
- Beta Blockers: Manage HTN before surgery (required after using alpha and calcium blockers)