mcb 102 mt 1
1/145
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
146 Terms
what makes carbon special?
can form very stable tetrahedral covalently bonded structures with many elements
extremely adaptable electronically - orbitals can hybridize to bond to other atoms and form planar/linear molecules that posses double and triple bonds
sp3
3 (2p1) + 1 (2s1)
4 hybrid orbitals with 4 unpaired electrons
sp2
2 (2p1) + 1(2s1)
3 hybrid orbitals with 3 unpaired electrons + 2p orbitals with 1 unpaired electron
sp1
1(2p1) + 1(2s1)
1 hybrid orbital with 1 unpaired electron + 2 2p orbital with 1 unpaired electron each
stereoisomers
isomer that cannot interconvert into one another without breaking some covalent bond
stereochemical configuration is conferred by..
double bonds (no rotation)
presence of a chiral center
geometric isomers
isomers derived from double bonds
cis = same side of double bond
trans = different sides of double bond
enantiomers
stereoisomers that are mirror images
chiral molecule
rotated molecule cannot be superimposed on its mirror image
s stereochemistry
counterclockwise
aka L
r stereochemistry
counter clockwise
aka R
conformation vs configuration
conformations are interconvertible by rotation around single bonds
biological relevance of H bonds
between hydroxl groups of alcohol + water
between carbonal group of ketone + water
between peptides in polypeptides
between complementary bases of DNA
hydrophobic effect
the tendency of water-hating molecules to gather together when placed in water
favored by a negative change in free energy
non-covalent interactions
h bonds between neutral groups & peptide bonds
ionic interactions: attraction/repulsion
hydrophobic interactions
vander waals
exergonic reaction
ΔG<0
system loses free energy
spontaneous
endergonic reaction
ΔG>0
system gains free energy
not spontaneous
enzyme-mediated catalysis
not consumed in the reaction
generates huge rate enhancements
occurs under mild physiological conditions (pH 7. 37 C, aq, low reactant conc)
involves precise reactions: highly selective for certain substrates, stereospecific products formed in high yield
subject to metabolic regulation
how do enzymes work?
enzymes accelerate reactions by facilitating approach to and formation of the transition state
enzymes stabilize the transition state by providing a favorable environment that lowers the energy required to reach that state, thus accelerating the overall reaction without altering the thermodynamics of the substrate and product.
binding energy:
enzyme active site is designed to be complementary to the transition state of the substrate rather than to the substrate itself.
when a substrate binds to the enzyme, the enzyme can effectively lower the activation energy required to reach the transition state.
By stabilizing the transition state, enzymes facilitate the approach and formation of this state, accelerating the reaction.
this complementary binding allows the enzyme to exert forces that stabilize the transition state, thereby reducing the energy barrier (TS E) that must be overcome for the reaction to proceed.
enzymatic catalysis
enzymes increase reaction rates (k) by decreasing the free energy of the transition state
at t=298K, ΔΔG≠ = -5.7 kJ/mol implies a 10-fold increase in k
06 kJ/mol is the free energy change involved in trading 1 hydrogen bond with water for one hydrogen bond with an enzyme
how do enzymes lower the activation energy barrier?
1) active site affinity: enzyme’s active site has a strong affinity for parts of the substrate.
Affinity compensates for the +ΔH associated with removing solvation water from both the active site and the substrates.
2) molecular complementary: geometry of active site fits substrates, maximise van der waal
3) desolvation of substrate: enzyme-substrate interactions replace H bonds between substrate and water
H bonds and salt bridges formed between the substrates and the active site are stronger in the absence of water
4) Proximity Effect: By binding substrates A and B, the enzyme brings them into close proximity, effectively increasing their local concentrations. This enhances the likelihood of interaction between the substrates.
5) Correct orientation: Binding at the active site holds the substrates in a precise orientation (promoting productive encounter)
6) transition state complementarity: active site of the enzyme is most complementary to the transition state of the substrate (induced fit)
helps the enzyme stabilize the substrate at this critical high-energy point.
binding energy between the enzyme and the substrate is used to induce strain or polarization in the bonds of the substrate - pushes substrate toward TS (Haldane-Pauling Hypothesis.)
Lock and key hypothesis
Emil Fischer: enzyme's active site is considered to have a specific shape that exactly fits the substrate, similar to how a lock fits a specific key
Induced fit model
enzyme's active site is flexible and changes shape to fit the substrate upon binding, rather than being a rigid structure.
flexibility allows the enzyme to adjust and mold itself around the substrate for a tighter fit
conformational change in the enzyme’s structure helps to better position the catalytic groups within the active site, making the chemical reaction more efficient
catalytic antibodies
antibodies with enzymatic properties that can catalyze chemical reactions by stabilizing the transition state of a reaction.
created using transition-state analogs (antibodies that look like molecule during the transition state)
Haldane-Pauling hypothesis
enzymes catalyze reactions by binding most tightly to the transition state of the substrate, thereby lowering the activation energy and facilitating the reaction.
This binding often involves inducing strain or polarization in the substrate's bonds.
covalent catalysis
enzyme forms a transient covalent bond with the substrate, providing a new pathway with a lower activation energy for the reaction to proceed.
The enzyme is regenerated at the end of the reaction, making the process repeatable.provides a new pathway with a lower TS to reach products
types of enzymes

Michaelis-Menten Equation
describes how the reaction rate of an enzyme depends on substrate concentration, enzyme concentration, and the affinity of the enzyme for the substrate (given by Km)
Km: substrate concentration at which the reaction rate is half of its maximum value.
low Km: enzyme has a high affinity for the substrate
high Km suggests lower affinity.
[S]≪Km: reaction rate is proportional to the substrate concentration (first-order kinetics).
[S]=Km: reaction rate is half of Vmax (1/2vmax)
[S]≫Km, reaction rate approaches Vmax and the reaction becomes independent of substrate concentration (zero-order kinetics) (Vo=vmax)
![<p>describes how the reaction rate of an enzyme depends on substrate concentration, enzyme concentration, and the affinity of the enzyme for the substrate (given by Km)</p><p><strong>Km:</strong> substrate concentration at which the reaction rate is half of its maximum value. </p><ul><li><p>low Km: enzyme has a high affinity for the substrate </p></li><li><p>high Km suggests lower affinity.</p></li></ul><ul><li><p><strong>[S]≪Km</strong>: reaction rate is proportional to the substrate concentration (first-order kinetics).</p></li><li><p>[<strong>S]=Km</strong>: reaction rate is <strong>half</strong> of Vmax (1/2vmax)</p></li><li><p><strong>[S]≫Km, </strong> reaction rate approaches Vmax and the reaction becomes independent of substrate concentration (zero-order kinetics) (Vo=vmax)</p></li></ul><p></p>](https://knowt-user-attachments.s3.amazonaws.com/e589869b-c0de-4ef9-a9bb-7693f076e725.png)
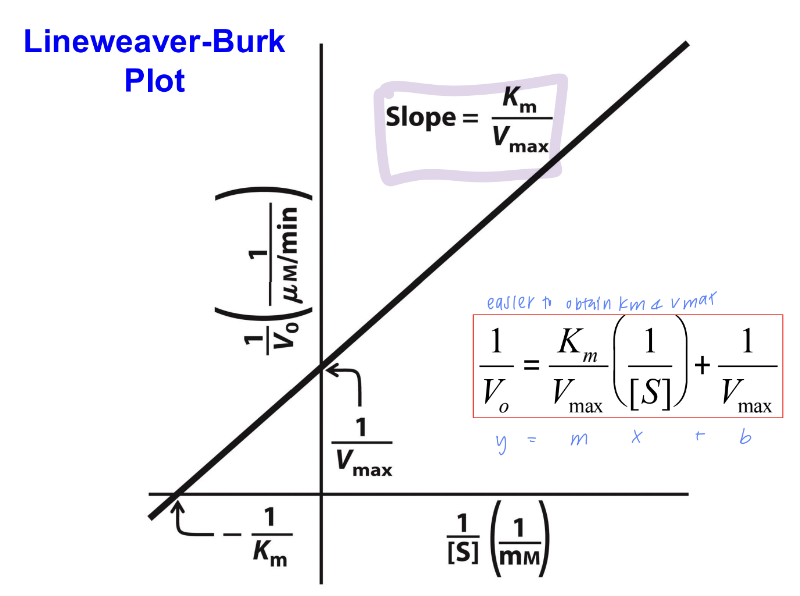
lineweaver-burk plot
graphical representation of the Michaelis-Menten equation, designed to linearize the relationship between enzyme activity and substrate concentration.
Slope: Km/Vmax
Y-intercept: 1/Vmax
X-intercept: −1/Km
catalytic efficiency constant
kcat/Km is a measure of how efficiently an enzyme catalyzes a reaction at low substrate concentrations.
The higher the kcat/Km, the more efficient the enzyme is.
The diffusion limit sets the upper boundary for catalytic efficiency, around 10⁸ to 10⁹ M⁻¹ s⁻¹ - enzyme’s efficiency is limited by how fast substrate molecules can diffuse to it.
rate constant can never be larger than the rate of diffusion between E and S
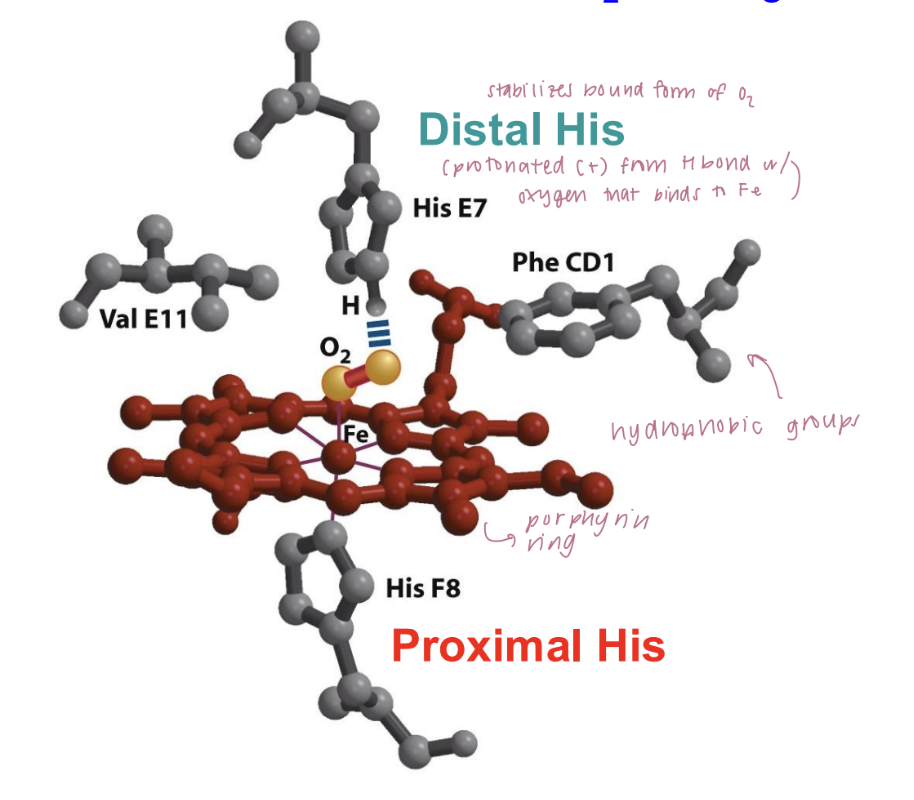
myoglobin structure and function
bind oxygen - intramuscular oxygen transporter
8 alpha-helical segments connected by loops + heme group (porphyrin ring containing iron in center)
iron binds to O2
pucker conformation: deoxygenated state
flat conformation: When O2 binds to iron, it is pulled back into the plane of the porphyrin ring.
movement of iron pulls the proximal histidine with it, inducing changes in the structure of the entire protein.
distal histidine:
located near the iron but is not directly bonded to it
stabilize the bound oxygen molecule through hydrogen bonding.
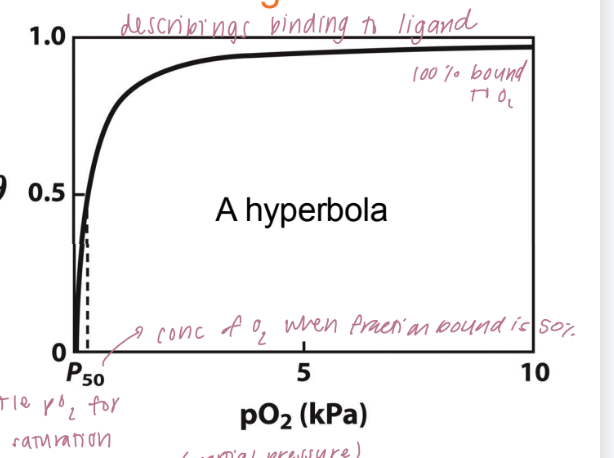
binding isotherm
curve that describes how myoglobin binds oxygen as a function of partial pressure of oxygen (pO₂).
θ (theta) is the fraction of myoglobin molecules that are bound to oxygen at a given oxygen pressure.
As the oxygen concentration (measured in partial pressure, pO₂) increases, the fraction of myoglobin bound to oxygen rises and eventually levels off at 1.0 (100% bound). This creates a hyperbolic curve.
p₅₀ is the partial pressure of oxygen at which 50% of the myoglobin is saturated with oxygen (θ = 0.5)
low value of p₅₀ suggests that myoglobin binds oxygen with high affinity
quantitive description of binding
P (Protein) and L (Ligand) bind reversibly to form a PL complex.
kinetics described by the association rate constant ka or the dissociation rate constant kd
equilibrium: association and dissociation rates are equal
The equilibrium composition is characterized by the equilibrium binding constant Kaasoc = ka/kd
measure of the affinity between the protein and ligand
binding in terms of bound fraction
θ (theta) represents the fraction of binding sites on the protein (P) that are occupied by the ligand (L):
The relationship between θ and ligand concentration follows a hyperbolic curve: as ligand concentration increases, θ approaches 1 (full saturation).
dissociation constant kd: represents the ligand concentration where 50% of the binding sites are occupied and is a measure of how tightly the ligand binds to the protein.

graphical analysis of binding
the fraction of bound sites depends on the free ligand concentration and the equilibrium dissociation constant (Kd, concentration of L at which the protein is half saturated)
Kd can be determined experimentally by analyzing the binding curve of θ versus [L] (graph) or via least-squares regression
hemoglobin structure and function
quaternary structure: alpha2 beta 2 heterotetramer
each unit has 1 porphyric ring = 4 O2 binding sites on each molecule
O2 affinity varies with pO2 (cooperative binding)
molecule can exist in 2+ states (high and low affinity) and transition between the states depending on the ligand concentration
low pO2 = high affinity state
high pO2 = low affinity state
Hb: sigmoidal binding curve
molecular level explanation cooperativity
no ligand: segments of protein are flexible, few conformations facilitate ligand binding ( low affinity)
ligand bound to one subunit: binding stabilizes a high affinity conformation of the flexible segment. polypeptide takes up a higher affinity conformation
second ligand bound to second subunit: binding occurs with higher affinity than binding of the first molecule (positive cooperativity)
multi-chain proteins are common and often involved in cooperative interactions
quantitative description of cooperativity
Proteins that display binding cooperative have multiple binding sites
If a protein that has n binding sites were infinitely cooperative then only 2 species would exist: fully bound or fully empty (all or none)
no protein is infinately cooperative
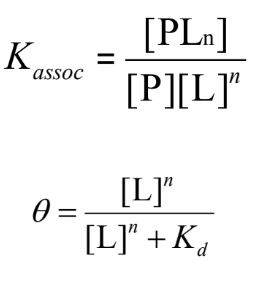
the hill coefficient
a measure of the degree of cooperativity
Finite cooperativity (nₕ < n) means the protein does not exhibit infinite cooperativity but can still show cooperative binding to some degree.
nₕ = n : infiniate cooperative
nₕ = 1: protein binds ligands without cooperativity (non-cooperative binding).
hyperbola like myoglobin
high/low affinity state
nₕ > 1: positive cooperativity - binding of one ligand increases the affinity for additional ligands
sigmoid
nₕ < 1: negative cooperativity - binding of one ligand decreases the affinity for additional ligands
hyperbola
Value of nₕ determines the sigmoidicity of the curve
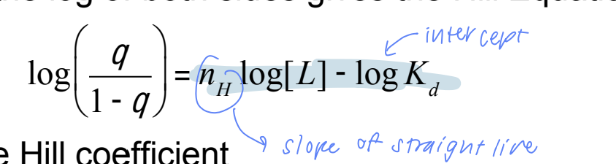
Monod-Wyman-Changeaux or Concerted Model
describes hemoglobin’s cooperative oxygen binding as a transition between two states: the T state (low-affinity, majority component in low pO2) and the R state (high-affinity).
As more oxygen binds, the equilibrium shifts toward the R state, which has a higher affinity for oxygen, leading to cooperative binding.
The transition between these states is concerted, meaning all subunits of hemoglobin transition together from the T to R state as oxygen binding increases, providing a mechanism for hemoglobin’s efficient oxygen transport
KR >> KT (binding constants)
effect of pH on oxygen affinity of Hb
Lower pH (acidic conditions, such as in tissues) decreases hemoglobin’s affinity for oxygen, promoting oxygen release where it’s needed most.
Higher pH (basic conditions, like in the lungs) increases hemoglobin’s affinity for oxygen, making it easier to pick up oxygen.
allosteric effectors
factors that bind and can change the strength of interactions in the T or R states that change the cooperativity
Bohr effect
H+ and CO2 promote oxygen release from Hb
salt bridge requires protonation of His146 and stabilizes the T state
stabilization reduces hemoglobin’s oxygen affinity, facilitating the release of oxygen where it’s needed most
2,3-bis-phosphoglycerate (BPG)
2,3-BPG binds to the central cavity of hemoglobin, stabilizing the T state (low oxygen affinity) and promoting oxygen release in tissues.
When 2,3-BPG is bound, hemoglobin has a lower affinity for oxygen, which is essential for oxygen delivery in conditions where tissues are deprived of oxygen (cooperative properties)
allows for O2 release in the tissues and adaptation to changes in altitude
R state: narrow cavity, 2,3-BPG can’t bind - allows for high oxygen affinity and efficient oxygen binding in the lungs.
properties of peptide bonds
tautomerization (resonance) of amide nitrogen lone pair to the carbonyl oxygen allows for greater electron delocalization, giving every peptide bond:
a dipole moment (charge separation)
planarity (partial double bond character of the amide C-N linkage)
how do steric constraints contribute to determining protein structure
physical hindrance between atoms can prevents certain conformations of a molecule due to spatial crowding
Trans: groups around the peptide bond are spaced out, minimizing steric clashes —> CAN ROTATE, more favored
Cis: groups around the peptide bond are closer together. —> restricts the # of conformations the peptide bond can adopt, less favorable
** X-Pro bonds are an exception! - cyclic structure introduces steric hindrance in both trans & cis, more likely to be found in Cis than other AA forms
forces dictating protein structure
weak non-covalent interactions
hydrogen bonds
electrostatic interactions
van der waals
hydrophobic effect (entropy changes in H2O)
degrees of freedom in amino acid residues
ϕ (phi) angle: rotation about the bond between N and the Cα
ψ (psi) angle: rotation about the bond between Cα and the carbonyl carbon (C).
These angles give the backbone its two degrees of freedom per residue, allowing the polypeptide to fold into its specific shape
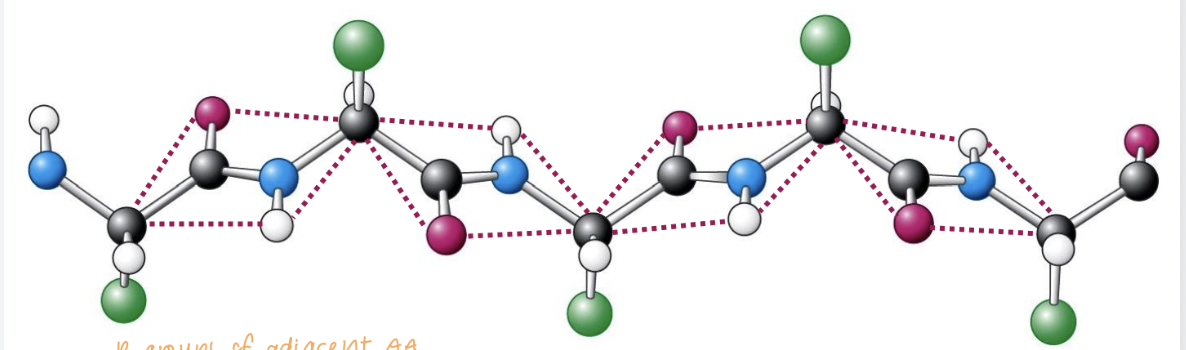
polypeptide backbone
The backbone of a polypeptide chain can be thought of as a sequence of planes.
These planes are connected to each other by "swivel joints" that allow rotation.
The peptide group is the rigid unit connecting each amino acid: atoms within the peptide group cannot move out of the plane
When the chain is in its completely extended configuration, the torsional angles ϕ and ψ are ±180°, which means the backbone forms a flat, fully extended plane
side chain constraint in protein structure
the side chains of each amino acid cannot collide with the main chain
positions are restricted.
creates a constraint on the possible ϕ and ψ angles.
side chains must be opposite one another to avoid steric clash
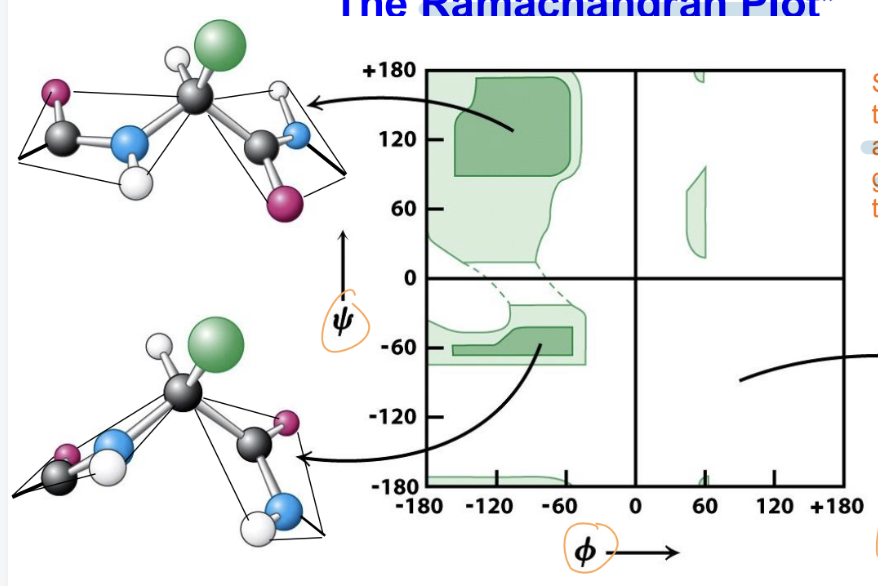
Ramachandran Plot
plot shows the relationship between the ϕ (phi) and ψ (psi) torsional angles of the polypeptide backbone
found that there is steric hindrance between the carbonyl oxygen and side groups, preventing the adoption of certain conformations
the angles ϕ = 90° and ψ = -90° are specifically noted as disfavored due to severe steric clashes
α-helix
most energetically and sterically favorable of allowed conformations (25% of all aa in proteins)
Ala is most common
side chains do not have b branch fit better than those that do Ile, Val, Thr less common)
right-handed is common, left-handed is rare
i to i+3 Hydrogen Bonding: each carbonyl oxygen accepts an H bond from the amide group 3 residues away
side groups face out and up towards N terminus to minimize steric clash
3.6 residues/turn
height/turn (periodicity): 5.4 Å
specific torsional angles ϕ = -57° and ψ = -47°
β-strand
accommodates rigid planar peptide bonds
"up-down" orientation of adjacent side chains minimizes steric clash of the R groups
Bulky and β-branched side chains are well accommodated and are favored
forms hydrogen bonds with other strands to gain stability, forming β-sheets (often seen as barrels)
usually 4 residues required for a β-turn that reverses chain direction
parallel: ϕ = -139° and ψ =135°
anti-parallel: ϕ = -119° and ψ =113°
organization of secondary structures —>
secondary structures organize in motifs —> tertiary structures (3D)
Secondary structures like α-helices and β-strands are the building blocks of motifs (recognizable patterns in a protein's three-dimensional structure)
β-α-β
β-hairpin: 2 β-strands connected by a tight turn
αα: 2+ α-helices twist around each other, forming a superhelix.
The Anfisen Experiment
native ribonuclease treated with denatured using urea (distrupts covalent bonds) and β-mercaptoethanol (reduces disulfide bonds)
rapid dilution —> incorrectly folded, inactive scrambled ribonuclease
When urea is slowly removed through slow dialysis —> protein can refold into its thermodynamically stable structure by forming correct disulfide bonds —> renatured ribonuclease (properly folded and fully active)
found that protein folding is determined by the primary amino acid sequence and that proteins can refold into their native, functional state under the right conditions.
factors affecting protein stability
1) configurational entropy: single configuration —> random coil with many accessible configurations —> high entropy state
2) hydrophobic effect: hydrophobic groups cluster inside the protein during folding, less H2O molecules need to be ordered, increasing overall entropy (DOMINANT FORCE)
hydrophobic effect for protein folding and stability
hydrophobic groups in proteins cause water molecules to organize around them, leading to a decrease in entropy (ΔS).
This ordered structure of water (cavitation) is energetically unfavorable.
However, when hydrophobic groups are buried inside the protein (as in protein folding), water molecules are less ordered, which increases entropy and helps stabilize the protein's folded state
exolains why folded proteins have a “greasy core” and expose their polar residues to the solvenet
configurational entropy effect on protein folding and stability
upon unfolding, polypeptide chain goes from single configuration —> random coil with many configurations (high entropy)
Entropy (S) is given by Boltzmann's equation: S=kB*ln*W
kB:Boltzmann’s constant
W: number of accessible configurations.
In proteins, folded conformations have fewer accessible configurations (lower W) than unfolded conformations = unfolded proteins have higher entropy.
The increase in configurational entropy upon unfolding is highly destabilizing for the folded protein, as it favors the unfolded state. Proteins should, theoretically, be more stable in the unfolded state due to the higher entropy. (not bc of other interactions - hydrophobic effect)
equation for electrophoresis in a free solution (no gel)
v=Ez/ξ
v is the rate of migration of a particle.
E is the electric field strength (in volts per centimeter).
z is the net charge of the particle.
ξ is the frictional or drag coefficient, which depends on the particle’s shape and size.
The mobility (μ\muμ) of a particle, which represents how fast it moves in an electric field: μ = vE = z/ξ
mobility depends on the charge of the particle (z) and the drag coefficient (ξ).
The drag coefficient (ξ\xiξ) for spherical particles is given by Stokes’ equation:
ξ=6πηr
r is the hydrodynamic radius of the particle.
η is the viscosity of the medium.
role of sodium dodecyl sulfate in electrophoresis (SDS)
SDS is a detergent that binds to proteins and causes them to denature (unfold).
Each SDS molecule attaches to the protein, giving it a negative charge.
Gives every protein the same charge-to-mass ratio and the same linear shape (since SDS denatures them).
uniformity ensures proteins separate based on their molecular weight, not their original charge or shape.
polyacrylamide GE (PAGE)
technique used to analyze protein composition by separating proteins by size
The protein sample is mixed with bromophenol blue dye
When voltage is applied, proteins move through the highly cross-linked polyacrylamide gel towards the positive electrode
smaller proteins moving faster than larger ones.
The dye moves ahead of the proteins, indicating when to stop the process.
marker proteins are used to estimate the polypetide MW
analysis of protein homogeneity by mass spectrometry
a protein solution is passed through a charged needle kept at high voltage, which sprays it as a mist of charged micro-droplets (ESI)
The charged droplets evaporate, leaving charged protein molecules in the gas phase.
charged molecules pass through a vacuum interface into the mass spectrometer, where they are analyzed based on their mass-to-charge ratio (m/z)
Various different methods to put macromolecules into the gas phase:
MALDI (Matrix-Assisted Laser Desorption/Ionization): Uses a laser to ionize molecules.
ESI (Electrospray Ionization): Converts proteins into gas-phase ions by spraying them into a high-voltage field.
how to solve for protein mass
solve for n: n = [(m/z)n+1 - 1]/[(m/z) n - (m/z)n+1
mass (M) = n [(m/z)n – 1]
(m/z)n = (M + n)/n → n (m/z)n = M + n
![<ol><li><p>solve for n: <span>n = [(m/z)n+1 - 1]/[(m/z) n - (m/z)n+1</span></p></li><li><p><span>mass (M) = n [(m/z)n – 1]</span></p></li></ol><p><span>(m/z)n = (M + n)/n → n (m/z)n = M + n</span></p>](https://knowt-user-attachments.s3.amazonaws.com/629d44a3-ff89-4cb3-9eea-bfea7f36dc95.png)
how to identify purified protein
We can get more information about a protein’s identity, and more accurate masses, if we chop it up into smaller peptides
Peptide Mass Fingerprinting (PMF):
Proteins are digested into smaller peptides, and their mass spectra are analyzed.
The resulting pattern of peptide masses is compared to known databases to identify the protein.
Database Search Using Fragment Ion Masses from MS/MS Data:
In tandem mass spectrometry (MS/MS), proteins are fragmented further into ions.
The fragment ion masses are compared against a database to match sequences and identify the protein.
Sequence Tags from MS/MS Data:
Short peptide sequence fragments (tags) are obtained from MS/MS data.
These sequence tags are used to search databases to identify the corresponding protein.
how to identify if a known protein is present in a sample
western blotting / immunoblot and enzyme-linked assay is a technique used to detect specific proteins in a sample.
Protein Separation by SDS-PAGE: (size separation)
Separated proteins are then transferred from the gel onto a membrane (usually nitrocellulose or PVDF).
membrane is blocked with a solution (like milk or BSA) to prevent non-specific binding of antibodies.
a primary antibody that specifically binds to the target protein is added to the membrane.
a secondary antibody (linked to an enzyme or dye) that recognizes the primary antibody is added.
enzyme catalyzes a reaction that produces a detectable signal (chemiluminescence, fluorescence, or color change), revealing the presence of the protein.
reasons for multiple bands on a gel
naturally occuring post-translational modifications of protein
phosphate: P-Ser, P-Thr, P-Tyr, P-His, P-Asp
carbohydrate (Asn-linked: Ser, Thr-linked)
acetyl groups (Ac-N-termnal-NH2, Ac-εN-Lys)
methyl groups (Lys, Arg, His, Asn, Glu)
sulfate (OH-Tyr)
ubiquitination (εN−Lys)
ubiquitaination
refers to the attachment of ubiquitin, a small protein, to the epsilon-amino group of a lysine (Lys) residue.
The isopeptide bond is formed between the C-terminal glycine (G) of ubiquitin and the ε-amino group of a lysine (K) residue on the substrate protein
Monoubiquitination: A single ubiquitin molecule is attached to a single lysine residue.
Multi-monoubiquitination: Multiple ubiquitin molecules are attached to different lysine residues on the substrate.
functions of post-transcriptional modification: phosphate (P-Ser, P-Thr, P-Tyr
involved in cell signaling.
helps regulate metabolism and cell growth, turning these processes on or off by activating or deactivating proteins, particularly in signaling pathways.
functions of post-transcriptional modification: acetyl groups (Ac-εN-Lys)
involved in gene expression regulation.
leads to turning gene expression on by loosening the interaction between histones and DNA, making the DNA more accessible for transcription
functions of post-transcriptional modification: methyl groups (Lys, Arg)
affects gene expression.
typically turns gene expression off, contributing to tighter DNA packing and reduced accessibility to transcription factors.
functions of post-transcriptional modification: ubiquitination
targets the protein for destruction by the proteasome, marking it for degradation and recycling within the cell.
practical uses for bioconjugation to proteins
Common uses include fluorescent dyes, immobilizing proteins on surfaces, and creating diagnostic tools or therapeutic agents.
reasons for modification of proteins in vitro
Lysine (Lys):
Modified using NHS-esters.
reacts with the amino group of lysine residues to form a stable amide bond, often used to label or cross-link proteins.
Aspartic Acid (Asp) and Glutamic Acid (Glu):
Modified using carbodiimides and amines.
Carbodiimides activate the carboxyl group (-COOH) of Asp and Glu to react with amines, typically forming amide bonds. This is commonly used for cross-linking or coupling proteins to other molecules.
Tyrosine (Tyr):
Modified using diazonium salts, which react with the hydroxyl group (-OH) on tyrosine residues.
used for tagging or conjugating proteins.
Cysteine (Cys):
Modified using maleimides and iodoacetamides. These reagents react with the thiol group (-SH) of cysteine residues to form thioether bonds.
used for site-specific labeling or cross-linking of proteins.
Tryptophan (Trp):
Modified using metallocarbenoids, which are reactive species generated from diazo compounds.
used to alter the aromatic side chain of tryptophan.
amino acid residue
amino acid that has been incorporated into a protein or peptide chain, minus the parts lost during the formation of the peptide bond.
names end in “yl”: alanyl, histidyl, glutamyl, tyrosyl
isoelectric point (pI)
the pH at which the protein has no net electrical charge.
pH=pKa
if a protein has pI > 7, at physiological pH the protein is positively charged - basic protein
if a protein has a pI < 7, at physiological pH the protein is negatively charged - acidic protein
signs of proteins
sign = pI - pH
pI > 7: positively charged at pH 7 = basic protein
pI < 7: negatively charged at pH 7= acidic protein
if n is large, then the number and nature of the ionizable side chains in a protein dominates its overall charge properties (rather than just the terminal groups)
personality traits of proteins
conferred by the collective properties of its constituent residues
proteins have a more complex ionization behaviour that that of a single amino acids
how to distinguish one protein from another
mass
shape
polarity
charge
density
hydrophobicity
ligand-binding
stability
etc
physiochemical properties exploited for protein purification
methods for fractioning protein mixtures rely on differences among proteins
mass
shape
polarity/solubility
charge
density
hydrophocity
ligand-binding specificity and affinity
special features (heat stability, added tags, etc)
column chromatography for protein purification
involves passing a mixture of proteins (mobile phase) through a column packed with a solid porous matrix (stationary phase), where the proteins interact differently with the matrix, allowing for their separation.
ion exchange chromatography for protein purification
separates proteins based on charge
stationary phase matrix has charged groups that interact with oppositely charged proteins.
anion exchangers (diethylaminoethyl / DEAE): positively charged - positive proteins move faster
cation exchangers: (carboxymethyl CM) negatively charged - negative proteins elute earlier
Interaction strength is controlled by changing the pH or salt concentration
Size exclusion chromatography (SEC)
Separates proteins by size
protein mixture added to column containing cross-linked polymer
Larger proteins move faster through the column as it moves around polymer
Smaller proteins enter the pores of the matrix and move more slowly.
Affinity chromatography
Separates proteins based on specific binding interactions.
protein mixture is added to column containing a polymer-bound ligand specific for protein of interest so unwanted proteins are washed through column
solution of ligand is added to column so protein of interest is eluted.
fusion with GST tag
glutathione s-transferase (GST) is often genetically fused to protein of interest, and the combined fusion protein is easily purified in one step.
The GST-tagged protein binds to glutathione sephrarose in an affinity column, allowing the protein to be purified while other proteins wash away. The protein is then eluted by adding free glutathione, and the GST tag can be removed if needed.
Tagging a recombinant protein with a GST tag for expression and purification
a gene for a target protein can be fused with a GST tag, expressed in a host cell, and collected in a cell extract for further purification steps.
Gene Fusion: The gene for the target protein is fused to the gene for GST.
Transcription: The fused gene is transcribed into mRNA, which is then translated into a fusion protein inside the host cell.
Expression in Cells: The host cell (such as a bacterial cell) expresses the fusion protein, which now contains both the target protein and the GST tag.
Cell Extract Preparation: After expression, a cell extract is prepared from the host cells. This extract contains the fusion protein as part of a mixture of other cellular proteins.
Glutathione-beads: Protein fusion mixture is added to column, glutathione anchored to matrix binds GST tag
Purification: solution of free glutathione is added to column, fusion protein is eluted
what is a biochemical assay
A quantitative or qualitative method for detecting the presence and measuring the amount of a biological substance in a complex mixture of other biomolecules.