Exam 1 Pharm COMBINED
1/89
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
90 Terms
Pharmacodynamics vs. Pharmacokinetics
Pharmacodynamics: what the drug does to the body; the study of the biochemical and physiological effects of drugs and their mechanisms of action
Pharmacokinetics: what the body does to the drug; the study of the absorption, distribution, metabolism and excretion of drugs.
Define Pharmacogenomics
Study of genomic influence on drug response and side effects
Therapeutic action vs Toxic Action
Therapeutic action: the dosage at which the drug reaches its desired effect
Toxic action: the dosage at which an undesirable effect occurs; can be due to supra-therapeutic doses, genetic predispositions, inappropriate use, non-selective actions
Pharmacognosy
The study of drugs that come from plant and animal sources, alternative medicine
Drug Development Phases
Phase 0: the first clinical trial where sub-therapetic doses are administered to 10-15 people for a limited duration <7 days
Phase 1: drug is tested on 20-100 healthy and diseased individuals to test for efficacy, side effects, optimal dose and safety
Phase 2: Trial conducted on several hundred individuals with the disease to test for efficacy and side effects
Phase 3: Designed to assess the drug’s efficacy compared to the gold standard in 300-3000 individuals
Phase 4: FDA tests the drug on hundreds to thousands of patients
The Ideal drug must have what characteristics
Specific size, shape and charge to be able to interact with a specific receptor
The necessary properties to travel to the site of action
The ability to be easily inactivated and excreted by the body
Very small drug vs Very Large drug
Very small drug results in poor selectivity while a very large drug has poor absorption and distribution

Ex. Alteplase is way too big to be distributed so it has to be directly distributed to the vascular compartment
You want it not big, not small, just right; goldilocks
What role do the types of bonds play in pharmacodynamics?
Covalent Bonds: strong and stable/irreversible
Used when you want the drug to be long-acting and with broad effects
Electrostatic: weaker than covalent but reversible (most used)
Used when you want a short and specific effect
Hydrophobic: weakest

Explain the nature of drugs relating to Enantiomers
R vs S configurations may have different effectiveness
Warfarin S is bioactive and Warfarin R is less active
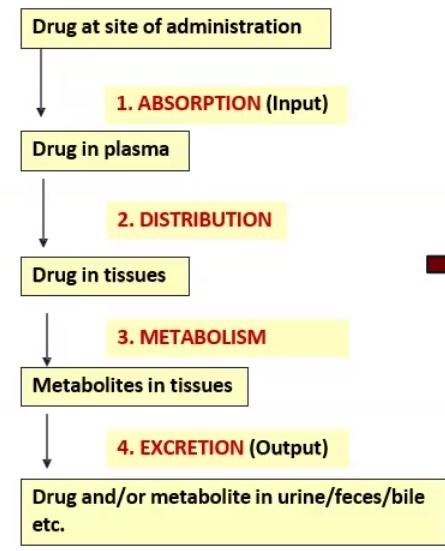
4 Stages of Pharmacokinetics
Absorption
Distribution
Metabolism
Excretion
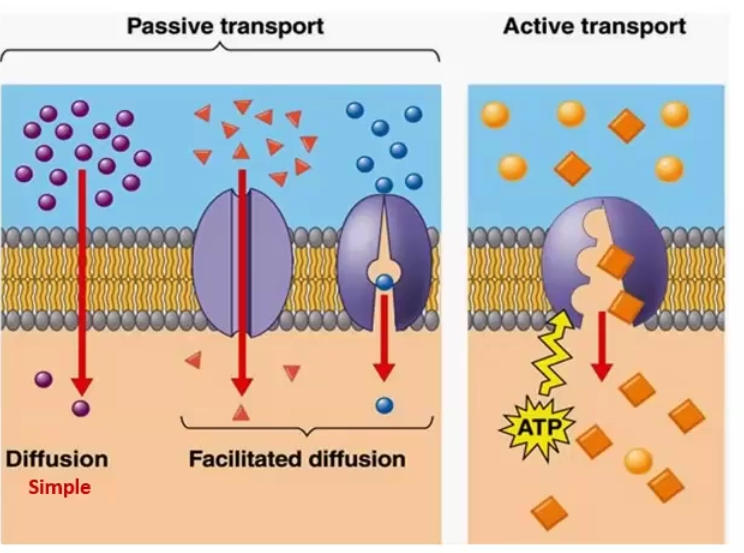
Types of Permeation Mechanisms
Aqueous diffusion: water soluble drugs diffuse through aqueous pores, governed by Fick’s Law
The more ionized, the more water soluble
Lipid diffusion: lipid soluble molecules move through the membrane
The greater partition coefficient the greater lipid solubility and faster diffusion
The less ionized (neutral), the more lipid soluble
Carrier-mediated Transport: for molecules that are too large or lipid insoluble, a carrier or transmembrane protein/channel will help it get through
Passive through concentration gradient
Active when it uses ATP directly to carry against the concentration
Exocytosis
Fick’s Law of Diffusion
Rate of diffusion is directly proportional to the concentration of the substances on both sides → gradient moves from high concentration to low concentration
Drugs are absorbed faster from organs with large surface (ex/ Intestines) areas and thinner membranes

Degree of Ionization
An acid in an acidic environment favors ______, while an acid in an alkaline environment favors ________.
A base in a basic environment favors _______, while a base in an acidic environment favors ________.
Degree of Ionization
An acid in an acidic environment favors absorption, while an acid in an alkaline environment favors excretion.
A base in a basic environment favors absorption, while a base in an acidic environment favors excretion.
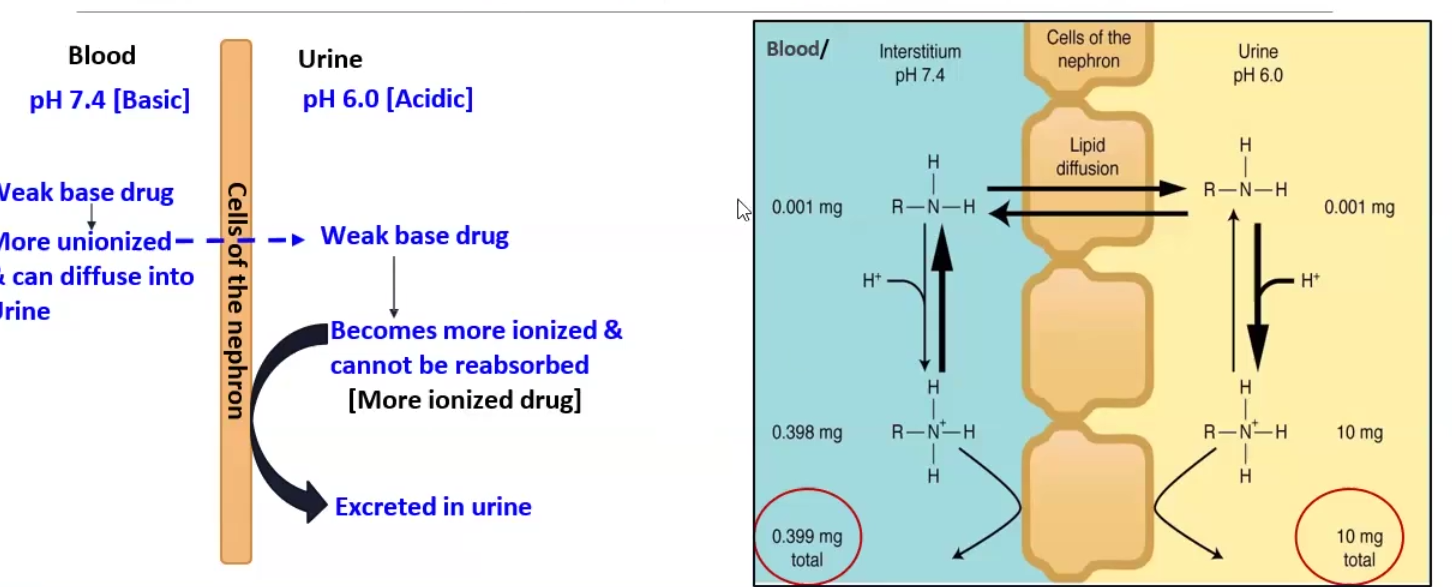
Henderson-Hasselbalch Equation
Ion trapping
FOR WEAK BASE → pH-pKa = log Un-ionized/ Ionized
FOR WEAK ACID → pH-pKa = log Ionized/ Unionized
Ion trapping: we make the environment the opposite of the drug so it can be excreted
Ex. methamphetamine (basic) can be forced to be excreted by making the urine very acidic, which stops reabsorption
Multidrug Resistance Protein Type 1 (MRP1)/ P-Glycoprotein
A very common drug transporter that pumps drugs out of the cell (REVERSE TRANSPORT and uses ATP)
Used by fetus to transport drugs back into maternal blood
Intestines use it to transport drugs into intestinal lumen
Brain limits access to brain-blood barrier by pumping back into circulating blood
Cancer cells use it to pump chemotherapy drugs out of the cell
In regards to ion trapping
______ ______ is used to alkalinize urine in patients presenting with a weak acid overdose
______ ______ is used to acidize urine in patients presenting with a weak acid overdose
In regards to ion trapping
Sodium Bicarbonate is used to alkalinize urine in patients presenting with a weak acid overdose
Ammonium Chloride is used to acidize urine in patients presenting with a weak base overdose
Endocytosis and Exocytosis
Endocytosis: transport of solid matter or liquid into the cell utilizing a coated vacuole or vesicle
Bioavailability and ROA
The fraction of the administered dose that reaches the systemic circulation following administration by any route
Bioavailability may be different depending the Route of Administration (ROA)
IV has the highest (100%) bioavailability
ROA Classification
Systemic:
Enteral: directly into the digestive tract
oral, sublingual, and rectal
Parenteral:
Transdermal or Injections (IV, IM, SC, IT)
Topical/Local
Skin, eye, mucosa, inhalational
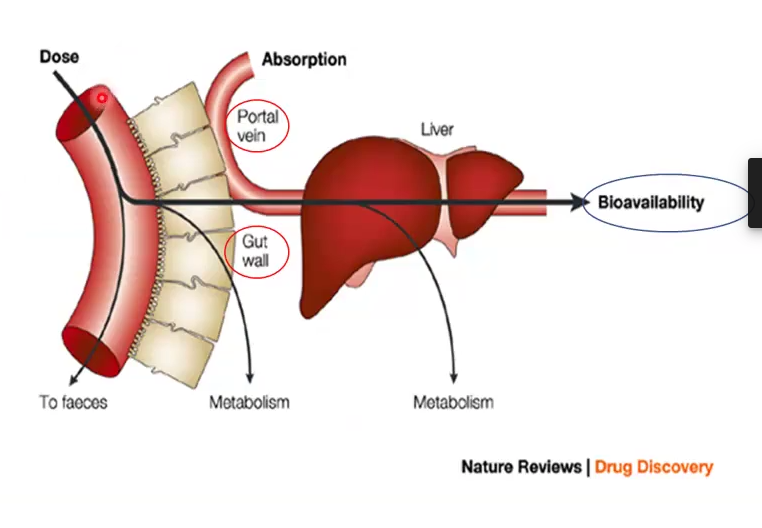
First Pass Effect
The reduction of bioavailability of the administered dose due to metabolism by the gull wall enzymes and liver
ROA: Oral (PO)
Advantages
Disadvantages
Absorption can occur at any point of the digestive tract but must cross the digestive and vascular endothelium
Advantages: safe, most convenient and economical
Disadvantages: limited absorption, destruction of drug by digestive enzymes, first pass effect, patient compliance
ROA: Sublingual (SL)
Advantages
Disadvantages
Absorption occurs through oral mucosa under tongue (or buccal route)and goes into blood
Advantages: fast absorption, avoids first pass effect
Disadvantages: limited to certain drugs, may lose effect if swallowed, irritation on site
ROA: Rectal (PR)
Advantages
Disadvantages
Rectal suppository
Advantages: Less first pass effect than oral, ideal for comatose or vomiting patients
Disadvantages: erratic and variable absorption, irritation
ROA: Intravenous (IV)
Advantages
Disadvantages
Absorption not required, goes into circulation right away
Advantages: most rapid onset, ideal for large doses, emergencies, noncompliant patients
Disadvantages: May increase adverse effects, Must be slowly injected, aseptic techniques needed, unsuitable for oily substances
ROA: Intramuscular (IM)
Advantages
Disadvantages
Injected into a muscle
Advantages: ideal for moderate volumes of a drug, good for self-administering patients, suitable for oily vehicles and irritants
Disadvantages: can cause intramuscular hemorrhage and contraindicated in anticoagulant
ROA: Subcutaneous (SC)
Advantages
Disadvantages
Injected below dermis, the hypodermis
Advantages: suitable for slow release drugs and poorly soluble suspensions
Disadvantages: smaller volumes administered and may be painful
ROA: Transdermal
Advantages
Disadvantages
Placed on top of the skin in like a patch
Advantages: avoids first pass effect, convenient duration of administration, convenient and painless
Disadvantages: some are allergic
ROA: Inhalational
Advantages
Disadvantages
ROA: Inhalational
Advantages: avoids first pass effect, rapid onset, less systemic side effects
Disadvantages: route for abuse, effect dissipates quickly
ROA: Mucosal
Sites
Sites: nasopharynx, oropharynx, vagina, urethra
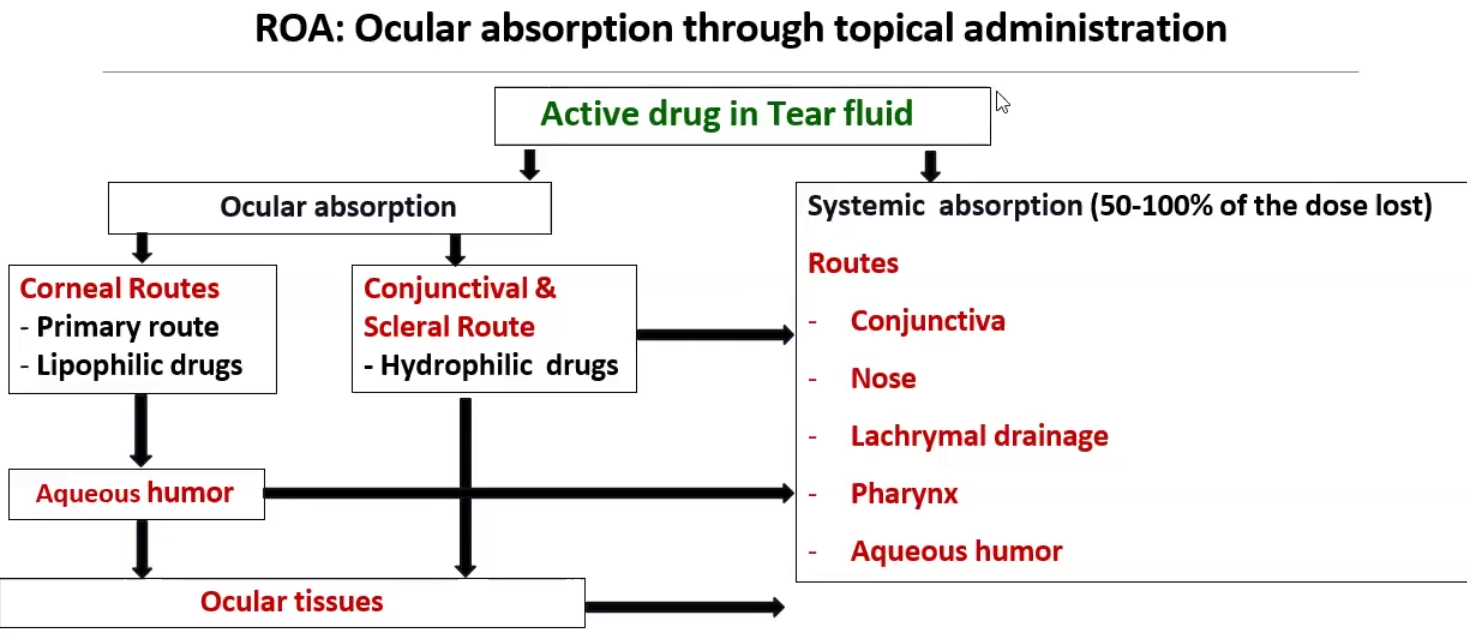
ROA: Ocular
Very difficult to have good bioavailability because there are many ways to absorb the drug
Pharmacogenomics vs Pharmacogenetics
Pharmacogenomics: study of variations in the whole genome (DNA and RNA) as related to drug response
Pharmacogenetics: study of variations in DNA sequence of 1-2 genes as related to drug response
Intracellular receptors
Define
Example
Intracellular receptors have lipid-soluble ligands that can cross the cell membrane to bind the receptor (proteins, NA, enzymes such as guanylyl cyclase, transcription factors)
Ex: Glucocorticoid/steroid receptor that needs heat shock protein (hsp90) to cross the membrane

Ligand-regulated enzyme receptors
Define
Usually the receptors have what kind of activity
Example
Ligand-regulated enzyme receptors have the ligand bind the extracellular domain of the transmembrane protein and trigger a reaction within the cytoplasmic domain
Usually they are tyrosine kinases
Ex. the Epidermal Growth Factor Receptor (EGF) and the Insulin receptor
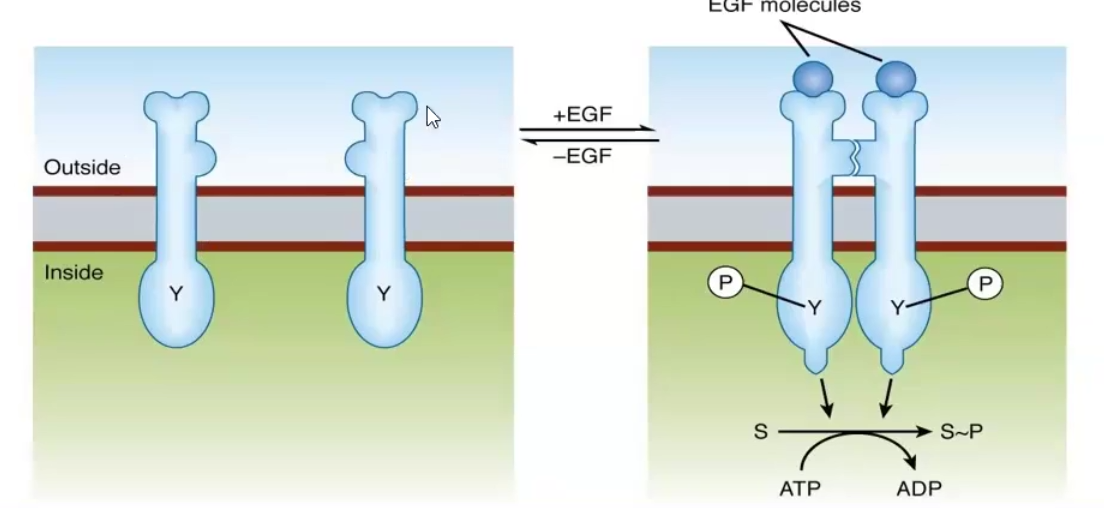
Tyrosine kinase associated receptors/Cytokine receptors
Define
Example
Tyrosine kinase associated receptors are transmembrane proteins whose cytoplastic domain is associated with tyrosine kinase units
Ex. Cytokines act as the ligand to the receptor and the receptor activates the JK STAT pathway of the tyrosine kinase
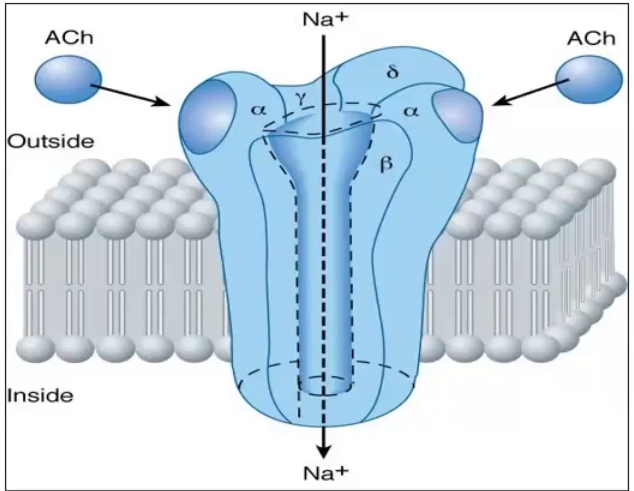
Receptors linked to ion channels
Define
Examples of endogenous ligands that bind to this type of receptor
Receptors linked to ion channels are those that are ligand gated, voltage gated or 2nd messenger regulated and only open by changing conformation once it binds the ligand
Ex. Acetycholine, GABA-A, Glycine, Glutamate (not adrenergic or muscarinic)
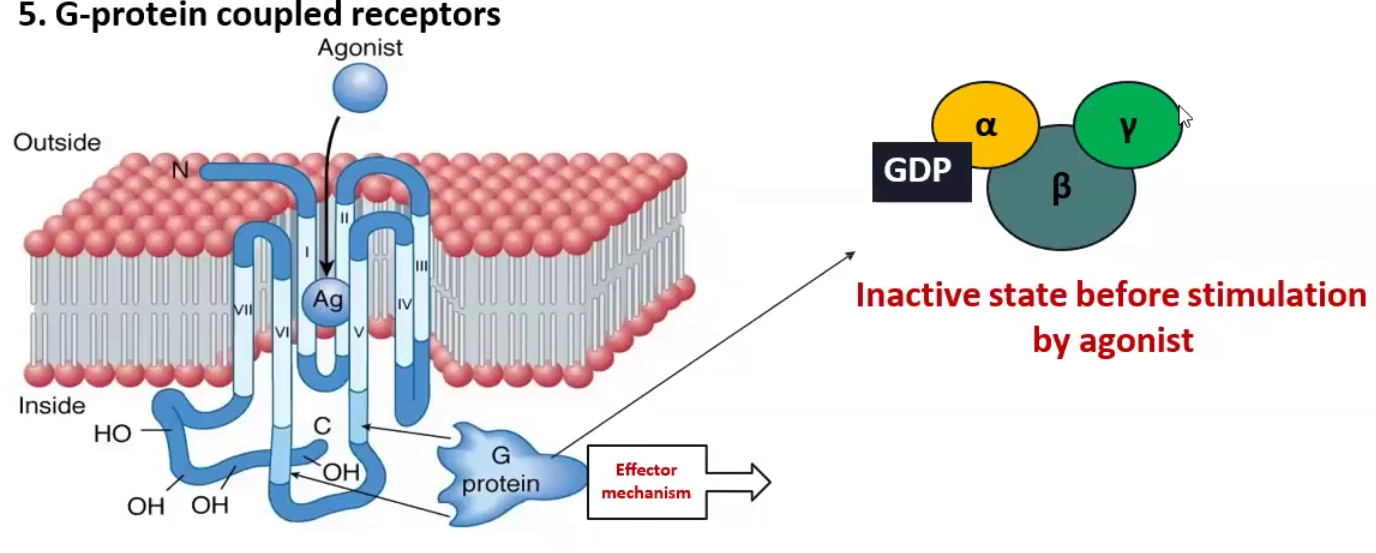
G- coupled Receptors
Define
Example
G-coupled receptors are the most abundant transmembrane receptor in humans, these receptors have a membrane subunit and a G protein attached to the cytosolic component, once the ligand binds to the outside, the G protein becomes activated and causes an effect inside
Alpha-1 adrenergic (NE) receptor is coupled to Gq protein and increases Phospholipase C, which stimulates DAG 2nd messenger
Causes Vasoconstriction
Beta-1 adrenergic (NE) receptor and Histamine-2 receptor are coupled to Gs proteins and increases Adenylyl cyclase, which stimulates cAMP 2nd messenger
Causes contraction of heart
Define
Signal Transduction
Desensitization
Ex.
Tachyphylaxis
Ex
Pharmacodynamic Tolerance
Signal Transduction: A chemical message sent that leads to a physiologic response
Desensitization: when the receptor response to a drug decreases due to being given continuously or repeatedly
Can be due to receptor downregulation, loss of function or reduction in receptor density
Tachyphylaxis: rapid and reversible decrease in receptor responsiveness
Ex. Nitroglycerin for angina
Pharmacodynamic Tolerance: seen in chronic drug abuse, a gradual decrease in receptor responsiveness, resulting in a higher dose needed for the same magnitude effect
Define
Affinity
KD
Fractional Occupancy
Affinity: the ability of a ligand [L] to bind its receptor [R] and become [LR]
KD : Equilibrium dissociation constant that quantifies affinity, defined as the concentration at which 50% of the receptors are bound to ligand [LR]
The lower KD, the greater affinity
Fractional Occupancy: the percentage of occupied receptors [LR] compared to the total amount of receptors
[LR] / { [R] + [LR] } or B / Bmax
Ps: sigmoidal = cooperativity
![<ul><li><p>Affinity: the ability of a ligand [L] to bind its receptor [R] and become [LR]</p></li><li><p>K<sub>D </sub>: Equilibrium dissociation constant that quantifies affinity, defined as the concentration at which 50% of the receptors are bound to ligand [LR]</p><ul><li><p>The lower K<sub>D</sub>, the greater affinity</p></li></ul></li><li><p>Fractional Occupancy: the percentage of occupied receptors [LR] compared to the total amount of receptors</p><ul><li><p>[LR] / { [R] + [LR] } <strong>or</strong> B / Bmax</p></li></ul></li></ul><p>Ps: sigmoidal = cooperativity</p>](https://knowt-user-attachments.s3.amazonaws.com/8349fdca-786e-4036-a698-4e004da3ace7.png)
Graded Dose Response
Define
EC50 / ED50
Emax
Potency
Efficacy
Graded Dose Response: the effect of various doses of a drug in a single individual
EC50 / ED50: The concentration or dose at which half the maximal response is produced
Emax: the maximal response
Potency: measure of strength, the concentration needed to reach EC50 / ED50
Efficacy: The ability of a drug to produce a biological response; an index of the maximal response Emax
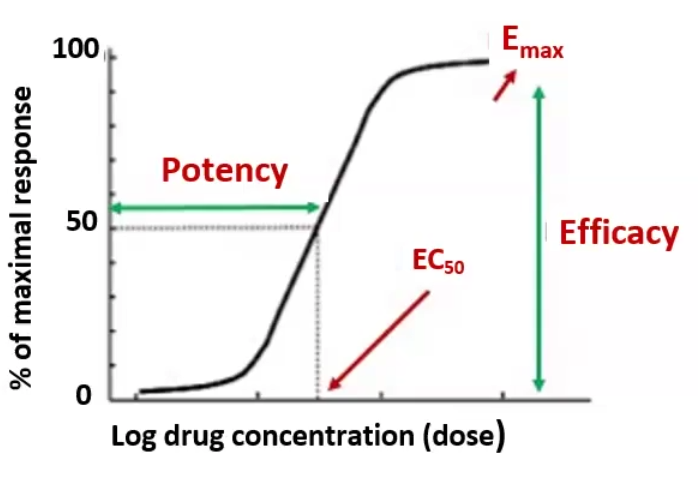
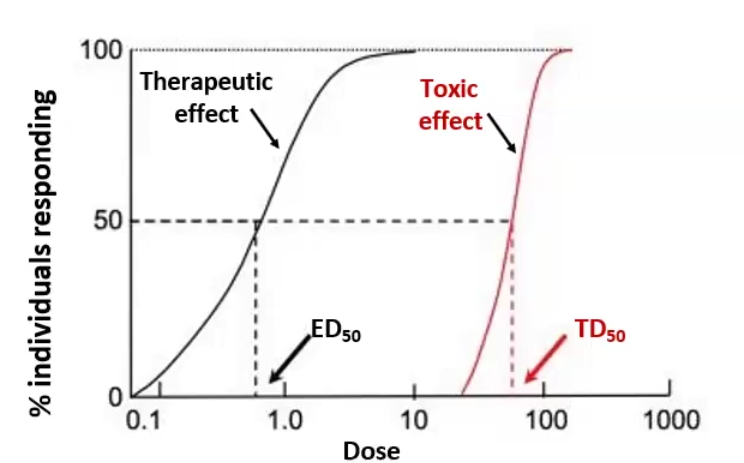
Quantal-Dose Response
Define
ED50
TD50
TI
Quantal-Dose Response: the concentrations of a drug that produces a certain effect in 50% of the population
ED50: Effective dose, the dose at which 50% of the population receives a therapeutic effect
TD50: Median Toxic dose, the dose at which 50% of the population receives toxic effects
TI: Therapeutic index, the distance between ED50 and TD50, you want it to be as high as possible → TI = TD50/ED50
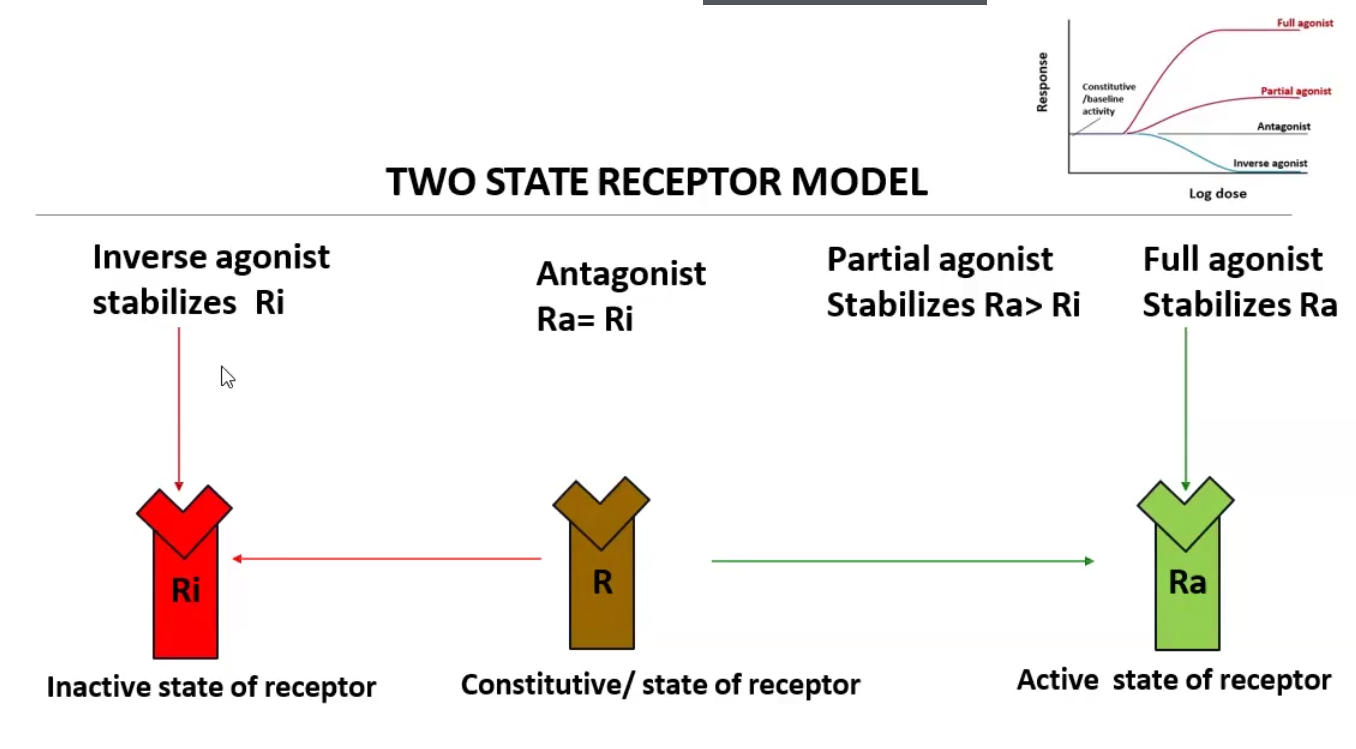
Agonist Classifications
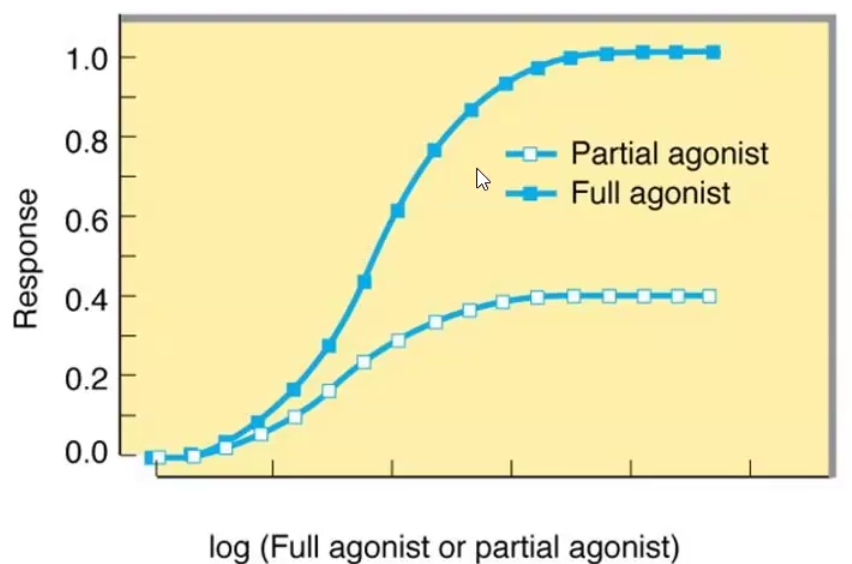
Full agonist: Binds and stabilizes the active state of the receptor (Ra)
Partial agonist: Binds and stabilizes the active state of the receptor (RA) but at a lower maximal response than a full agonist at a similar receptor occupancy
Inverse agonist: Binds and stabilizes the inactive state of the receptor (Ri), thus producing the opposite effect of a full agonist
Antagonist Classifications
Receptor antagonists: bind receptor and stabilize Ra and Ri equally, thus creating a constitutive state
Active site binding antagonists:
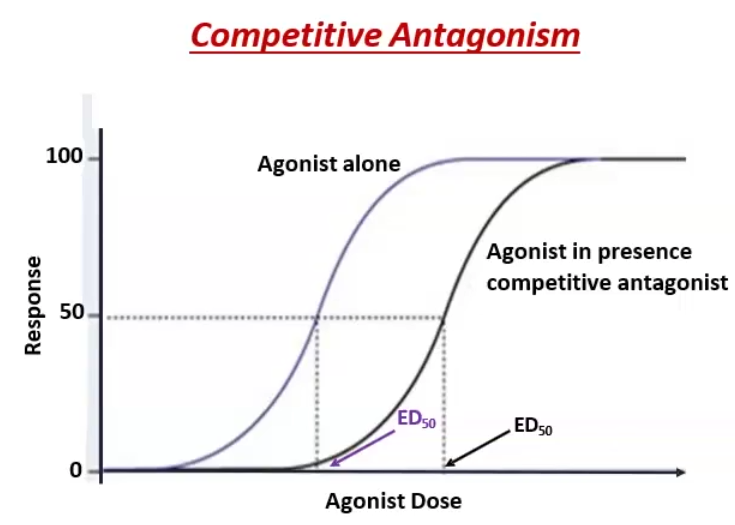
Reversible antagonists (competitive): can be overcome by increasing agonist concentration → ED50 (potency) decreases, shifting curve to the right, but Emax (efficacy)stays the same
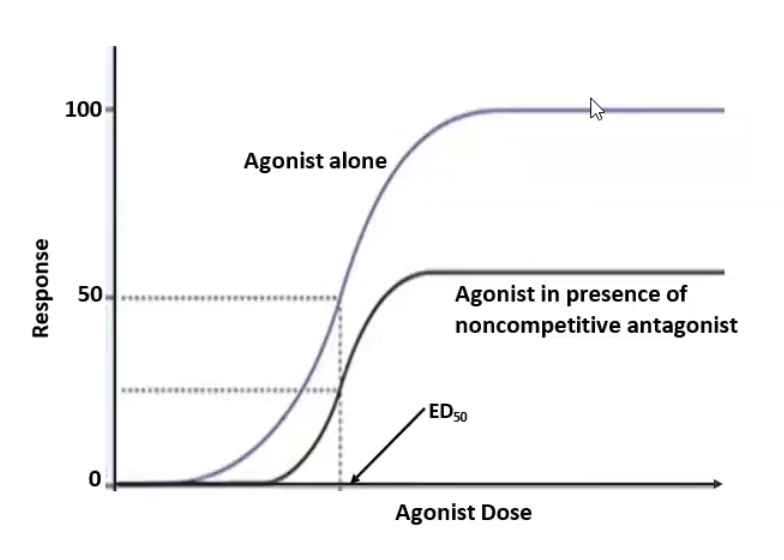
Irreversible antagonists (non-competitive): binds to the receptor active side via covalent bond; antagonist cannot be outcompeted by increasing concentration → Emax (efficacy) decreases while ED50 (potency) stays constant unless spare receptors are present
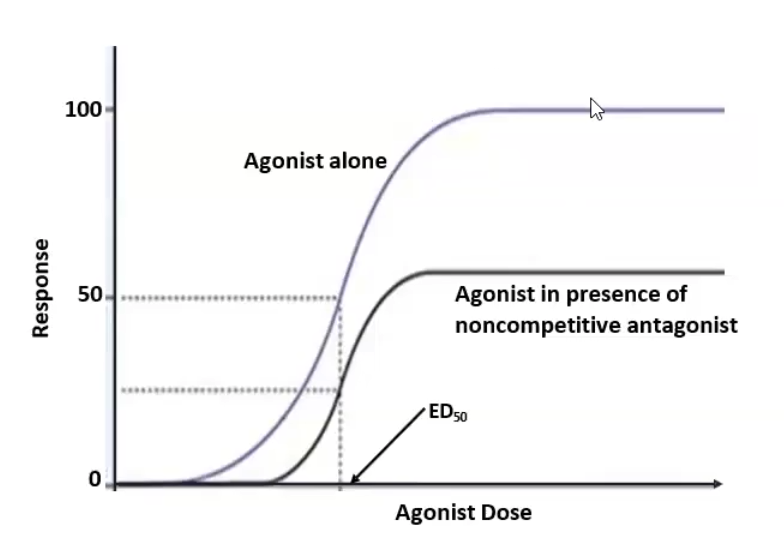
Allosteric site binding (non-competitive): bind somewhere other than the active site and inhibits agonist → Emax ((efficacy) decreases while ED50 stays constant
Non-receptor antagonists:
Chemical antagonist: a drug or endogenous ligand that interacts with another drug to block its activity
Physiologic antagonist: blocks the activation of a receptor by an agonist
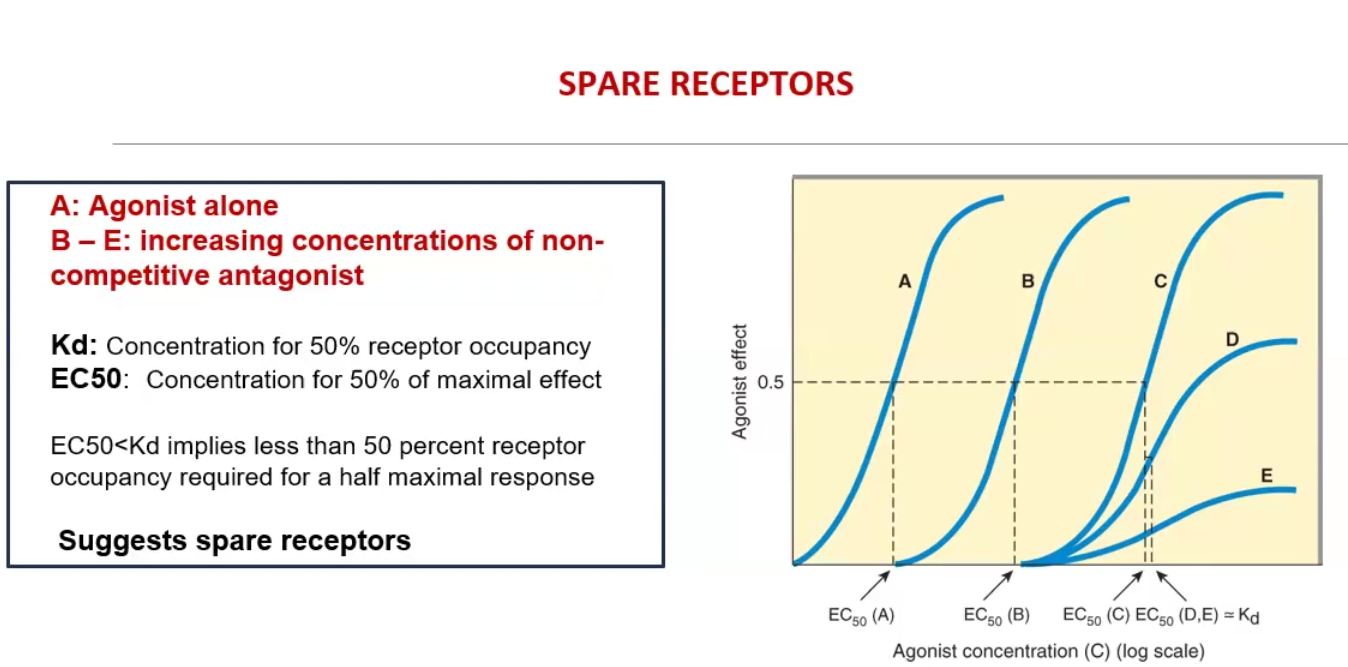
Spare Receptors
what does it mean for the Kd vs EC50
Receptors that do not have to be occupied to achieve maximal effect
EC50 < Kd implies less than 50% of the receptors are needed for a half maximal response
Intrinsic activity (IA)
Define
Difference between antagonist and agonist IA
Intrinsic activity (IA): the ability of drug-receptor complex to produce functional response
Agonists have both affinity and IA, but antagonists only have affinity
Full agonist IA = 1, Partial agonist IA >0 but <1, Inverse agonist IA = -ve
Define the following
Additive effect
Synergism
Potentiation
Additive effect: two drugs that when taken together are equal to the sum of the effects of the two drugs taken separately → 1 + 1 = 2
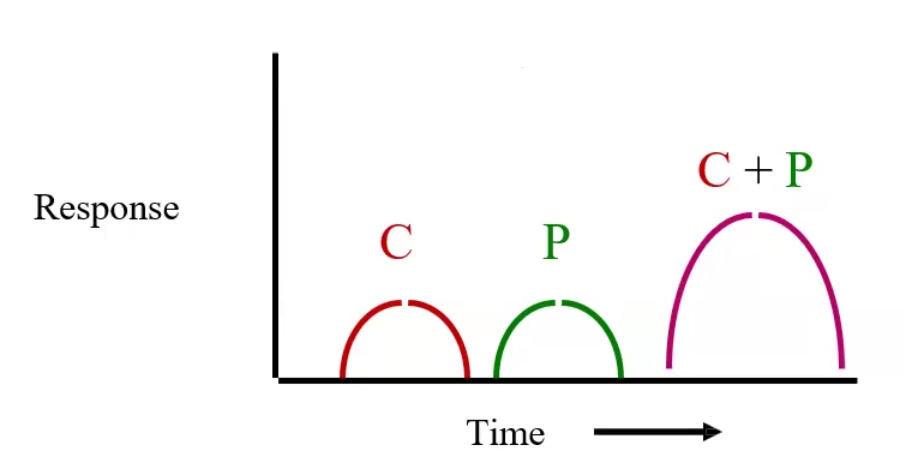
Synergism: The effect of two drugs taken together is greater than the sum of their separate effect at the same doses → 1 + 1 = 4
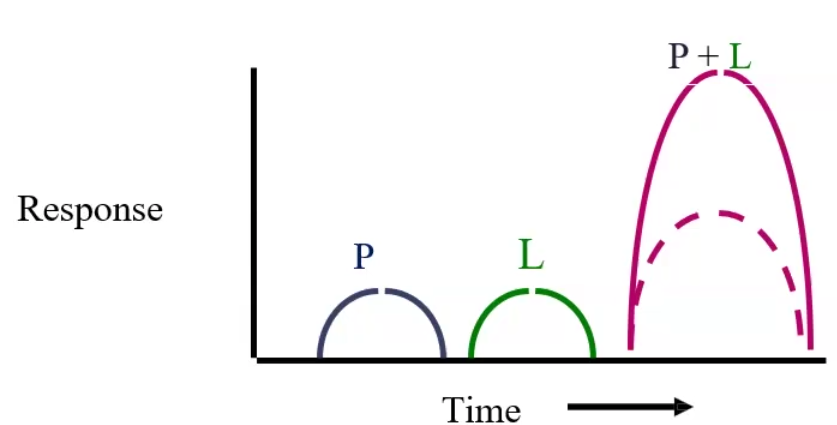
Potentiation: a drug that by itself has no effect can potentiate another drug when taken together → 0 + 1 = 2
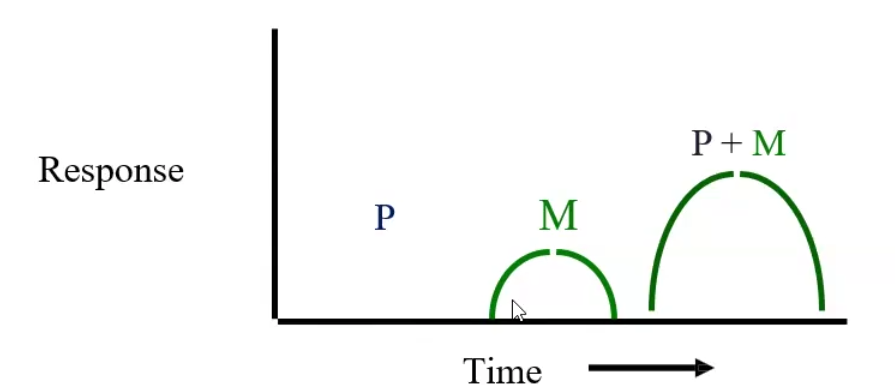
60/40/20 Rule
Estimates the volume of fluid in each body compartment based on total body weight
60% of total weight → total body water
40% of total weight → intracellular fluid (ICF)
20% of total weight → Extracellular fluid (ECF) → 75% interstitial and 25% intravascular
Drugs are absorbed faster from organs with large ______ _____ and ________ membranes
Drugs are absorbed faster from organs with large surface areas and thinner membranes
1st Pass or Pre-systemic Metabolism
Process through which ORAL drugs lose most of their bioavailability due to pre-systemic metabolism by the gut wall or liver
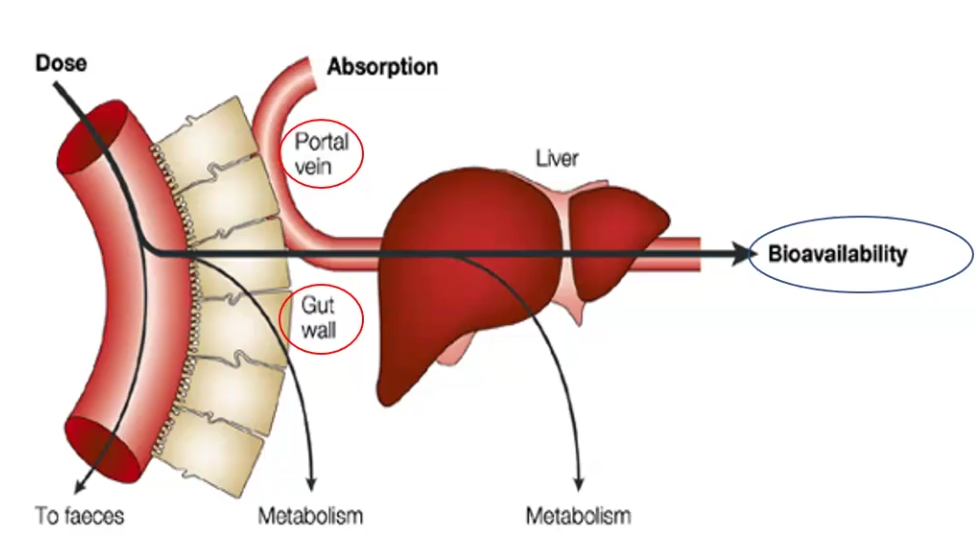
Bioavailability
The fraction of the given dose that reaches the systemic circulation following administration by any route
Bioavailability of Major Routes of Administration
Intravenous
Intramuscular
Subcutaneous
Oral
Mucous membranes
Rectal
Inhalation
Transdermal
Sublingual
Intravenous 100%
Intramuscular 75-99%
Subcutaneous 75%-99%
Oral 5-99%
Mucous membranes ECTREMELY LOW
Rectal 30-99%
Inhalation 5-99%
Transdermal 80-100%
Sublingual EXCELLENT
Factors Affecting Oral Drug Absorption
Blood flow to the area, the more the better absorption
Gastric emptying time; can be increased by gastric stimulants, fatty foods, anticholinergic drugs and pyloric stenosis
pH of gut: affects ionization and stability of drug
Food particles that trap drug molecules
Surface area and thinness of area increases absorption
Factors Affecting Drug Distribution
Drug binding to plasma or tissue proteins decreases distribution and excretion
Storage of lipophilic drugs in ADIPOSE TISSUE
Blood flow to tissues and organs
Physical properties
Chemical properties: hydrophilic vs lipophilic
Capillary permeability
Ionization
Describe how plasma protein binding affects distribution
Why is this no longer an issue in MOST cases
Plasma protein binding such as albumin traps drug molecules in the vascular space and prevents them from reaching the site of pharmacologic action, as well as decreases clearance
Not an issue anymore because plasma protein # far exceeds #drug molecules → also as free drug increases so does clearance
Only a problem if drug clearance is greatly decreased due to kidney or liver disease OR if displaced drug has low TI
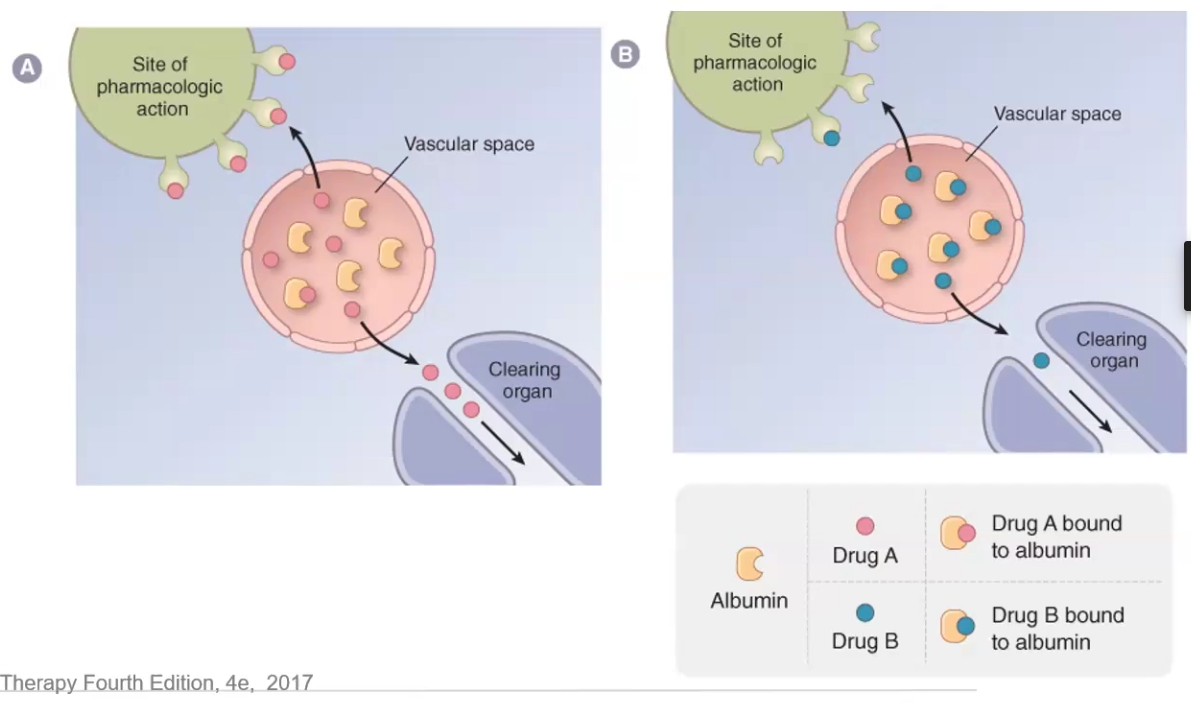
Describe Capillary Permeability in terms of drug acess
Liver endothelial cell junctions vs Brain endothelial cell junctions
Endothelial cells lining blood vessels in the liver have large fenestrations that allow free movement of substances between the blood and the interstitium
Blood vessels in the brain have endothelial cells with very tight junctions that only allow high lipid solubility substances to pass the BBB
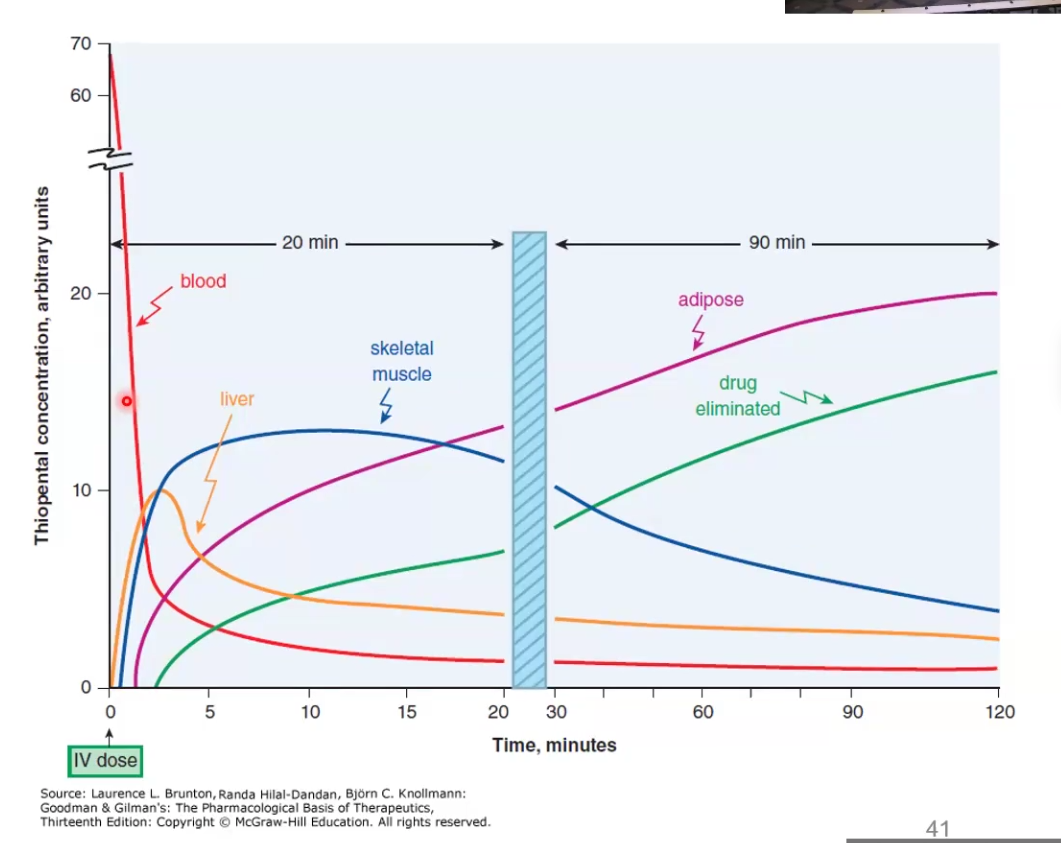
Explain how redistribution terminates drug action
While the drug may distribute initially to one place to have a pharmacological effect, redistribution to other tissues can terminate the effect
Ex. Thiopental acts rapidly crossing the BBB and causes anesthetic effect, but then it redistributes to muscle, fat, etc and the patient wakes up because the brain concentration decreased
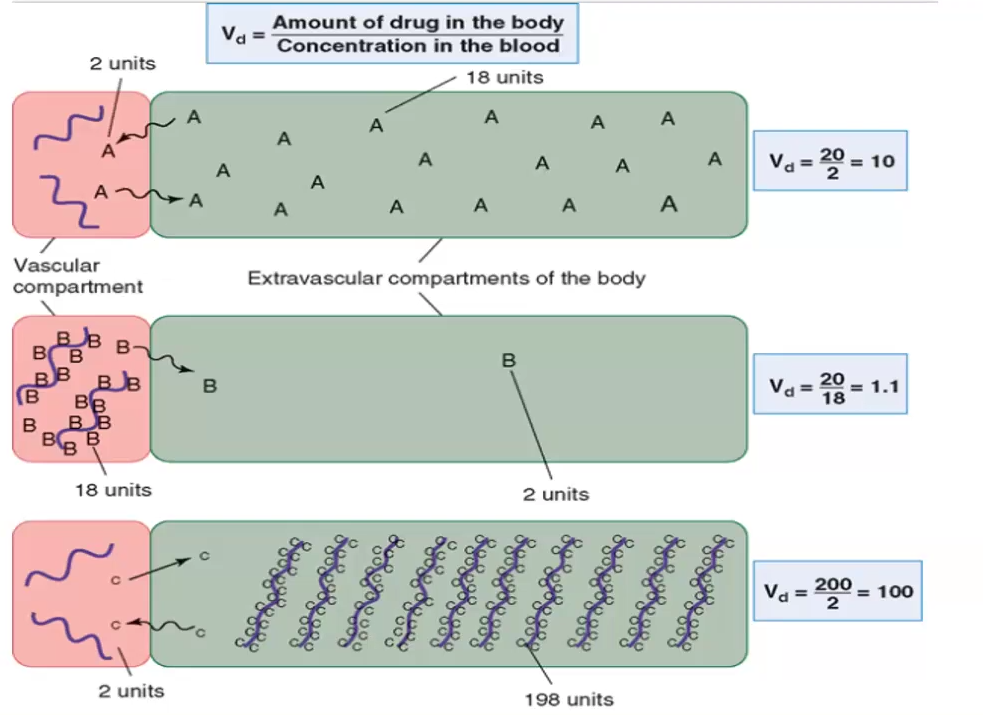
Volume of Distribution formula
Volume of Distribution (Vd) = Dose (D) / Plasma Concentration (Cp)
Xenobiotics
Define
How do drugs relate
Hydrophilic vs. Hydrophobic
Compounds foreign to the human body, we are exposed to them every day
Drugs are xenobiotics so they are metabolized by the same enzymes the body uses to get rid of foreign compounds
Hydrophilic xenobiotics undergo renal or biliary excretion, while lipophilic require metabolism by liver enzymes to become more hydrophilic
Describe the two main organs of elimination
Kidney: eliminates drugs with polar functional groups such as OH, SH, NH3 and drugs ionized/charged at physiological pH
Lipophilic drugs filtered by the glomeruli are not excreted, they are reabsorbed
Liver: biotransforms drugs from lipophilic to more polar metabolites
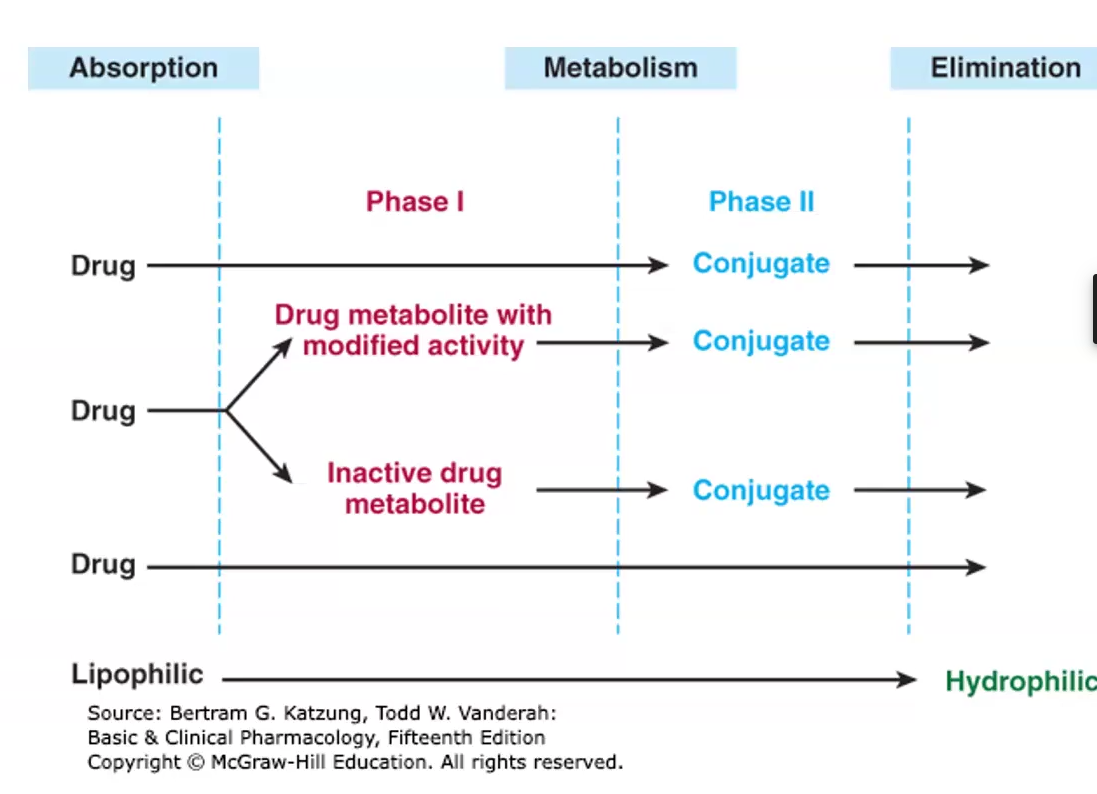
Biotransformation
The process of biochemical reactions transforming drug molecules into inactive, less toxic and more polar metabolites
Starts after drug is absorbed systemically but before it is eliminated
Occurs before absorption by gut wall enzymes or on 1st pass through liver
3 Different ways biotransformation alters a drug
Most cases: active drug is inactivated and converted into more water soluble metabolites
Some cases inactive drug (pro drug) is activated
Some cases active drugs are converted into other active or toxic metabolites
Phase I
Phase I: Functionalization reaction
Polar groups are added or hydrolysis reactions expose them or ester linkage
Metabolites are excreted in urine, but very slowly
Mainly oxidation, reduction, deamination and hydrolysis
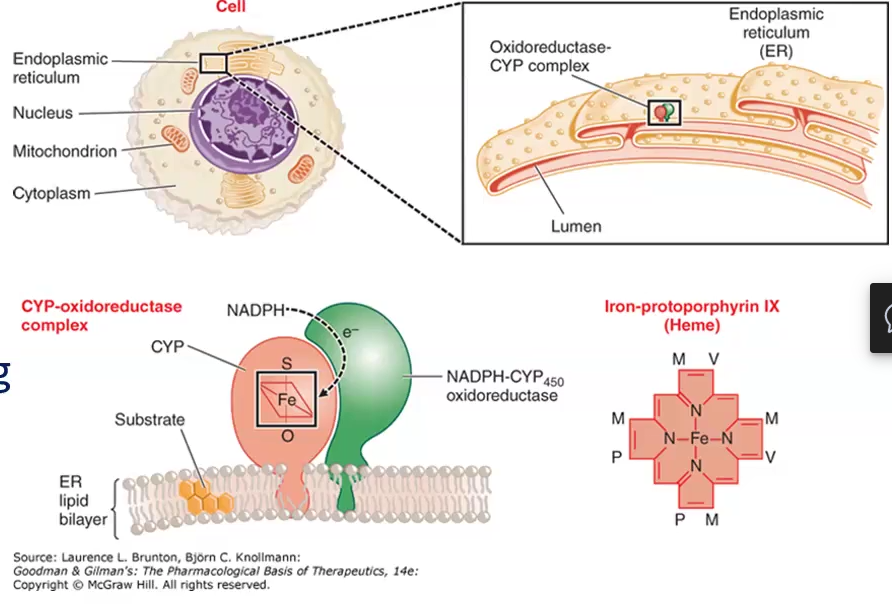
Cytochrome P450 Enzyme System (CYP)/ Microsomal enzyme oxidizing system (MEOS)
Define
Net reaction
Phase I P450 Oxidation
CYP P450 Nomenclature
System of drug metabolizing enzymes that are found in lipophilic membranes of of the smooth ER of the liver and other tissues
Net reaction:
RH (drug) + H+ + O2 + NADPH → NADP+ + H2O + ROH (oxidized drug)
Phase I P450 Oxidation: constitute about 95% of all phase I oxidation rxns by CYP
Not very selective, metabolize thousands of drugs
CYP P450 Nomenclature:
1st number: family
Capital letter: subfamily
2nd number: specific enzyme
CYP P450
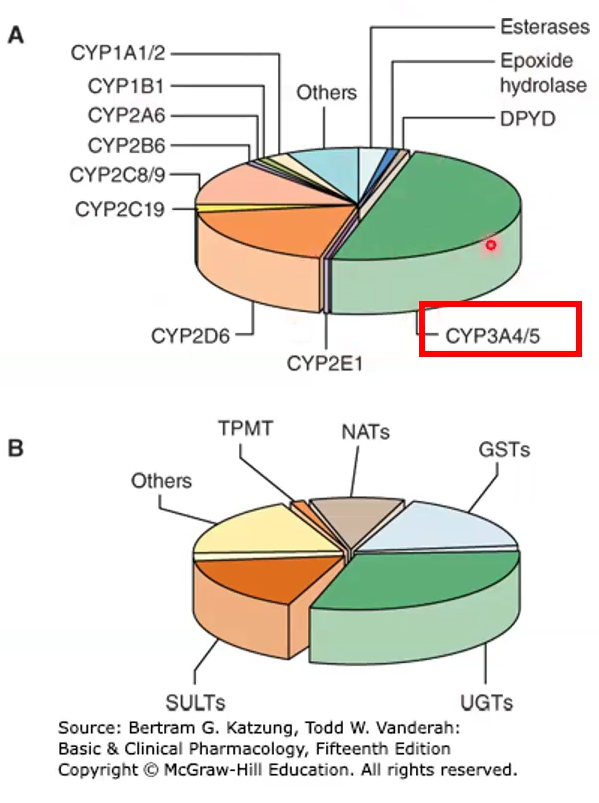
Most abundant type, what does it do
2nd most important
34A is the most abundant, it is found in the liver but also in the gut wall, where it can decrease bioavailability; metabolizes 50% of Rx drugs
Drugs that inhibit/induce 3A4 may also induce/inhibit P-glycoprotein bc they are on the same chromosome
2D6 is the second most abundant
Inducers vs. Inhibitors of CYP 450
Define
Decrease or increase active drug?
What must clinician do with the dose?
Inducers: Increase P450 gene transcription or prevent degradation, which will decrease the amount of active drug
Clinician must increase dose
Inhibitors: Competitively or non-competitively bind to active P450 and decreases its activity, thus increasing the amount of active drug in the system
Clinician must decrease dose
Grapefruit Juice (GJ) Facts
Contains furanocoumarin, an irreversible inhibitor of CYP 3A4 in the gut
Reduces pre-systemic metabolism and increases drug bioavailability
Contraindicated with many drugs by the manufacturer regardless route of administration
Substrates, Inducers and Inhibitors of each CYP 450 enzyme
Non-CYP Phase I Oxidations
Monoamine oxidase rxs: oxidative deamination of monoamines
Xanthine oxidase: oxidoreductase catalyzes the oxidation of purines
Alcohol and aldehyde dehydrogenase: oxidation of alcohols and aldehydes
Phase I can also activate drugs, how is this useful
Drug companies may use pro-drugs that are activated by phase I reactions in order to improve bioavailability
Phase II Reactions
Define
What kind of reactions are they
Examples of Transferases and Endogenous reactants for these conjugations
Usually follows phase I, but not always, some drugs skip phase I and go directly to phase II
Mainly conjugations that bring together 2 molecules into 1using energy and drastically increase solubility
Ex of Trans and End reactants:
UDP glucuronosyltransferase (UGT) and UDP glucoronic acid
MOST COMMON
N-acetyltransferase (NAT) and Acetyl CoA
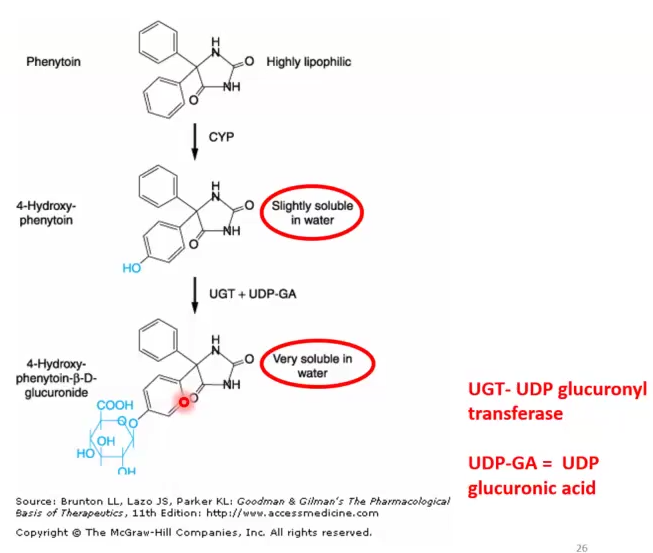
Phase II Conjugation of 4-OH-Phenytoin
Phase I: Phenytoin → 4-OH-Phenytoin )slightly soluble)
Phase II: 4-OH-Phenytoin + UPD glucuronyltransferase (UGT) → Very soluble compound
Some Phase II Reactions Precede Phase I
Due to the compound already being slightly soluble
Most lead to inactivation but sometimes some can lead to activation
Ex: morphine → morphine-6-glucoronide (more active)
Clinical Applications of Inducer and Inhibitors that can be beneficial
Ritonavir: inhibits 3A4 and can be used as booster to increase bioavailability of a substrate drug
Phenobarbital: inducer of UDP UGT in newborns with unconjugated hyperbilirubinemia, it conjugates it and eliminates it, prevents kernicterus
Explain how acetaminophen (APAP/Tylenol) can form toxic metabolites
Acetaminophen can be metabolized either by 34A or 2E1 but 10% can convert into toxic metabolite NAPQI which is toxic to hepatocytes
Excessive doses of Tylenol causes enzymes to be saturated and therefore more of NAPQI is produced
Alcohol consumption also increases production of NAPQI
Toxicity reversible within 8 hrs of reaching ER and being administered
Genetic Polymorphisms
In regards of drug effects
Genetic mutations with an incident of 1% in the population
Defects cause either too little or too much of the drug
Resemble drug interactions
Examples
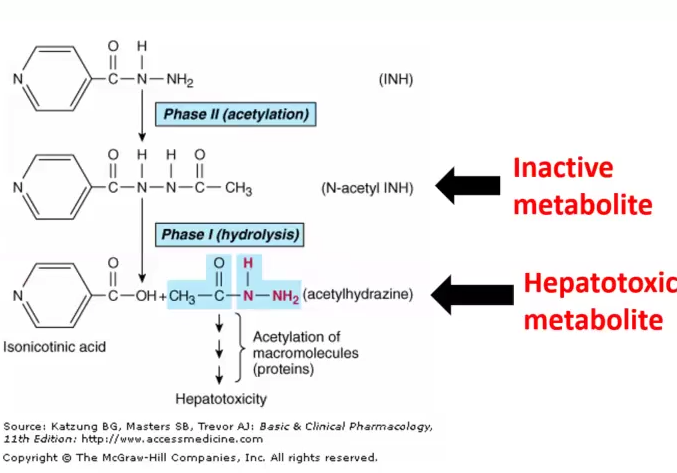
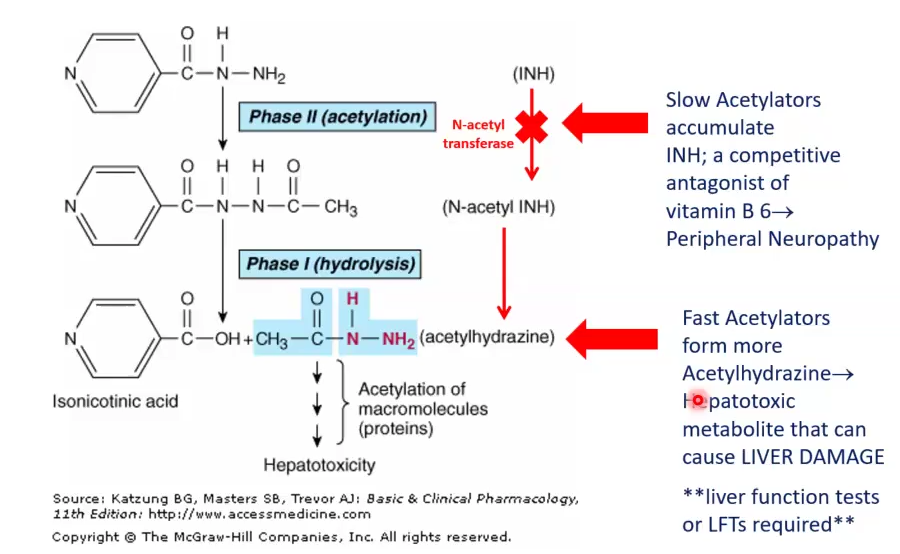
N-acetyl transferase enzyme deficiency:
Slow acetylators accumulate INH, which is a competitive antagonist of B6, causes Peripheral Neuropathy
Fast acetylators produce too much Acetylhydrazine, which is hepatotoxic
Butyrylcholinesterase deficiency:
Defective ester hydrolysis causes decreased metabolism of succinylcholine (anesthetic), which can cause apnea after surgery
Acetaldehyde dehydrogenase:
Defective aldehyde dehydrogenation can cause decreased metabolism of ethanol, causing flushing and hypotension
1st order kinetics vs Zero order kinetics
1st order: Drug concentration decreases by the same proportion per unit of time → Half life is a constant of 1st order kinetics
Zero order: Drug concentration decreases by the same amount but not the same proportion, thus the half life is constantly changing
Ex. Aspirin, Ethanol and Phenytoin
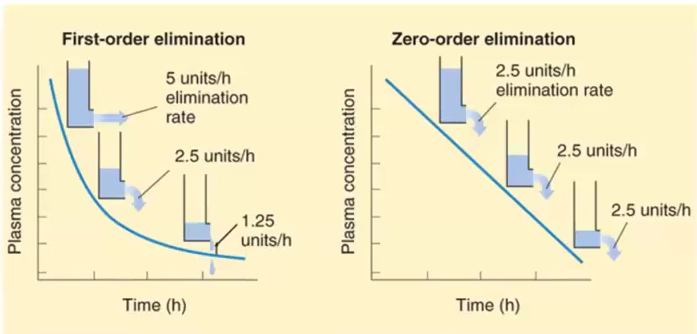
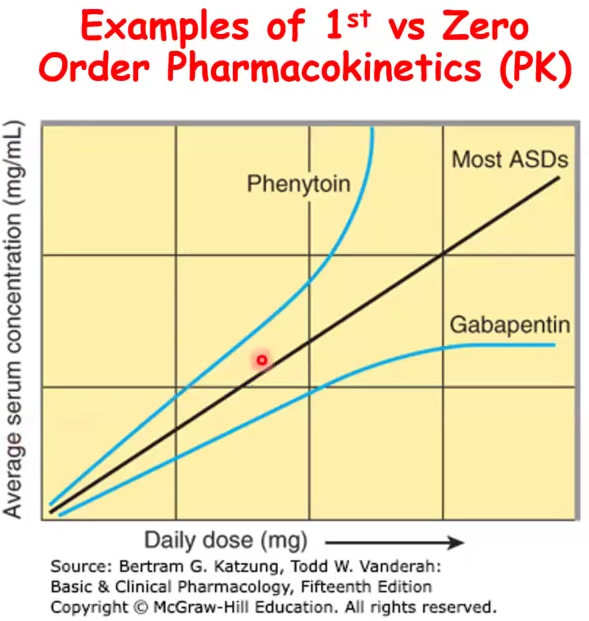
Phenytoin and Gabapentin as an example of Mixed order kinetics
Phenytoin: follows 1st order kinetics until a specific threshold where enzymes become saturated and then it becomes zero order
At threshold, the concentration keeps going up regardless of the dose of the drug
Gabapentin: follows 1st order kinetics at low doses then zero order is evident due to saturable absorption (gut transporter is saturated) and then serum concentration does not increase regardless of dose
Drug elimination occurs due to 3 processes
Liver Metabolism
Excretion removes active or inactive metabolites
Clearance by renal system
Drug excretion occurs in 3 ways
Renal, Biliary or fecal → can also be lungs, breast milk or sweat tears etc
Renal excretion
Glomerular filtration
Tubular secretion
Tubular reabsorption
Amount of drug entering depends on GFR, Cp, Extent of bound protein
What does low Vd vs high Vd mean
Vd = D/ Cp
High Vd means the drug is mostly in the extravascular compartments
Low Vd means the drug is mostly in the vascular compartment, likely tightly bound to plasma proteins
Clearance
Formula
Used to determine
The removal of the drug by the body
Clearance (Cl) = rate of elimination of a drug / Plasma drug concentration (Cp)
Used to determine maintenance dose
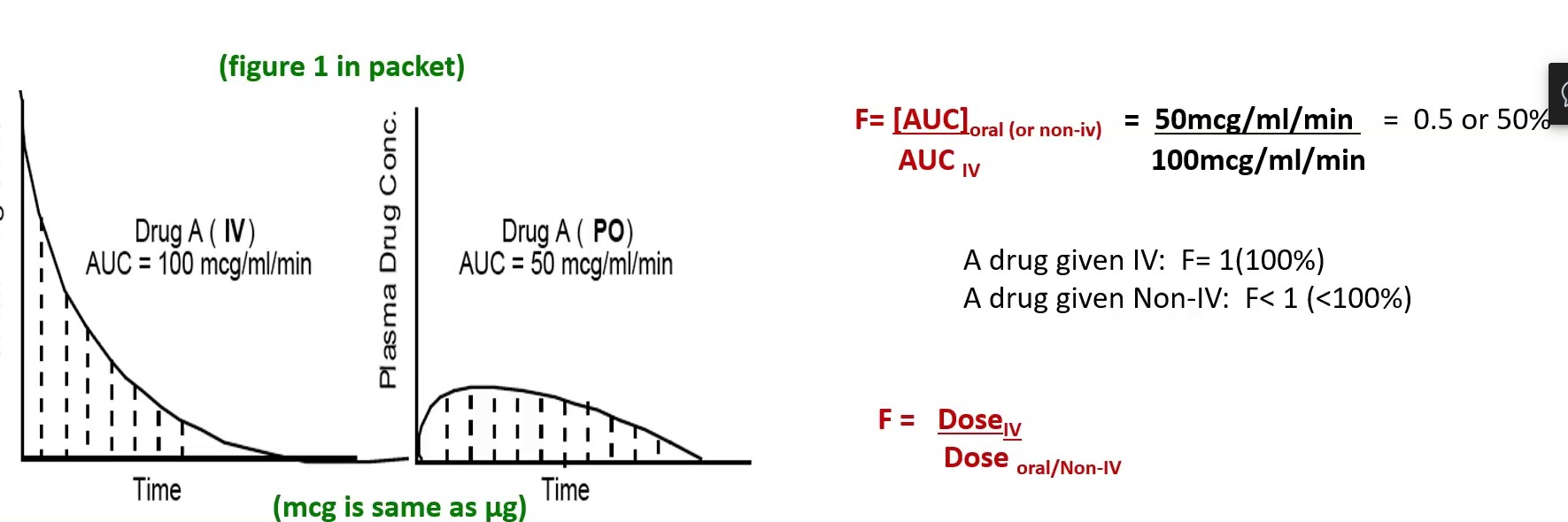
Bioavailability 2 Formulas
Bioavailability (F) = AUC (non-IV) / AUC (IV)
Bioavailability (F) = Dose (IV) / Dose (non-IV)
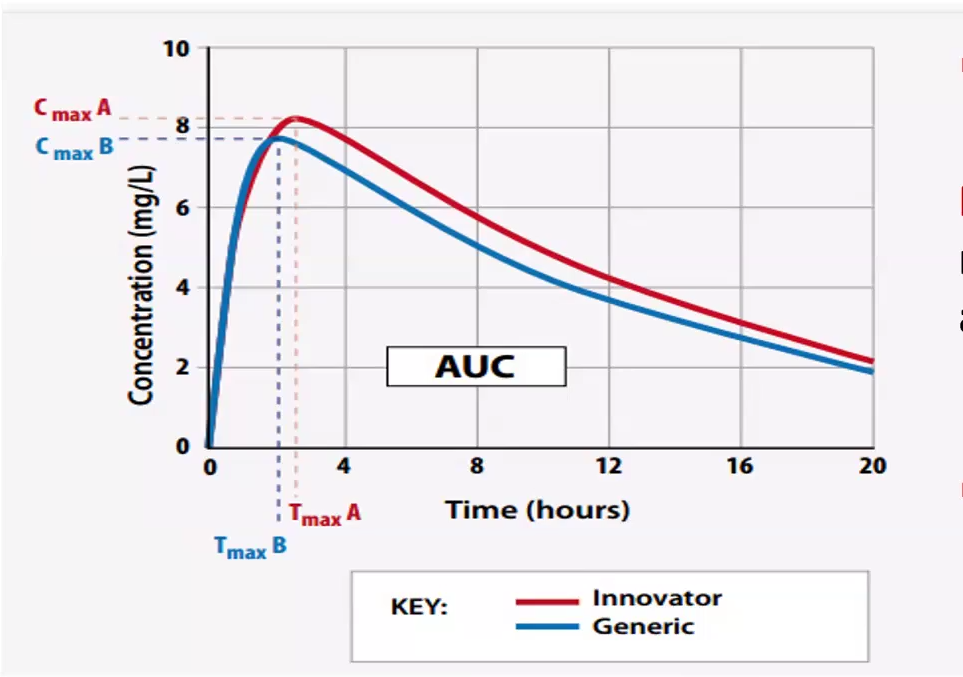
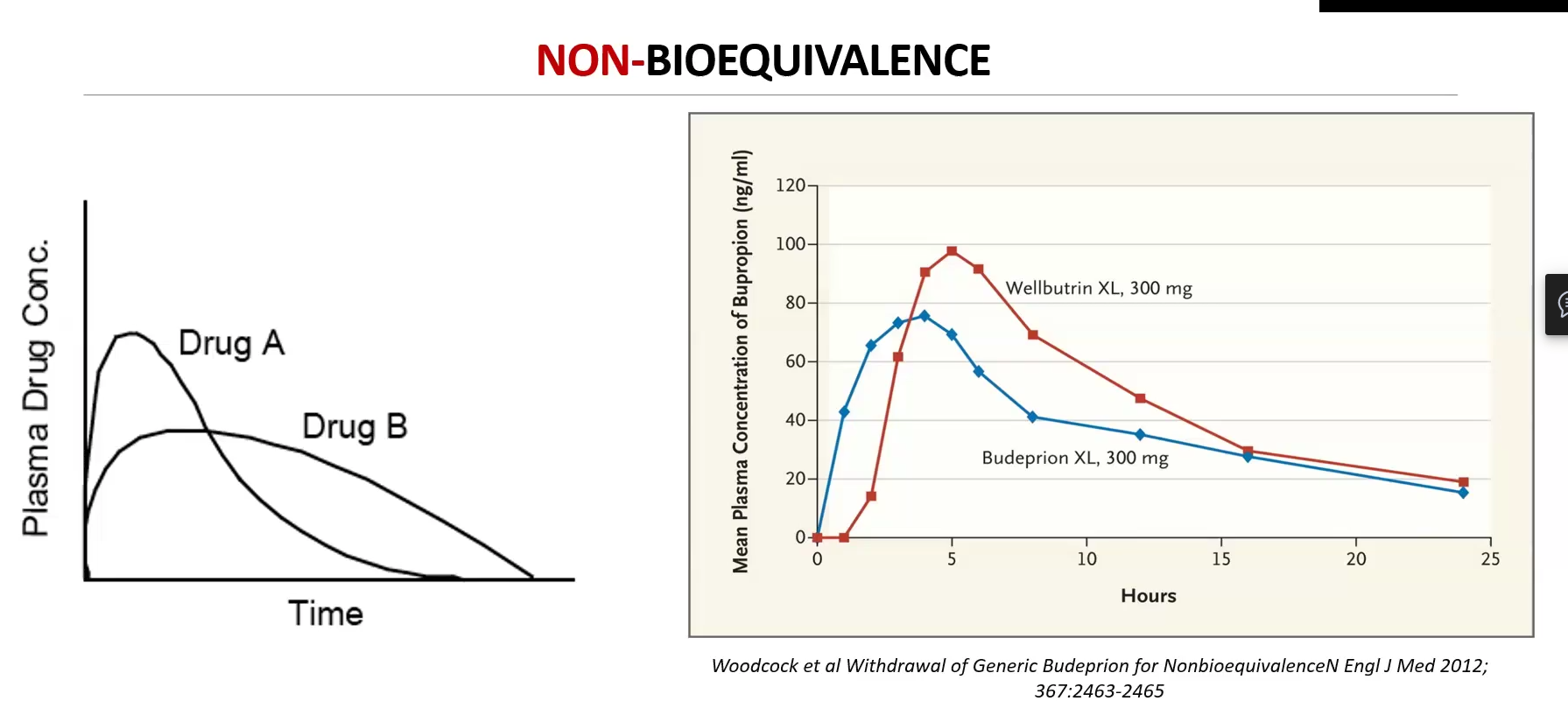
Bioequivalence studies compare two drugs (generic : innovator) and considers:
Two drugs are Bioequivalent IF:
Bioequivalence studies compare two drugs (generic : innovator) and considers:
Same ROA
AUC
Pharmacokinetic parameters such as Peak plasma concentrations (Cmax) and Time to Peak (Tmax)
Cmax and AUC most important and must be +-20% of branded drug
FDA requires 90% confidence interval of the AUC and Cmax ratios to fall between 80% and 125% of the branded drug
Two drugs are Bioequivalent IF:
They are pharmaceutically equivalent
Display comparable bioequivalence, peak plasma concentration and time to peak concentration
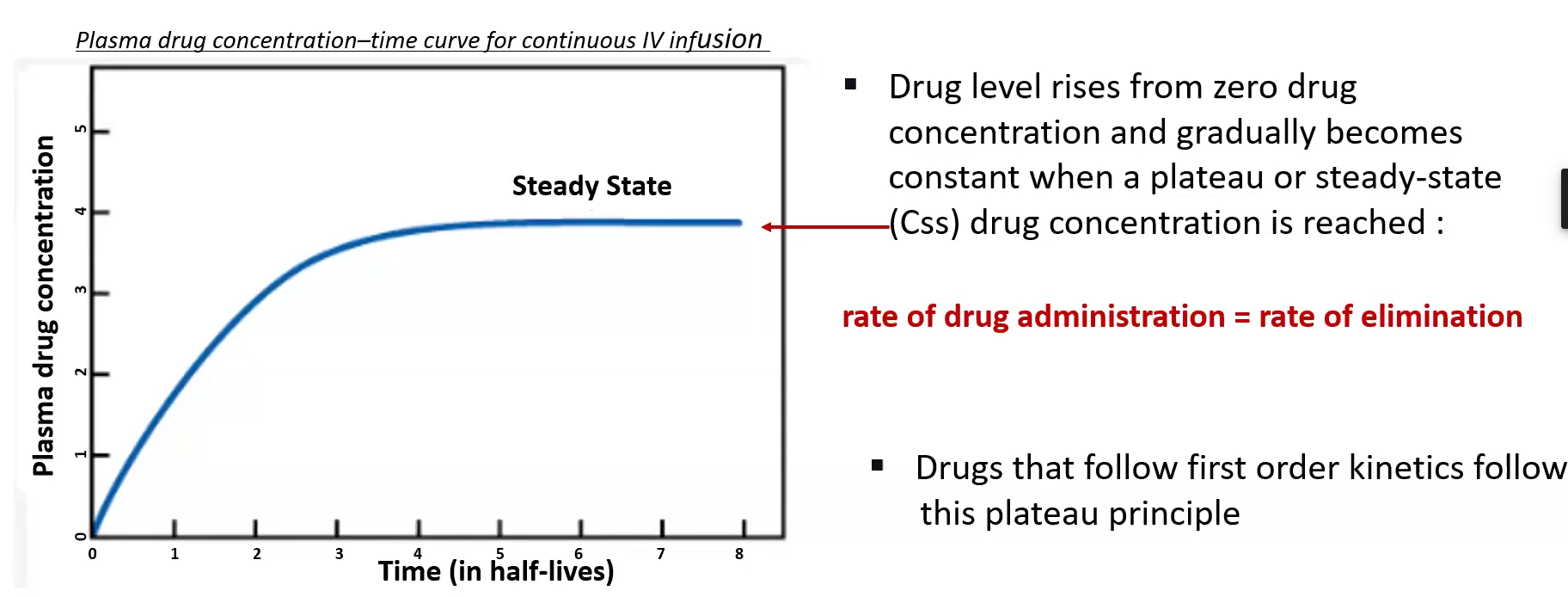
Steady State Concentration (Css)
Achieved when the dosing rate = clearance/rate of elimination
Css = rate of infusion / clearance (Cl)
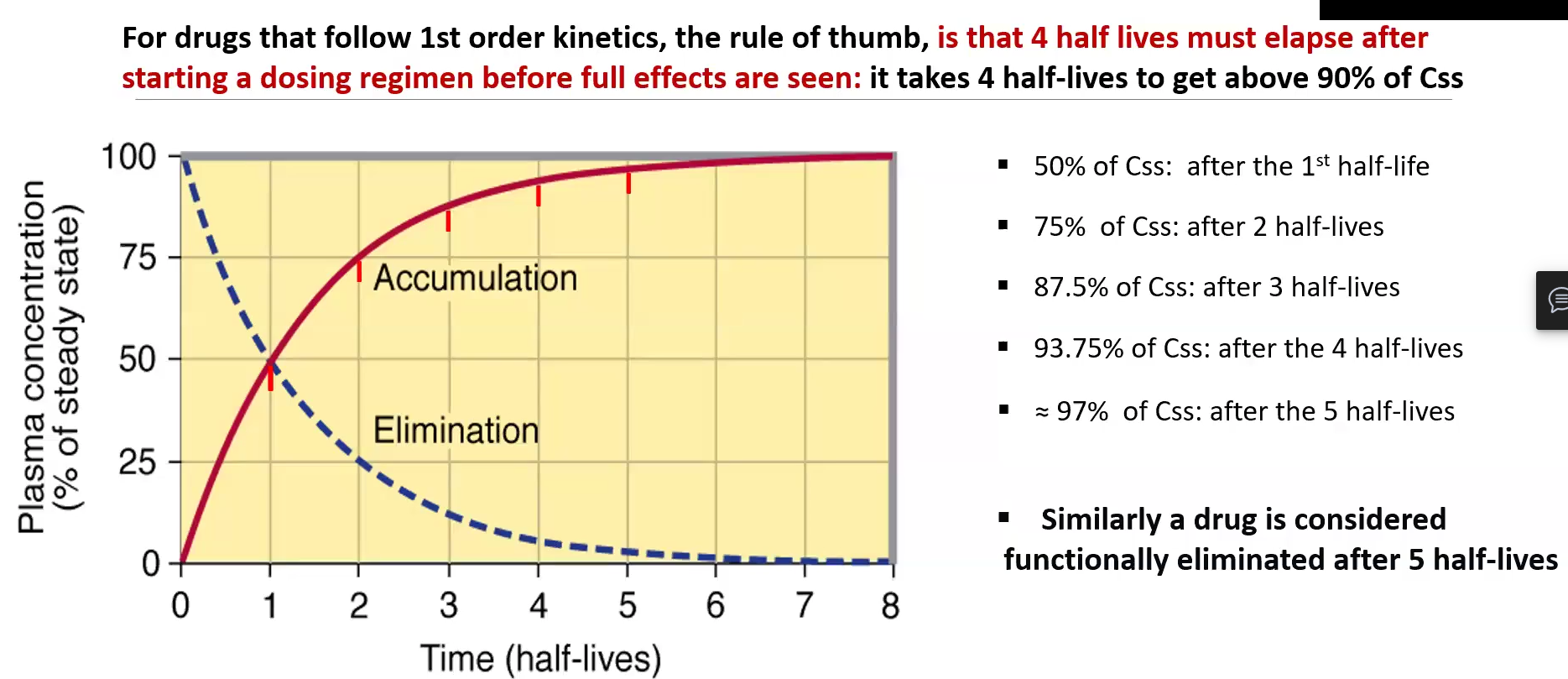
Steady State (Css) 2 Rule of thumbs
At least 4 half-lives must elapse after starting a dosing regimen before full effects are seen and 90% of Css is reached
1 hl = 50% Css
2 hl = 75% Css
3 hl = 87.5% Css
4 hl = 93.75 %hl
5 hl = 97% Css
At least 5 half-lives must elapse before functional elimination is reached
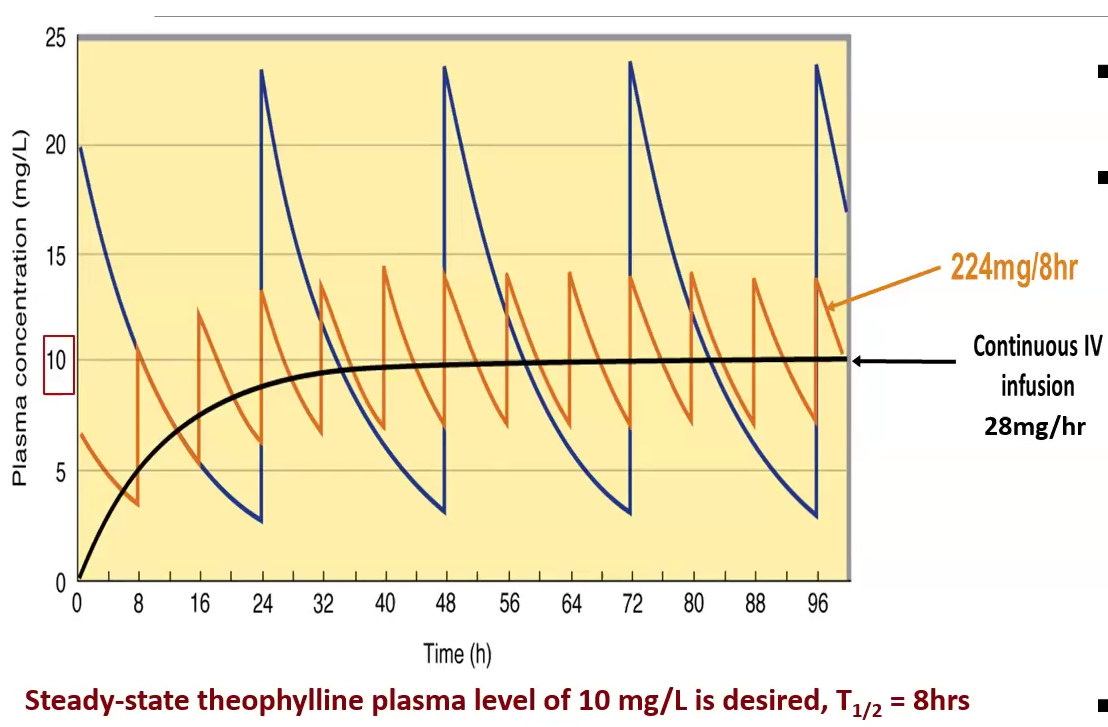
Single dose vs. Continuous dose
Single dose: has more fluctuations in concentration between doses, may be riskier if the therapeutic window is small
Constant: continuous injections or IV, safest and the best way to keep drug at target concentration
Loading dose vs Maintenance dose
Loading dose is the initial higher dose used to achieve the target concentration
Maintenance dose is the lower dose given to maintain steady state within the Therapeutic Window
Maintenance dose equation
Maintenance Dose = Dosing rate x Dosing Interval
Dosing rate IV = CL x (Cp or TC OR Css)
Bioavailability (F) oral = Dosing rate IV / Dosing rate oral
Dose adjustment calculation
Corrected dose = Average x creatine clearance/100 mL/min
Reasons for Therapeutic drug monitoring
Monitoring drugs with narrow therapeutic indices
Monitoring drugs with marked pharmacokinetic variability
Optimize therapy, especially if there is no easy marker for efficacy
Drugs known to cause adverse/toxic effects
Assessing patient compliance in taking medications