elshahawi - antiviral drugs medchem
1/62
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
63 Terms
viruses
the smallest human infectious agents
a virus is composed of:
a nucleic acid strand
associated proteins
it cannot reproduce on its own but attaches itself to the host cell and takes control
life cycle of a virus
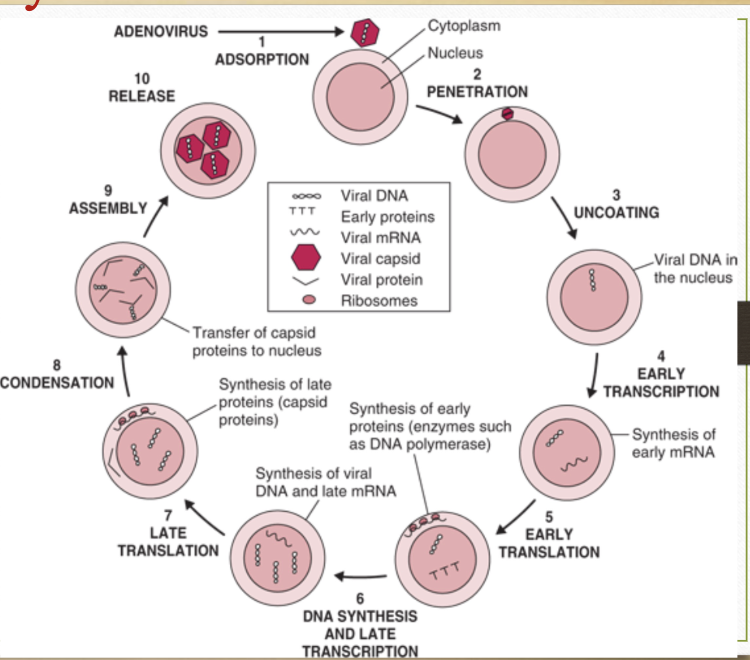
challenges associated with designing effective antiviral drugs
ability of viruses to undergo antigenic changes
latent period characterized by no symptoms
reliance on host enzymes and biochemical processes
massive virus proliferation
intracellular location of the virus in contrast to bacteria and fungi which can replicate independently
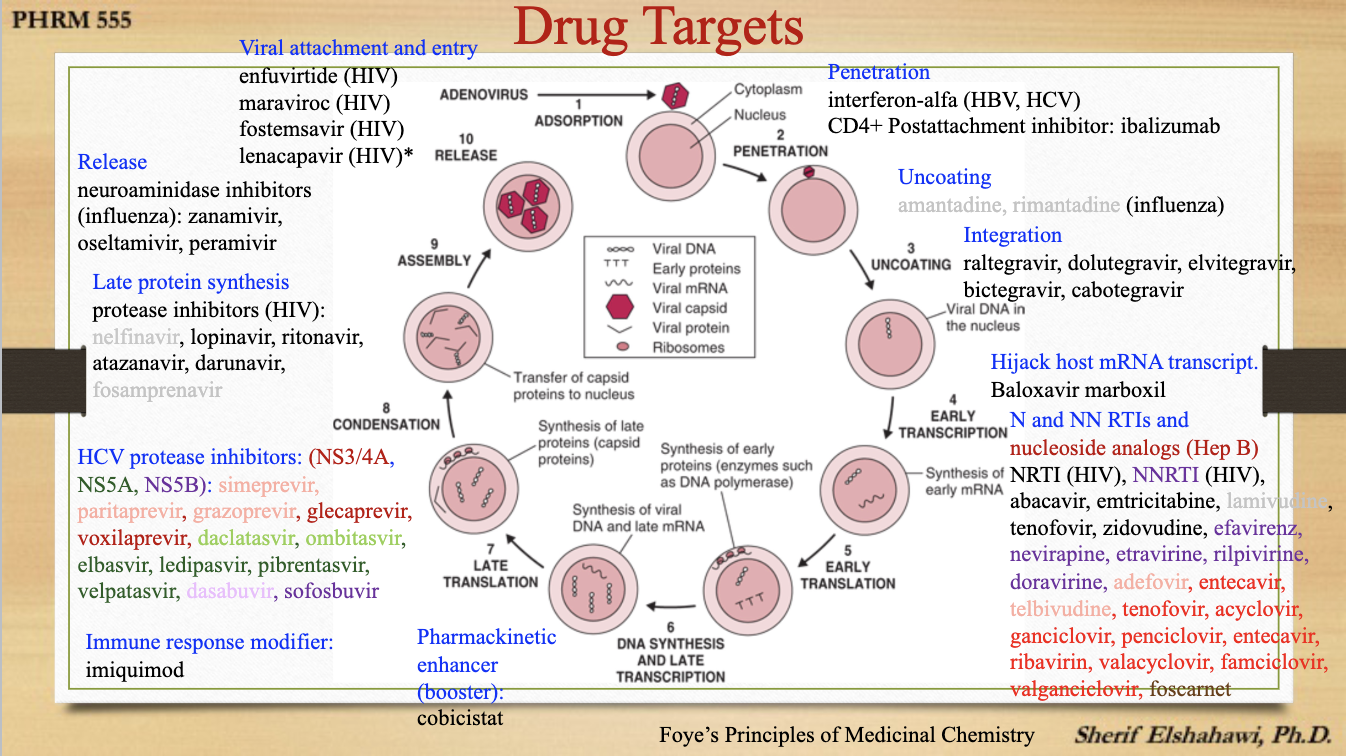
drug targets
influenza virus binding to a respiratory tract cell
influenza virus contains hemagglutinin protein, neuraminidase protein, M2 channel
hemagglutinin protein binds to sialic acid —> allows virus to enter the cell
neuraminidase protein hydrolyzes bond —> virus is free inside host cell
M2 channel ↑ protons and acidity —> needed for virus to release nucleic acids
influenza virus
genetic material is a single stranded RNA
types:
A (widespread influenza epidemics, occurs in two subtypes: H1N1, H3N2)
B (regional outbreaks)
C (low significance)
characterized by the presence of a Matrix-2 (M2) protein
M2 is a highly-selective protein channel protein that maintains pH
it is activated at low pH of the endoscope causing flow of protons into the virus
M2 inhibitors
Amantadine (Symmetrel, Symandine) and Rimantadine (Flumadine)
both are symmetrical tricyclic amine
they are the earliest effective flu drugs
they inhibit viral uncoating and target the viral ion channel protein M2, and thus prevents the acidic conditions required for viral membrane to fuse with the endosome —> blocks viral replication
active against Influenza A
should be given within he first 48h
antiviral resistance develops
CNS effects appear (as well as death due to drug overdose) but less with rimantadine
M2 inhibitors interactions with M2 channel
ionic interaction between protonated N & aspartate (negatively charged)
hydrophobic bonds between tricyclic amine and non polar residues
hemagglutinin (HA)
viral glycoprotein
spike-like objects
involved in adsorption, binds to the cellular glycoconjugates which contain sialic acid, allowing it to enter the host cell
neuraminidase (NA, Sialidase)
viral neuraminidase (NA) is a glycoprotein which catalyzes the cleavage of sialic acid (N-acetylneuraminic acid, Neu5Ac [terminal sugar molecule]) from glycoprotein and glycolipid
important in releasing virus from host cells after budding
hydrolyzes bonds between hemagglutinin and sialic acid conjugates
infection relies on a crucial balance between the rate of desialyation after budding vs the rate of attachment for adsorption
NA inhibitors cause viral aggregation by blocking release and prevent infection of new host cells
targets for flu vaccines
HA and NA are targets for vaccination
prevention via vaccination is the preferred method
neuraminidase inhibitors interactions w/ neuramindase
hydrogen and ionic interactions (hydrophilic)
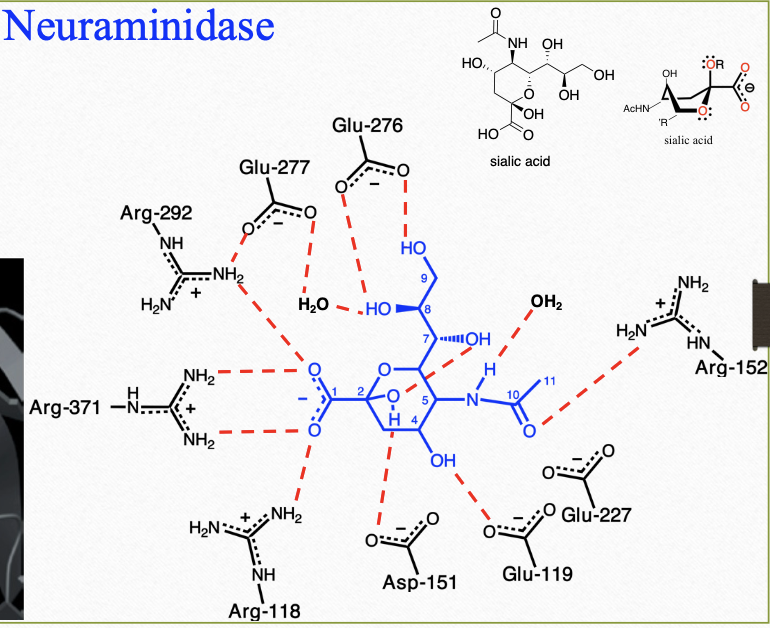
neuraminidase inhibitors
Zanamivir (Relenza), Oseltamivir (Tamiflu), and Peramivir (Rapivab)
oseltamivir is a prodrug —> ester is metabolized into carboxylic acid = active metabolite oseltamivir carboxylate
they act extracellularly and binds to an unoccupied area of influenza neuraminidase that results in competitive inhibition of the enzyme
they have also been shown to significantly inhibit the human sialidases NEU3 and NEU2 in the micro molar range which could account for some of the rare side effects in overdoses
zanamivir
(Relenza)
neuraminidase inhibitors
oral inhalation
excreted unchanged din the urine
t½ = 2.5-5 hr
oseltamivir
(Tamiflu)
neuraminidase inhibitor
oral
prodrug, metabolized to its active form via hepatic esterase to remove the ethyl ester
should be given within the first 48 hr of symptoms
t½ = 1-3 h (prodrug); t½ = 6-10 hrs (active form)
the active form is excreted unchanged in the urine
peramivir
(Rapivab)
neuraminidase inhibitors
IV
HIV life cycle-infection
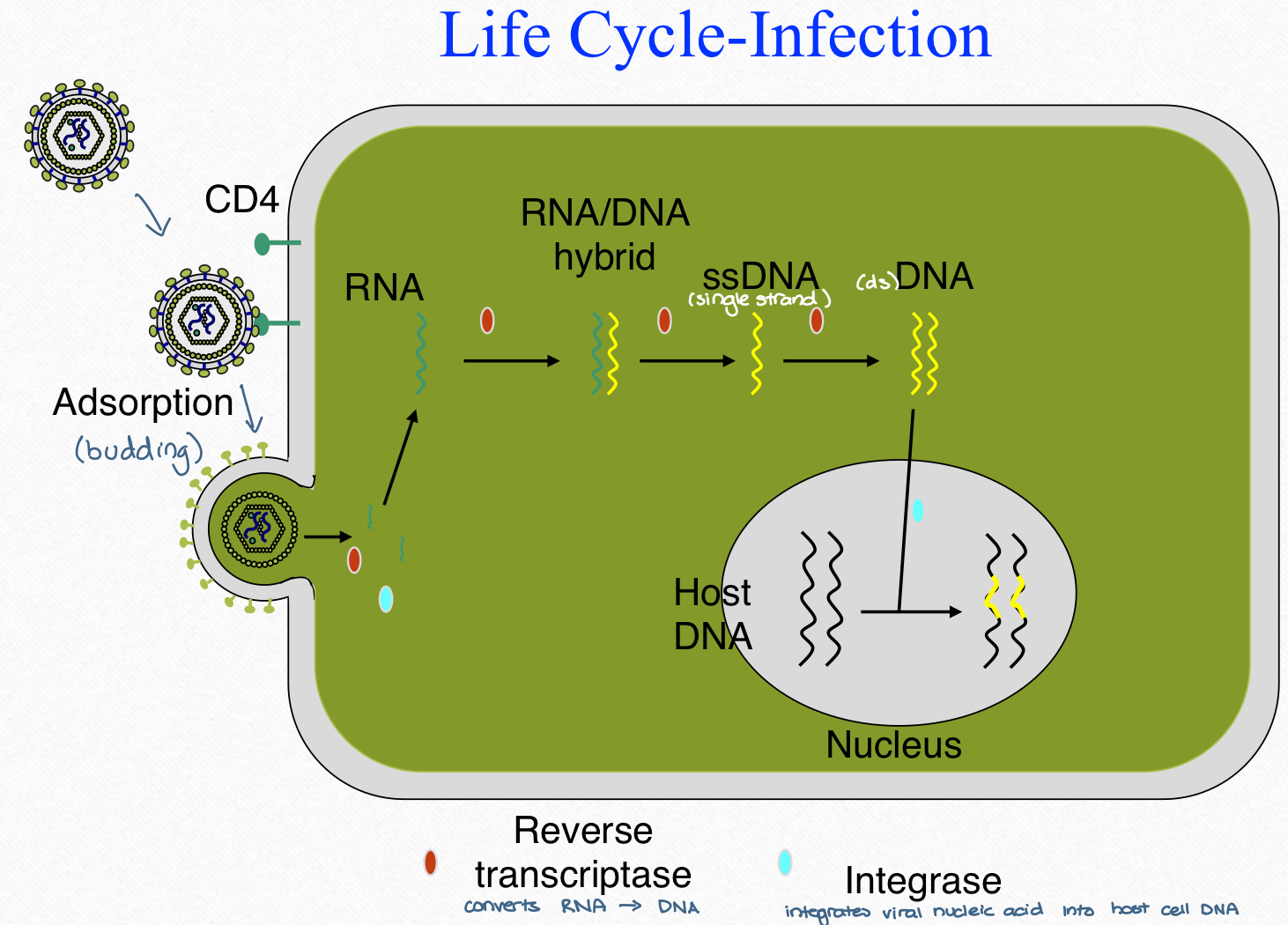
HIV life cycle-replication and release
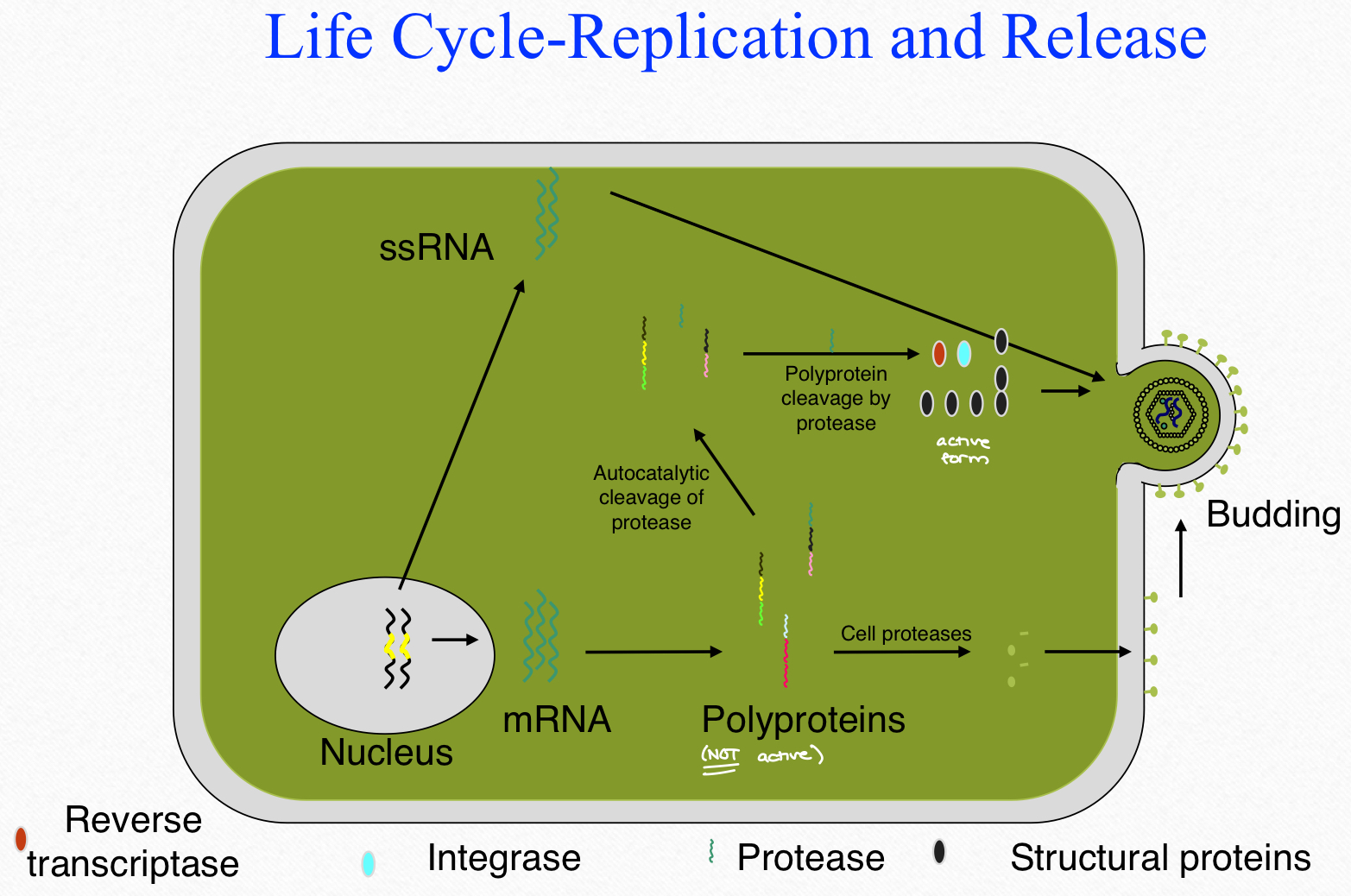
HIV protease
it is essential for the HIV life cycle
it cleaves newly synthesized precursor poly proteins at 9 cleavage sites to produce the mature protein components (active form)
inhibit of HIV protease interferes with the ability of the virus to replicate or infect additional cells
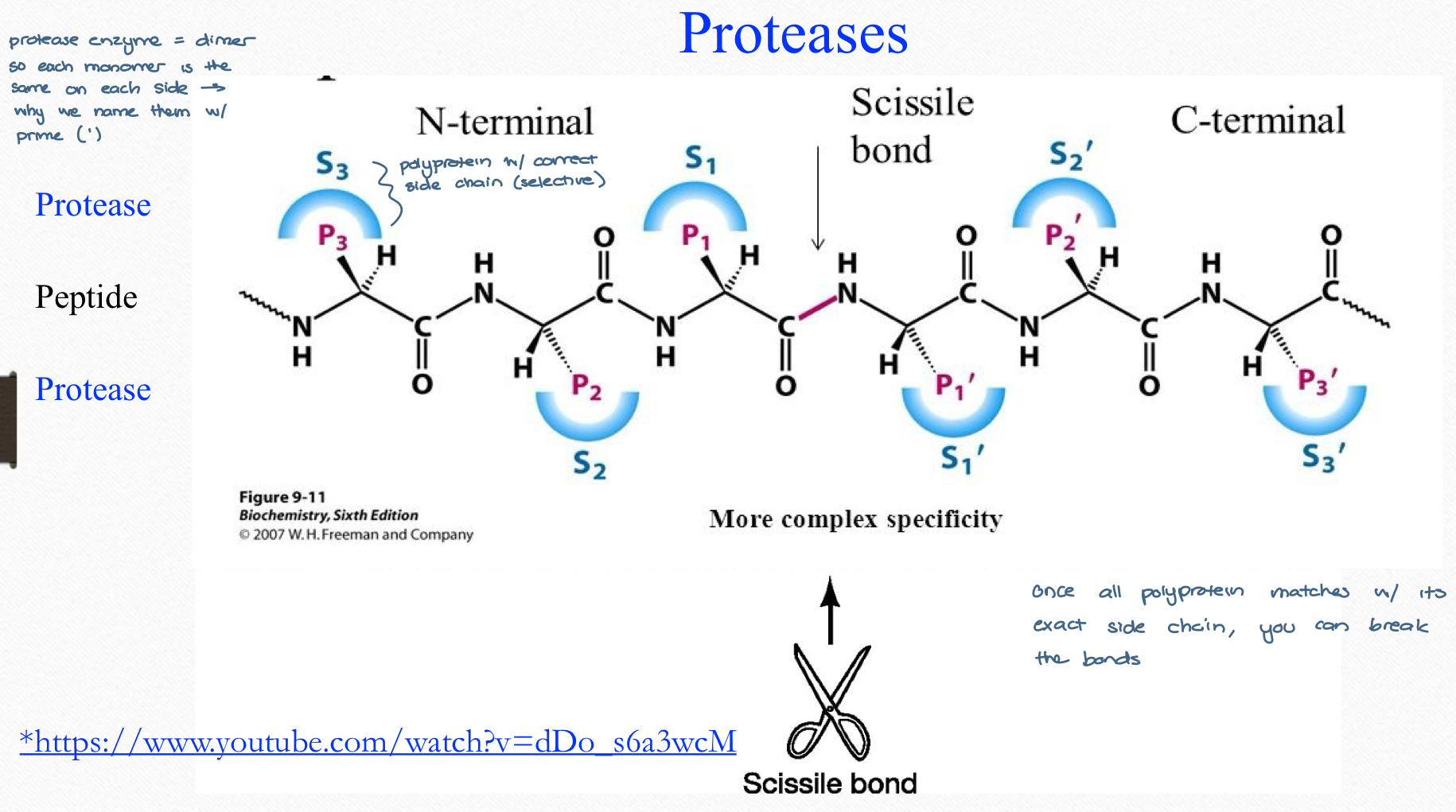
HIV proteases
symmetrical active site, unlike human proteases, thus selection inhibition might be possible
HIV proteases aspartic proteases
catalytic region = Asp-25 and Asp-25’ (one for each monomer)
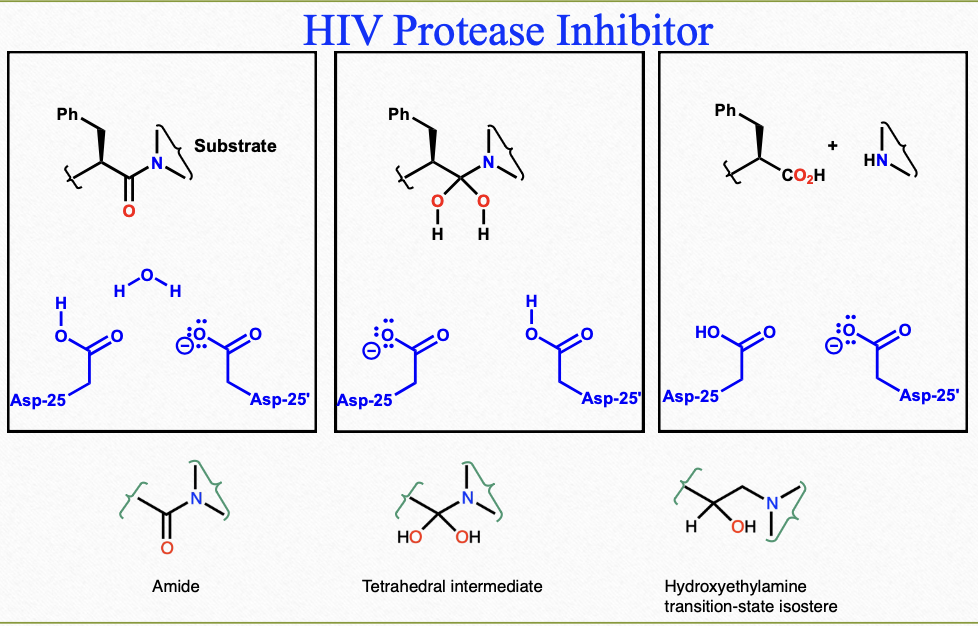
HIV protease inhibitors
amide (NOT stable), tetrahedral intermediate (NOT stable), hydroxyethylamine (transition-state isostere)
Phe fits in the S1 and S1’
R-OH forms 2 hydrogen bonds to both catalytic Asp residues
activity increases with bulky protecting groups
resistance to proteolytic degradation
pyridine (including other heterocyclic) and urea increase polarity and water solubility
urethanes improve plasma half life and potency
challenges with peptide drugs
high Mrwt, peptide bonds, poor water solubility
poor absorption
metabolic susceptibility
rapid excretion
limited access to CNS
high plasma protein binding
HIV protease inhibitors (agents)
-navir
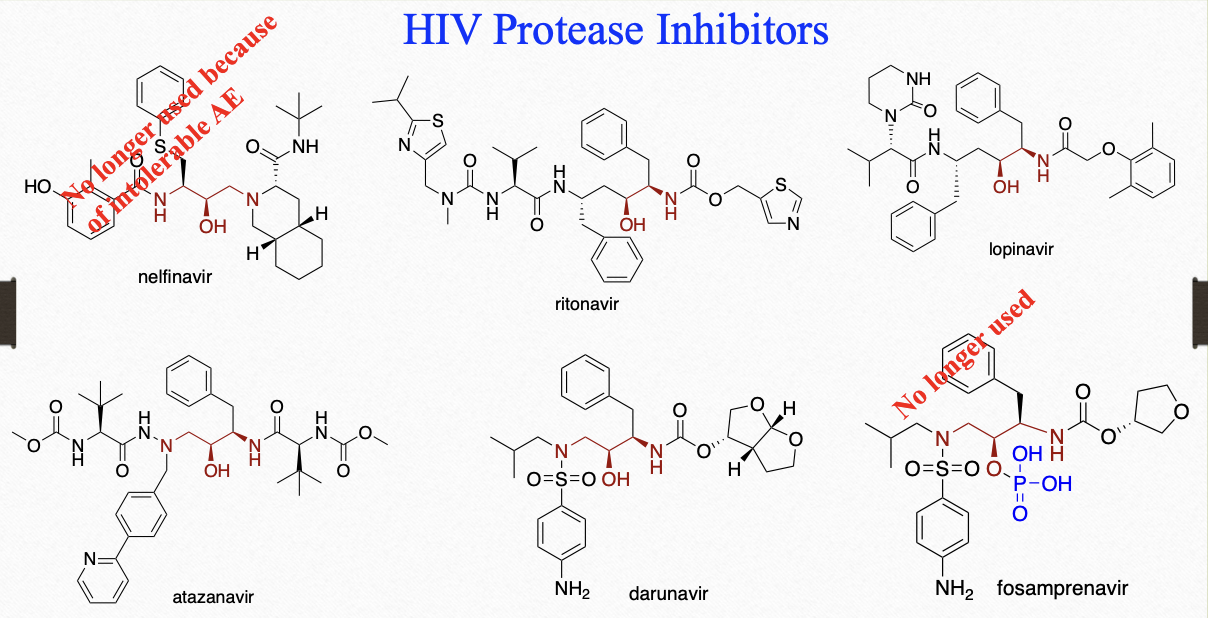
nelfinavir
decreased peptide character, lower Mr weight
hydroxyethylene transition state
t½ = 3.5-5 hrs
metabolized by CYP3A and CYP2C19 to hydroxylated analogs that also has antiviral properties
take with food as it decreases PK variability
atazanavir
extended subsequent to bind S1’ and S3’
metabolized mainly by CYP3A
t½ = 7 h
admin with food enhances bioavailability and reduces PK variability
plasma bound: 86%
fosamprenvair
aniline increases water solubility
tetrahydrofuryl carbamate gp (THF) is a good binding group for S2
phosphate prodrug (phosphates, longer t½ many other protease inhibitors)
the active form is metabolized in the liver by CYP3A4
good oral bioavailability (40-70%)
90% plasma bound
t½ = 7.7 h
darunavir
eis-tetrahydrofuryl gp is a better binding gp for and fills better the hydrophobic S2 ring oxygens forms hydrogen bond to the protein backbone
decreases HIV viral load and increases CD4 cell counts
absorption increases with food (30%)
metabolized mainly by CYP3A
coadministration with ritonavir inhibits CYP3A metabolism, increasing darunavir concentration 14-fold making t½ = 15 h
95% plasma bound
lopinavir
poor oral bioavailability and extensive biotransformation (needs to be taken with a booster)
admin with food increases absorption
binds plasma protein >98%
mainly eliminated in the feces
used together with ritonavir as Kaletra in a 1:4 ratio
ritonavir
5-thiazolyl nitrogen forms hydrogen bond with Asp
mutation of Val-82 to other residues causes resistance due to the disruption of the hydrophobic interaction
added in combination drugs as booster to inhibit CYP3A (recommendations to sue ritonavir-boosted combination therapies)
t½ = 3-5 h
mainly eliminated in the feces
used together with lopinavir as Kaletra in a 1:4 ratio
retroviral integrase
enables HIV to integrate its genetic material into the DNA of the infected cell
requires MG2+ for activity
Raltegravir (Isentress)
HIV integrase inhibitor
prevents the HIV DNA from interacting with the host DNA and subsequent HIV proliferation
has the diketo-enol system that binds the active site through:
hydrogen bonding: Asp128, Asp185, Glu221
van der waal interaction: Tyr221, Pro214
metabolized by glucuridation (UGT1A1)
t½ = 9 h
other drugs in this class include:
Doluegravir (Tivicay)
Elvitegravir (Vitekta)
Bictegravir (Biktarvy)
Cabotegravir
Fostemsavir (Rukobia)
approved July 2020
oral prodrug —> metabolized by phosphatase into Temsavir
first in its class attachment inhibitor
Temsavir is an attachment inhibitor that directly binds to the gp120 of the HIV envelope, locking it into a conformational state that prevents binding with the host CD4+ T cell receptor and entry into the host immune cell
it requires no dose adjustment when co-administered with protease inhibitors, boosters, or mild or moderately CYP including non-nucleoside reverse transcriptase inhibitors (NNRTIs)
Enfuvirtide (T20, Fuzeon)
cell entry inhibitors
a 36 amino acid peptide derived from and mimics the HIV-1 glycoprotein (gp)41 (responsible for the fusion of the virus to the cell membrane and subsequent subcellular uptake)
enfuvirtide inhibits protein formation needed for viral fusion with host cell membrane
plasma protein binding 92%
metabolized by proteolysis
t½ = 3.8 h
Maraviroc (Selzentry)
cell entry inhibitor
a chemokine receptor CCR5 antagonist
it disrupts the interaction of glycoprotein (gp)120 of HIV-1 and CCR5 receptors on uninfected cells, protecting healthy cells
76% bound to plasma protein
t½ = 14-18h
metabolized by CYP3A
AVOID St. John Wort as it induces CYP3A and thus will reduce the concentration of drug
Lenacapavir (Sunleca)
cell entry inhibitor
it is the first-in-class capsid inhibitor
approved Dec 2022
NO reported cross-resistance with existing anti-retroviral
it interferes with viral:
uptake
assembly
release
t½:
oral admin: 10-12 days
SC admin: 8-12 weeks
nucleic acid
3’ carbon of pentose has -OH that is needed to elongate the chain by attaching to 5’c carbon
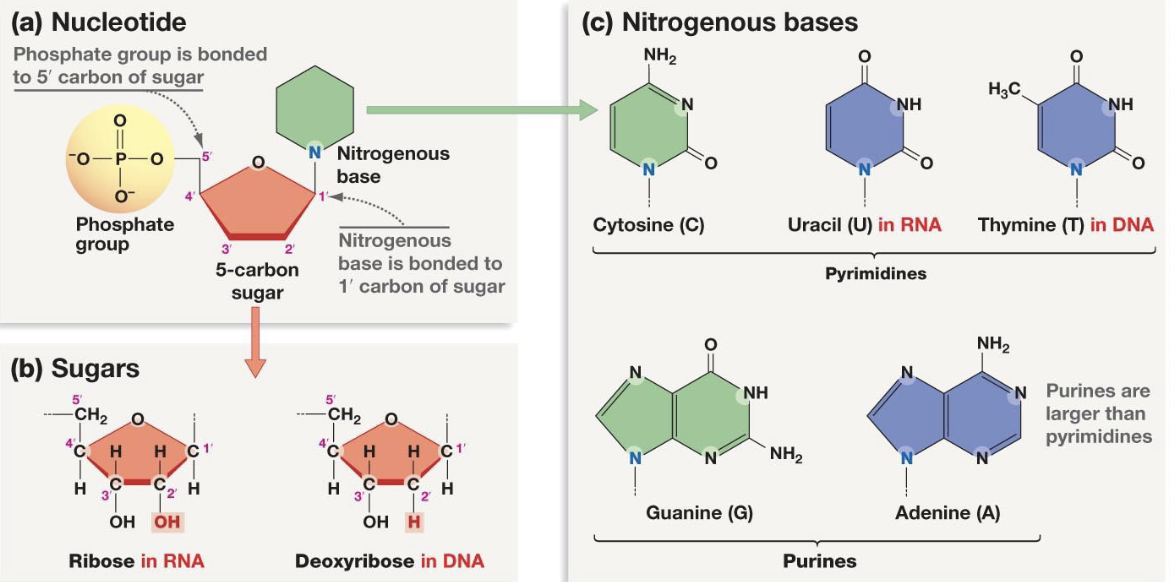
Lamivudine (Epivir, 3TC)
prodrug
nucleoside reverse transcriptase inhibitors (NRTIs, Nukes)
inhibits the reverse transcriptase of HIV and HBV via incorporation into viral DNA after phosphorylation to its 5’-triphosphate metabolite (L-TP) using HIV reverse transcriptase and HBV polymerase
structurally similar to nucleotides
Lamivudine is combined with zidovudine or abacavir for synergistic inhibition
t½ = 5-7 h
mainly excreted unchanged in the urine
excreted in human breast milk
food reduces absorption slightly
plasma protein binding < 36%
Emtricitabine
prodrug
nucleoside reverse transcriptase inhibitors (NRTIs, Nukes)
t½ = 10 h
mainly excreted unchanged in urine
plasma protein binding < 4%
Zidovudine
prodrug
nucleoside reverse transcriptase inhibitors (NRTIs, Nukes)
IV
t½ = 1.1 h
mainly excreted as the inactive glucurodinated ZDV
plasma protein binding ~35%
Abacavir
a carbocyclic nucleoside and requires cytosolic deaminase
prodrug
nucleoside reverse transcriptase inhibitors (NRTIs, Nukes)
plasma protein binding ~50%
metabolized to inactive:
5’-carboxylic acid metabolite by alcohol dehydrogenase
5’-glucuronide metabolite by glucuronosyltransferase
t½ = 1.54 h
nucleic acid synthesis inhibitors — acyclic nucleoside phosphonate prodrugs
Adefovir (Proven) and Tenofovir
prolonged antiviral activity
phosphonate are highly stable to serum esterase cleavage
activated by biphosphorylation (two additional phosphates are added)
the side chain in Adefovir dipivoxil and tenofovir diisopropoxyl fumarate increases bioavailability. the ESTER chain I released by esterase to give the drug
adenovirus (Prevon)
nucleic acid synthesis inhibitors
acyclic nucleoside phosphonate prodrugs
rarely used nowadays due to nephrotoxicity
tenofovir
tenofovir, tenofovir diisopropoxyl fumurate, tenofovir alafenamide (TAF)
tenofovr diisopropoxyl fumurate is more nephrotoxic compared to TAF
plasma binding proteins < 7%
t½ = 32 h
secreted in the urine by globular filtration and human organic anionic transporters
AE: bone toxicity and nephrotoxicity
non-nucleoside reverse transcriptase inhibitors (NNRTIs) — non-nukes
Nevirapine (Viramune), Efavirenz (Sustiva), Etravirine (Intelence), Rilpivirine (Edurant), and Doravirine (Pifeltro)
allosteric inhibitors of RT (non-competitive — binds to different site aside from the active site)
distorts active site without phosphorylation leading to chain termination
nevirapine (Viramune)
non-nucleoside reverse transcriptase inhibitors (NNRTIs) — non-nukes
plasma bound protein 60%
high bioavailability (> 90%)
metabolized by CYP3A4 to several hydroxylated analogs
t½ = 45 h
efavirenz (Sustiva)
non-nucleoside reverse transcriptase inhibitors (NNRTIs) — non-nukes
plasma bound protein >99%
metabolized by CYP450 system to several hydroxylated analogs
t½ = 40-55 h
AVOID alcohol consumption
rec. with empty stomach as food increases conc and AE
Etravirine (Intelence)
non-nucleoside reverse transcriptase inhibitors (NNRTIs) — non-nukes
plasma bound protein > 99%
metabolized by CYP3A4, CYP2C9, CYP2C19 to several hydroxylated analogs
t½ = 9-40 h
Rilpivirine (Edurant)
non-nucleoside reverse transcriptase inhibitors (NNRTIs) — non-nukes
metabolized by CYP3A4, CYP3A5
t½ = 34-35 h
take with food; absorption inc. 40% when taken with food
Doravirine (Pifeltro)
non-nucleoside reverse transcriptase inhibitors (NNRTIs) — non-nukes
plasma bound protein > 76%
metabolized by CYP3A4, CYP3A5
t½ = 15 h
acyclovir (Zovirax, Acyclo-G)
nucleoside analog — purine
used mainly against HSV-1
It binds 200 more times stronger to and 3 × 106 faster to HSV kinase than to host kinase. Thus, Acyclovir monophosphate is mainly and selectively produced in the infected cells.
Host kinases phosphorylate acyclovir monophosphate to acyclovir di- and triphosphate. Acyclovir TP is a competitive inhibitor of dGTP for the viral DNA polymerase and lacks 3’-OH gp and thus causes chain termination
the concentration of acyclovir TP is HSV infected cells is. 40-100 times that of non-infected cells
resistance develops due to mutation in viral thymidine kinase and DNA polymerase
t½ = 2.5-3 h
mainly excreted in the urine unchanged
other acyclic nucleosides
prodrugs require thymidine kinase or phosphotransferase for activation
they possess selective uptake by infected cells when compared to non-infected cells
ganciclovir cause bone marrow toxicity
peniciclovir produces higher triphosphate concentration in virus infected cells and has longer t½ when compared to acyclovir
entecavir is a guanosine analog but not a typical NRTI. It is phosphorylated intracellularly leading to competition with natural substrates
ribavirin (Virazole)
guanosine analog
inhibitor of RNA polymerase
ribavirin triphosphate inhibits ribonucleotide synthesis, RNA synthesis, or RNA capping in RNA viruses
ribavirin is phosphorylated intracellularly to mono-, di-, and triphosphate metabolites
once phosphorylated, ribavirin disrupts cellular purine metabolism by inhibiting inosine monophosphate dehydrogenase, which leads to decrease in guanosine triphosphate
ribaverin-5’-triphosphate is a selective competitive inhibitor
t½ = 120-170 h
acyclic nucleoside prodrugs
valacyclovir is the L-valyl ester derivative of acyclovir
famciclovir is the diacrtyl derivative of penciclovir precursor
valganciclovir si the L-valyl ester of ganciclovir, used against CMV. it has 10x greater bioavailability than ganciclovir
Baloxavir marboxil (Xofluza)
approved by FDA in Oct 2018
treatment of influenza A and B
a selective inhibitor of influenza cap-dependent endonuclease which prevents polymerase function and therefore influenza virus mRNA replication (cap snatching mechanism)
plasma protein binding 93%
t½ = 79 h
foscarnet
phosphoric acid derivative
mimics pyrophosphate that selectively inhibits the pyrophosphate binding site on viral DNA polymerase and inhibits RNA polymerase
protein binding 14-17%
t½ = 3.3-6.8 h
hepatitis C
it is an enveloped ssRNA virus
HCV enzymes NS3-4A protease is responsible for cleaving HCV poly protein at four sites to generate four peptides: NS4A, NS4B, NS5A, and NS5B
NS5A inhibitors
inhibits the phosphorylation of NS5A (dimer protein) which is involved in HCV replication, assembly, and secretion
-asvir
characterized by the presence of pyrrolidine and carbamate moieties
contains symmetrical elements
NS5B inhibitors
NS5B: RNA-dep RNA polymerase
suffix -buvir
block viral replication and thus subsequent translation
Sofosbuvir (Sovaldi)
nucleoside inhibitor
acts as a prodrug
Dasabuvir
non-nucleoside inhibitor
NS3/4A inhibitors
suffix - previr
NS3/4A protease cleaves polyproteins
inhibitors inhibit protein synthesis and prevent viral maturation
macrocyclic structure, contains N-(cyclopropylsulfonyl)acetamide group
can be used with interferon and ribavirin
viakirax is a combination of ombitasvir (NS5A), paritaprevir (NS3/4A), ritonavir (HIV protease)
imiquimod (Aldara, Zyclara)
toll-like receptor 7 agonist
cobicistat (Tybost)
pharmacokinetic enhancer (booster)
podofilox (Condylox)
a lignin found in podophyllin resin from the roots of podophyllum plants
remdesivir
treatment of SARS-CoV-2
prodrug that gets metabolizes into a nucleoside triphosphate
inhibits viral RNA-dependent RNA polymerase (RdRp) —> inhibits viral replication