PDTI Module 1 & 2: Introduction to Pharmokinetics, Pharmodynamics and Personalized Medicine
1/31
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
32 Terms
Pharmokinetics (PK)
• What the body does to the drug
• Study of the time course of its Absorption, Distribution, Metabolism, and Elimination (ADME).
• Often based on plasma/serum concentration time profiles.
Pharmocodynamics
• What the drug does to the body
• Study of the relationship (time course and intensity) between drug concentrations at the site of action and corresponding effect, including both therapeutic and adverse effects
Biopharmaceutics
• Effect of dosage form on PK
• Immediate release vs slow release formulations
Pharmagenomics
• Studies how a person's genes affect how the patient responds to medications
• Ex. Looking at gene mutations and SNPs (single nucleotide polymorphisms)
Plasma
• Derived from unclotted blood, contains no cells (heparin, EDTA)
Serum
• Supernatant of clotted blood
Therapeutic Goal
• Treat the patient with the right drug at the right dose and time to improve his/her status and ideally cure the disease.
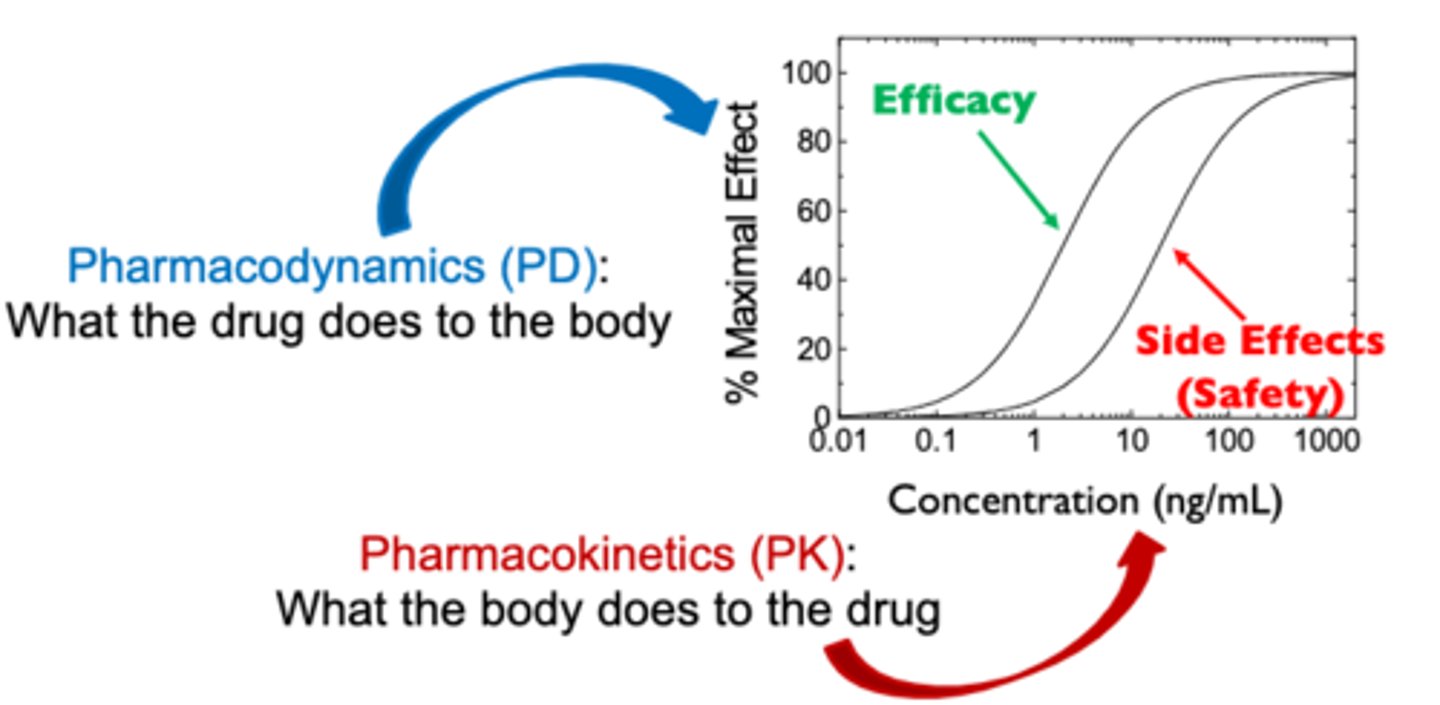
Why do we use serum/plasma for decision making?
• We cannot experimentally determine drug concentration in tissue, however, in plasma/serum it is done easily and there is a relationship where typically the tissue has 2x the concentration as seen in plasma/serum
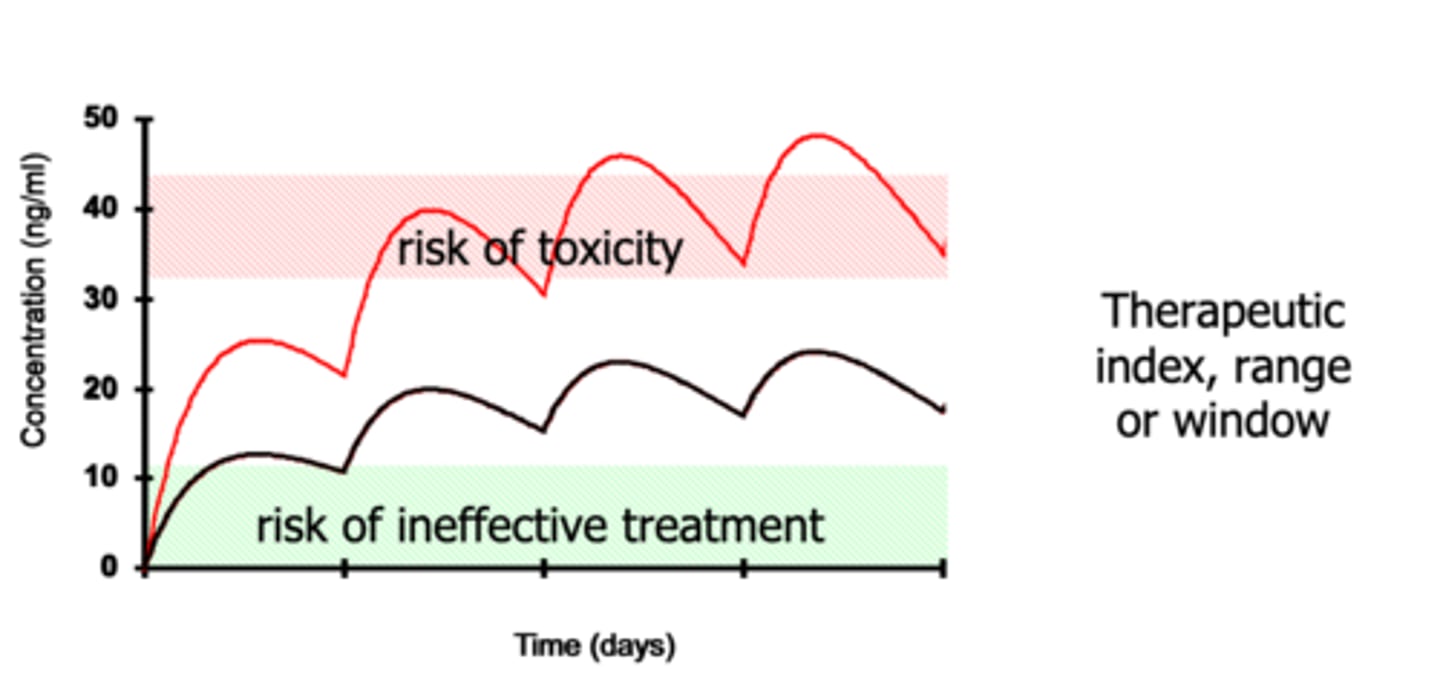
Therapeutic Index
• The ratio between the toxic and therapeutic concentrations of a drug
There is an average patient that can be used to determine the idea dose regimen of a drug. (True/False)
• False, there is no average patient, since patients differ in gender, race, body weight, etc. which all effect pharmcokinetics. Mutations may affect the pharmcodynamic side.
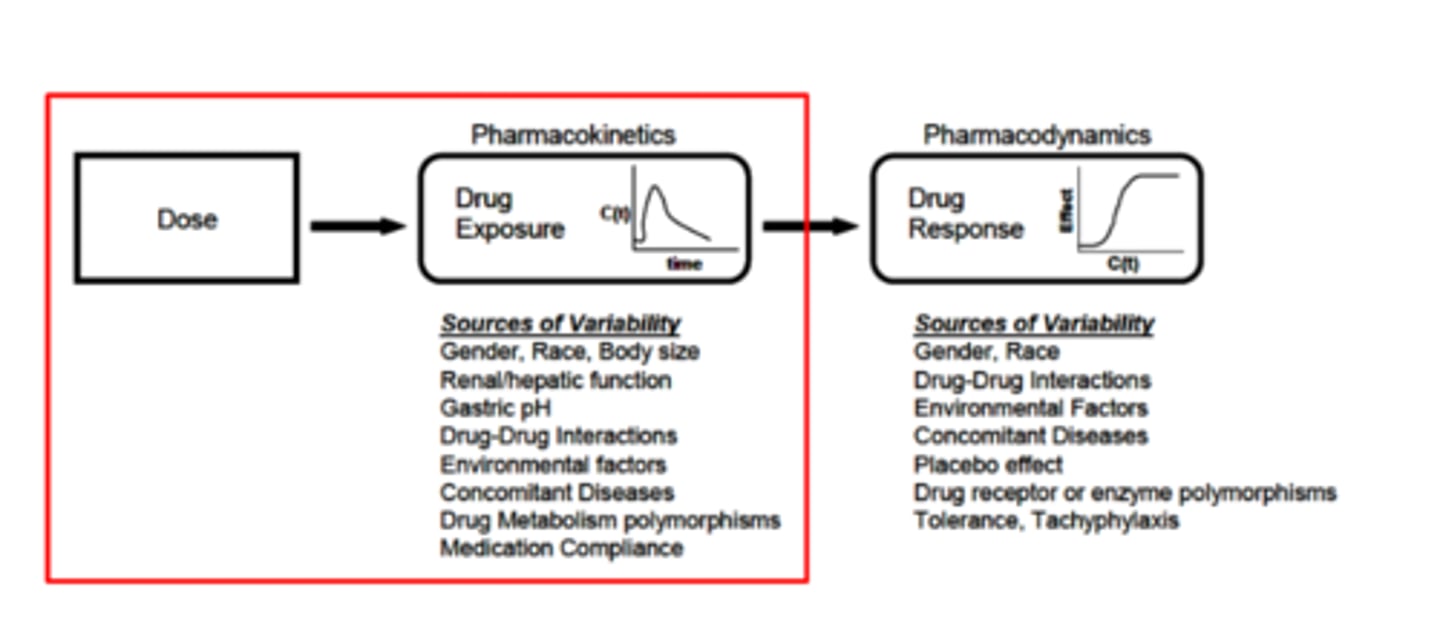
What are sources of variability for pharmacokinetics and pharmacodynamics?
• See Image
• Shared sources include:
Gender, Race, Drug-Drug Interactions, Environmental Factors, Concomitant Diseases
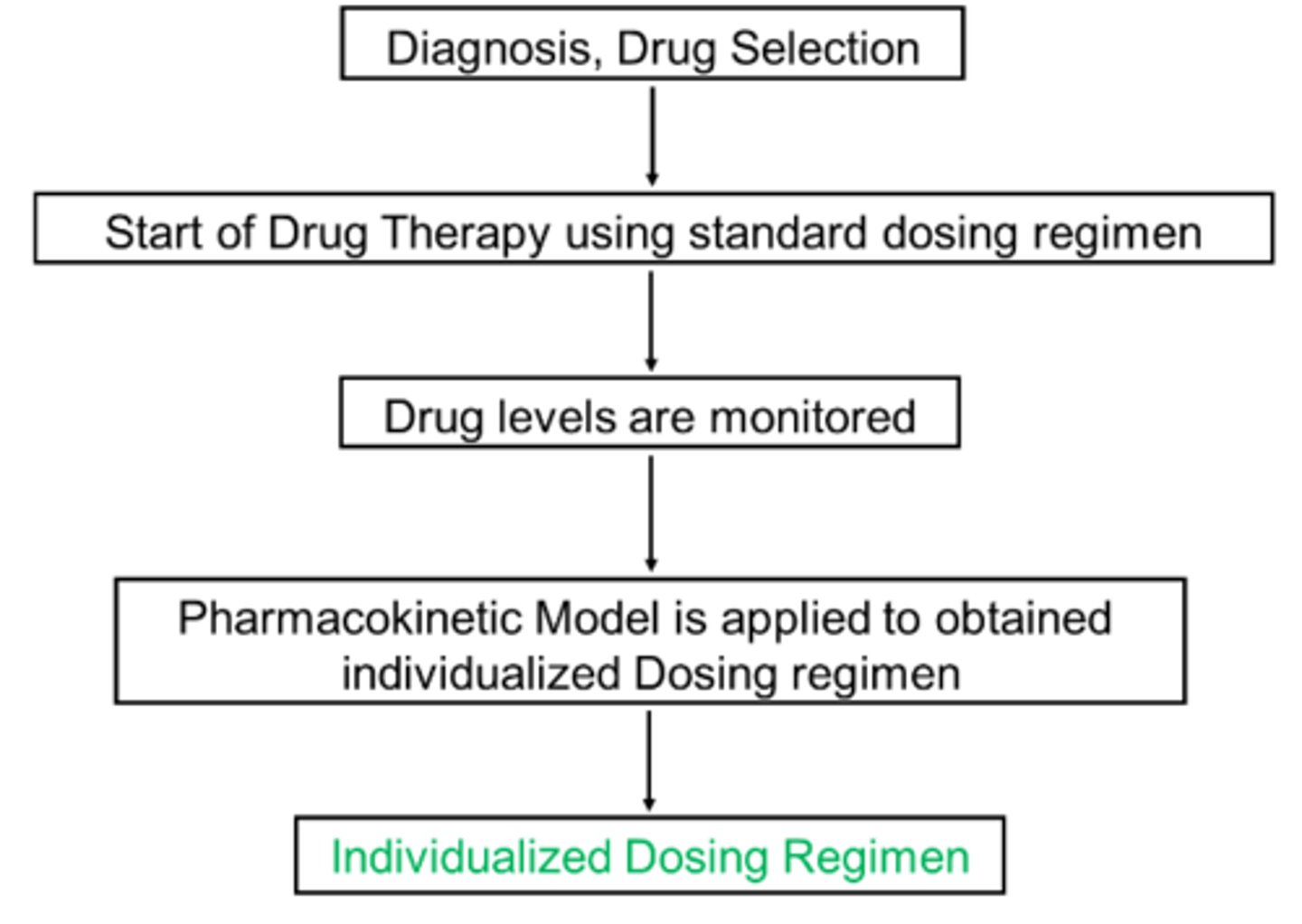
Retrospective dose adjustment
• Give a patient 2 - 3 doses, look at the plasma concentrations of the drug (which is therapeutic drug monitoring), and then adjust the drug regimen
Prospective dose adjustment
• Use pharmacogenomics to look at SNPs of enzymes, that may reduce or increase the efficacy of a drug, and decide the idea drug and dose based of that information
Therapeutic Drug Monitoring
• Used to monitor drug concentrations of narrow therapeutic index drugs (e.g., digoxin, aminoglycosides, phenytoin and to retrospectively adjust the dose if needed.
Pharmacogenetics (PGx)
• The term is meant to cover all types of investigations that provide information about a person’s genetic makeup that address questions about the choice of drug and drug doses that are likely to work best for that particular person
How to use PGx
• Define qualitative/quantitative differences between patient subgroups in terms of their PK and/or PD, based on pharmacogenetic information.
• Prospective selection of an optimal dosing and treatment regimen rather than retrospective adjustment based on patient’s genomic footprints( often based on specific diagnostic tests)
• Result: better and more cost-effective clinical outcomes
C₀ =
Dose/Vd
(applies to first order?)
Elimination rate constant
Varies from drug to drug and person to person, it is NOT the same thing as elimination rate
How do you calculate ke for a first order equation?
= [ln(C₂) - ln(C₁)]/(t₂-t₁)
Unit: 1/time; 1/h
![<p>= [ln(C₂) - ln(C₁)]/(t₂-t₁)</p><p>Unit: 1/time; 1/h</p>](https://knowt-user-attachments.s3.amazonaws.com/e365360c-e702-4c6e-97ad-27eb1a2b5ef9.jpg)
Half Life Formula t₁/₂ =
• = 0.693/ke
• = (0.693×Vd)/CL
First order process
• The actual amount of drug eliminated per time unit is changing (and is based on concentration), however the fraction of drug eliminated per time period is constant
• There is a constant t₁/₂
• Ke's unit is 1/time
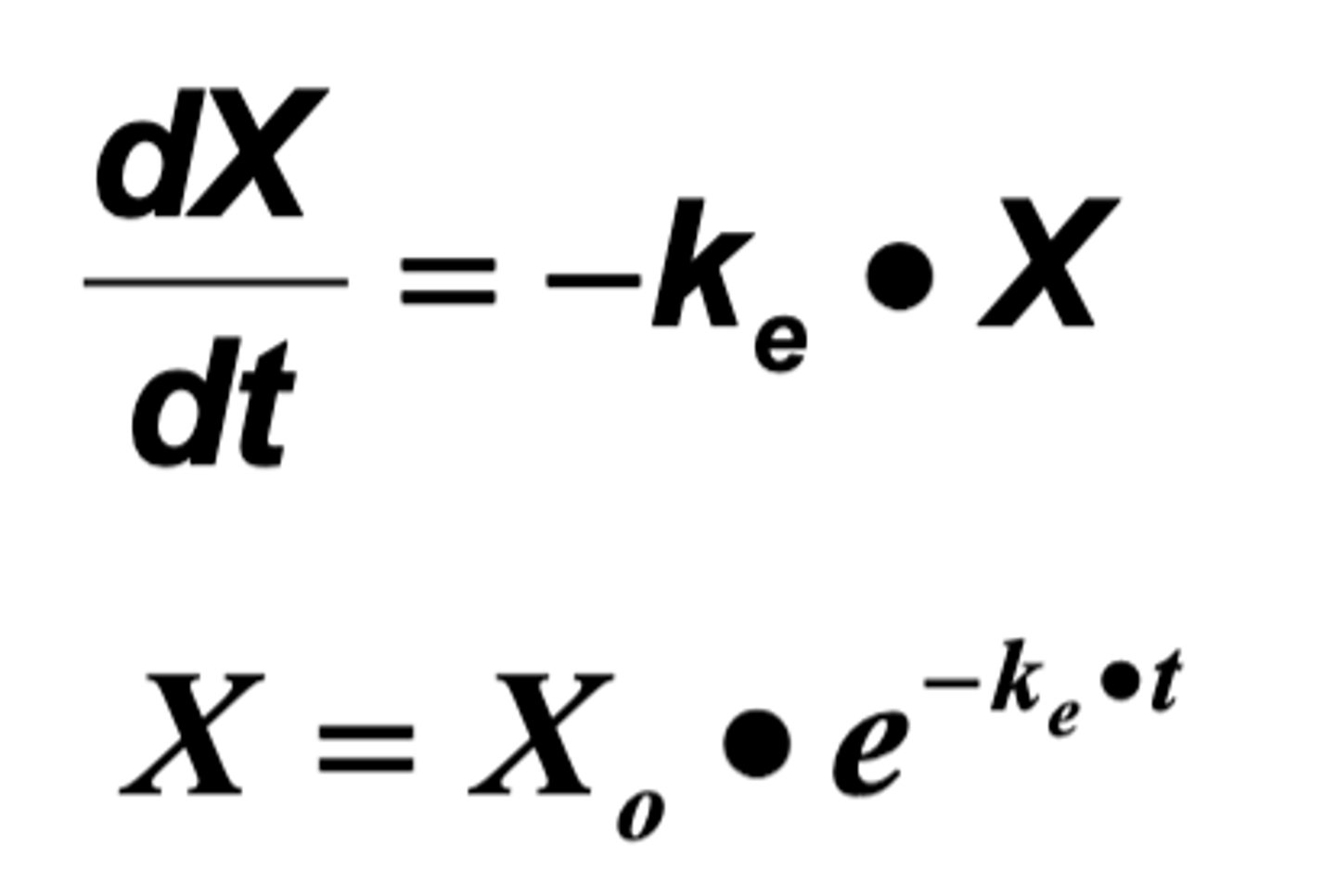
Concentration Formula for First Order
C = C₀×e^(-ke*t)
Ke's unit: 1/time; 1/h
e^(-ke*t) is the fraction eliminated per unit time
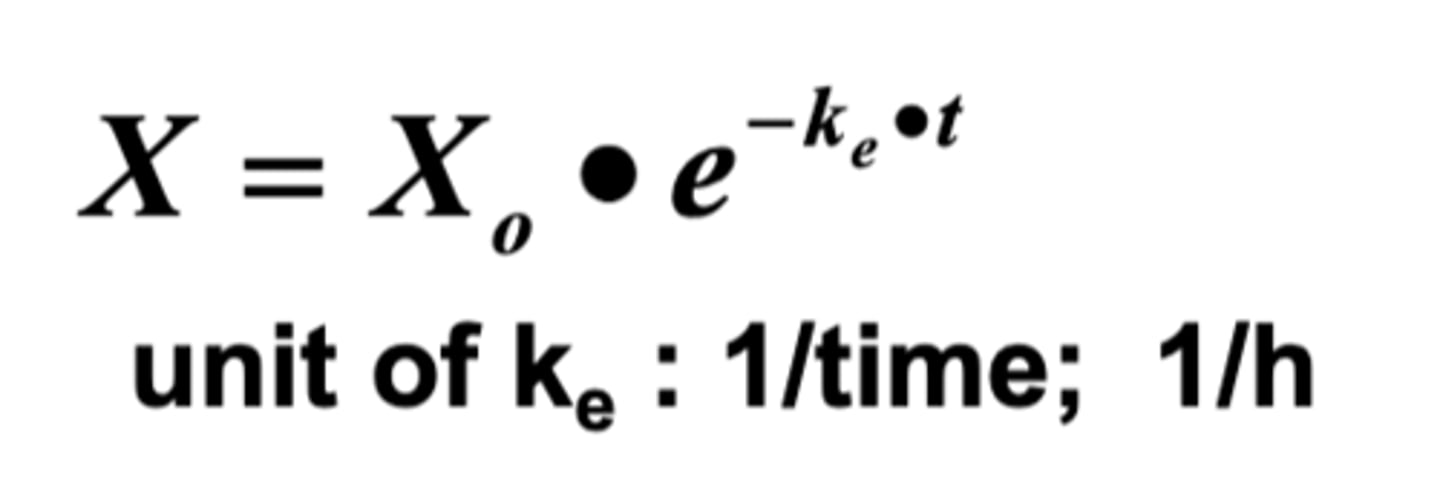
Excretion Rate of First Order
dC/dt = -ke*x
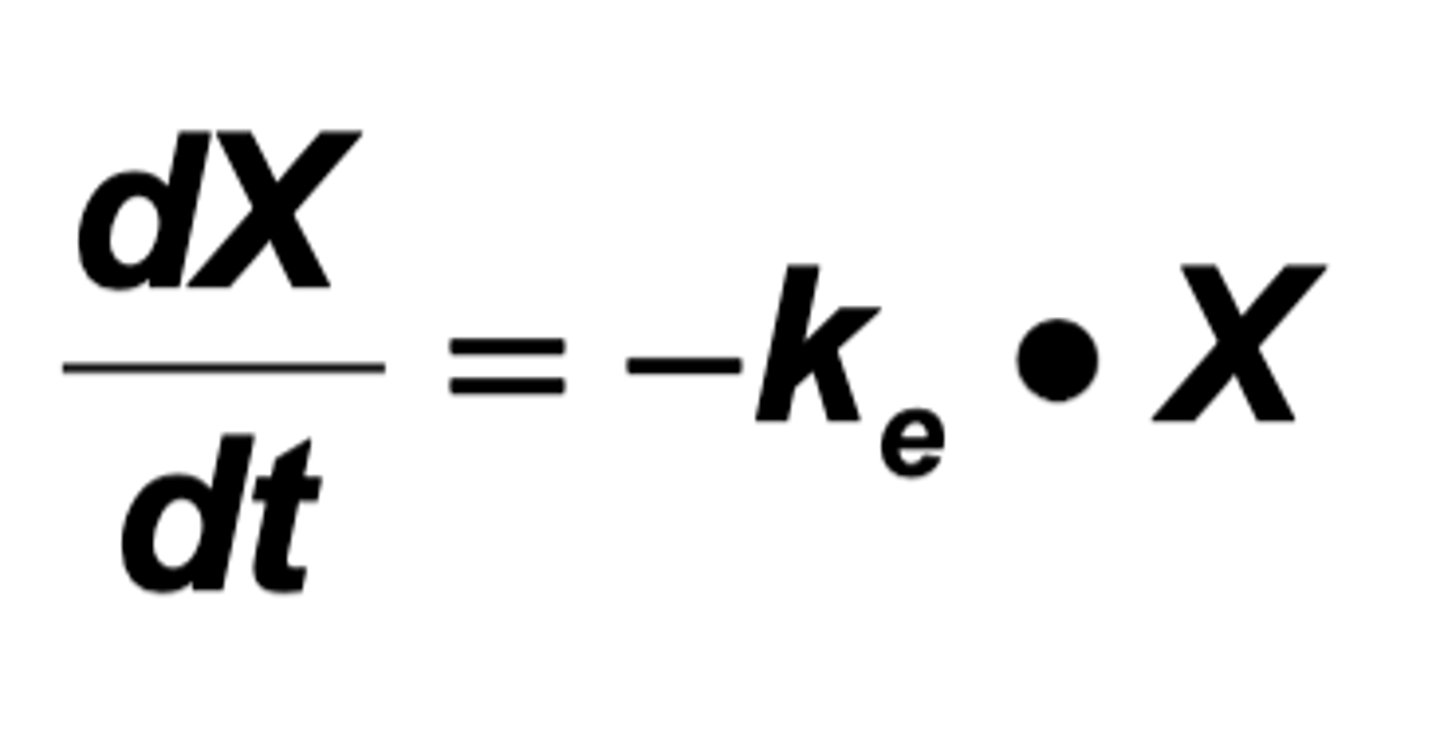
Zero Order Processes
• The rate of elimination is independent of amount of drug
• Occurs when drug is saturated
• The fraction is constantly changing and so is t₁/₂
• The amount eliminated is CONSTANT
• Ke's unit is amount/time
Concentration Formula for Zero Order
C = -Ke*t + C₀
Unit: Amount/time; mg/h
Area under the curve (AUC)
(for first order)
• Parameter telling us how much drug is present over time
• To assess how much drug entered the body (Bioavailability)
• Use definite integrals/trapezoid rule to deduce
Relationship between Cl, Dose and AUC
(for first order)
Cl = Dose/AUC
ke =
(for first order)
CL/Vd
Trapezoidal Rule
= [(C₂ + C₁)*(t₂ - t₁)]/2
• Be careful of the t₂ - t₁ changing as you move down the time-concentration profile chart
![<p>= [(C₂ + C₁)*(t₂ - t₁)]/2</p><p>• Be careful of the t₂ - t₁ changing as you move down the time-concentration profile chart</p>](https://knowt-user-attachments.s3.amazonaws.com/d81dd16d-4dca-4646-8db4-4fa57dbbb9d0.jpg)
Extrapolation to infinity
= Cx/ke
• Cx being the last concentration (C₂ that was used) to calculate AUC
• Then add this to the rest of the AUC that was calculated with the trapezoid rule
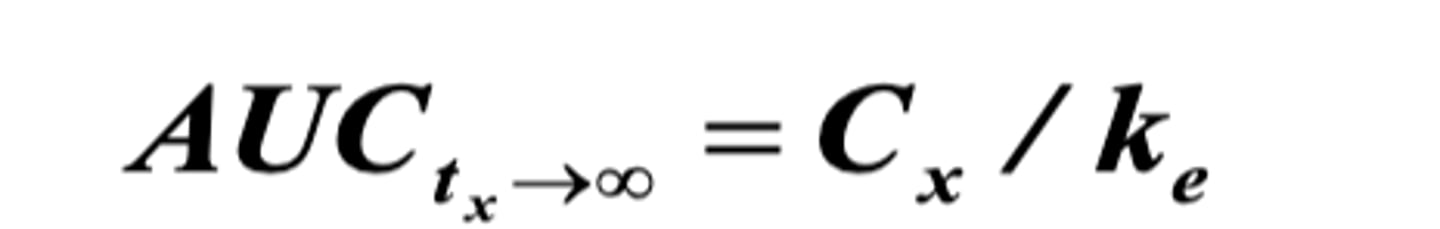
What are assumptions of the one compartmental model?
• Immediate Distribution
• Elimination is first order process
• Body acts like a big tank
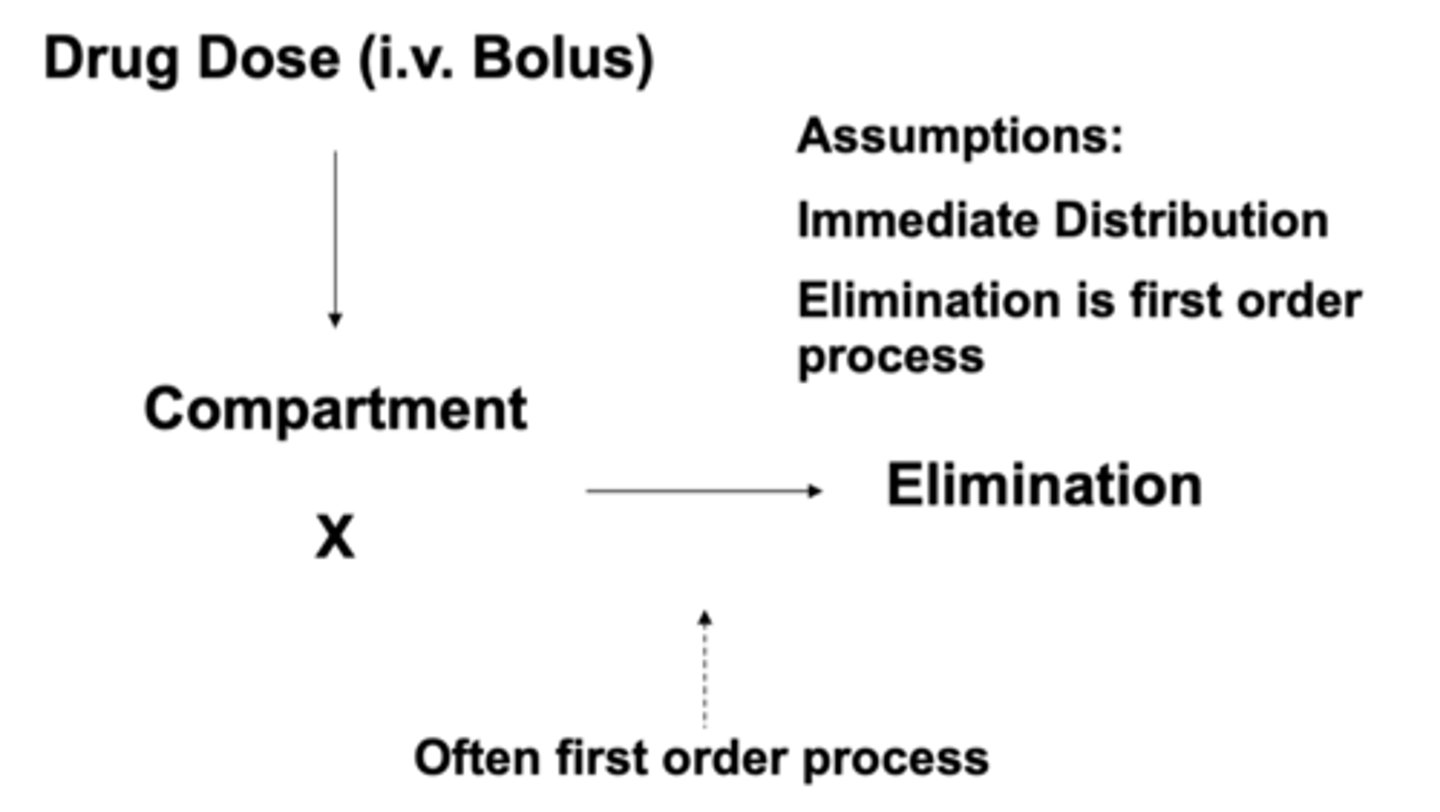
Clinical Relevance
• A number of Drugs (aminoglycosides, cephalosporins) display first order kinetics.
• The larger ke the shorter the half-life
• Ke and t1/2, Vd and CL do not change with dose (they are pt and drug specific properties) if we have first order processes
• Equations can be used to predict drug concentrations