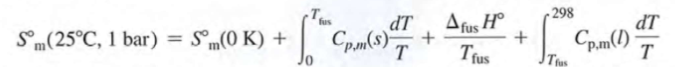Physical Chemistry Exam 1
1/66
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
67 Terms
Operational definition of energy
Energy is the capacity (of a system) to perform work, w
Work is
Force x displacement, amount energy transferred by performing work (kinetic, potential), integral of x1 to x2
force…
causes a body (matter) to accelerate, acceleration is proportional to force (Newton’s 2nd law)
1st law of thermodynamics
energy of the universe is conserved (constant)
Three idealized systems (universe dissected into system and surrounding), distinguished with respect to excheange of energy and matter between system and surroundings
Open: energy and matter are exchanged
Closed: only energy is exchanged
Isolated: no exchange of energy and matter
Sign covention for system
+ work done on system
- work done by system
heat
amount of thermal energy (randomized kinetic and potential energy of atoms and molecules) transfered
Work done on spring with Fex>0 (extension)
Work done on spring (system) positive
Integral
infinite sum of infinitesimally small summands (“limits”) commutative law of addition numbers rules of integration of sums of functions
w units
Nm = kg m²/s² = J (energy)
pressure units
Pa = N/m² = kg/ms²
pressure-volume work of compression
psys vs pex, x positive direction compression, w=(interal)Fexdx = -(integral)pexdV
only determined by external pressure
Irreversible vs reversible expansion
Reversible path can be reversed by an infinitesimal change of the variable that drives the process, implying psys=pex throughout psys = p (expansion stops when pex=p)
Reversible process (path)
process which can be reversed by means of infinitesimal change of a system property without increasing the entropy of the universe, during the entire reversible process the system is in thermodynamic equilibrium with its surroundings
work, w
energy transferred as a result of macroscopic forces, conservative process, pV-work in an electric field
dissipative process: macroscopic forces associated with frictional forces, work transformed into thermal energy
heat, q
energy which transferred as thermal energy (spontaneously from high → low T), defined by temperature change
heat (closed system)
transfer of infinitesimal amount of heat → T increase (dT), ration defines heat capacity (function of T)
C(T):=deltaq/dT → deltaq=C(T)dT
Heat capacities
Molar heat capacity, Cm = C/n (J/K*mol)
Specific heat capacity, c = C/m (J/K*kg)
intensive quantities
molar heat capacity at constant V or p: Cv,m; Cp,m
Cv,m<cp,m b/c pV-expansion work during heating constant p
Closed system first law of thermodynamics
Energy conserved, total energy universe constant, in thermodynamic process energy can be exchanged as q and q between system and surroundings, given a closed system (no exchange of matter) with internal energy U (energy within system) change of U resulting from process: change in U=q+w dU=deltaq+deltaw
(for isolated system change in U=0)
Energy can be transferred to/from system by: work
movement of an object against a force, ie mechanical work/expansion of gases
Energy can be transferred to/from system by: heat
random motion of atoms/molecules, heat transfer occurs when a temperature difference exists
Variables of state (Vos)
describe state of system, depend only on state and not on path used to reach, can be used to define new VoS: H:=U+pV
Total differential exists for Vos (change in Vos per cycle = 0, dU, dp,…) representing an infinitesimal change of the VoS
Extensive VoS
proprotional to the system mass (V, U, C)
Intensive VoS
indepenent of the system mass (T, p, Cm)
Path variables
q and w are not Vos, infinitesimal amount of q and w denoted as small delta q and w
Classical thermodynamics focuses on…
changes of VoS between states (time required not considered)
states define by values of VoS, final - intial
Energy closed transfer
change in U = U2-U1 <0, any path can be chosen, including reversible
Classification of path
1) Restriction of changes VoS
isobaric: constant p, change in p=0
isochoric: constant v, change in v=0
isothermal: constant T, change in T=0
2) restriction of a path variable
adiabatic: delta q = 0 (no heat exchange)
3) reversible vs. irreversible
entropy of universe doesn’t increase with reversible
4) cyclic: initial and final state identical
all change in VoS=0
Equations of State (EoS) relate VoS
EoS=constraints between VoS, dictate combinations of values set for set of all VoS exist
most frequently used type of EoS, p, V, T
EoS of solids and liquids
Compressibility is low, first approx: V constant, EoS is V(p,T) = V knot improved EoS from linear dependence on T and p → Vm=Vknotm[1+alpha(T-Tknot)][1-ki(p-pknot)]
alpha=coefficient volume expansion
ki=isothermal compressiblity
Eos Ideal gas
no interactions between gas molecules, pV=nRT
EoS ideal gas mixtures
partial pressure pi of component i, pi=niRT/B and p=sumipi, mol fraction xi=ni/sumi(ni) pi=xip
Eos molar heat capacity of monoatomic ideal gas
Cv=3/2R (constant V)
Cp=5/2R (constant p)
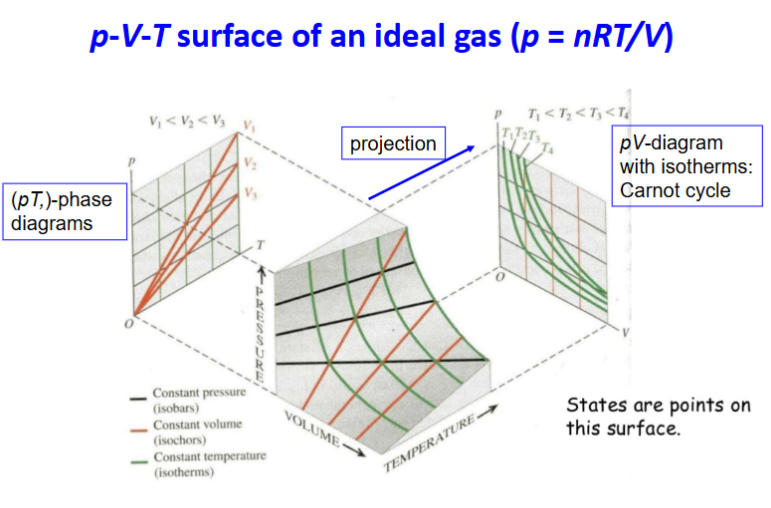
pVT surface ideal gas (diagram)
Equation of state: ‘real gas’
Deviate from ideal because of intermolecular force, volume occupied by gas molecules
Result in empirical VdW equation as approximate EoS: p=(nRT)/(V-nb)-(n²a)/V² → (p+(n²a)/V²)(V-nb)=nRT
a=(parameter model attractive forces between molecules), when a=0, b-(parameter to model volume correction bc intrinsic volume molecules) b=0 (p→0) = EoS ideal gas
new VoS, Enthalpy, H derivation
reversible isobaric process restricted to pV work, change enthalpy=amt reversible heat exchanged with surrounding

H, heat capacities for ideal gas, derivations
H=U+pV=U+nRT —> dH=dpq, rev
result in Cp=Cv+nR

Pure substances → phase transitions → chemical reactions, dependence U and H of pure substance on pVT
EoEoS related VoS p, V, T, for pure substances, compressiblity low:
Heat and cooling liquid water constant at atmospheric pressure, then Cp independent of T, and with molar capacity Cp,m, pV work can be neglected
deltaH approx delta U, Cp approx Cv

Pure substances (gases), pV work for processes with change V, but…
U of an ideal gas depends only on T (not p,V), heat capacities define T dependence U and H
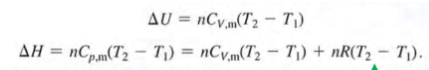
dependence U and H of pure substance
1) Reversible (p=pex) constant pressure expansion work wp, rev → wp=-p(V2-V1)=-nR(T2-T1)
2) Reversible isothermal expansion work

change U = change H = 0 for isotheran processes (pV=nRT=constant)
H=U+nRT for 1st law closed, change U=0 so qt,rev=-wt,rev → all heat transferred to system converted to work
Phase transitions of pure substance
phi, transition a→b: delta(phi)H=Hb-Ha (=qp only if rev pV work wp=-pdeltaphi at constant p
so then deltaphiU=deltaphiH-pdeltaphiV at const p
*HEAT CAPACITY INFINITY DURING PHASE TRANSITION
all latent heat used to drive transition

To obstain deltaphiU and deltaphiH at different T2, cucle constructed given U and H are VoS
Change of U involving gas, constant p
Difference between U and H due to pV work against constant p
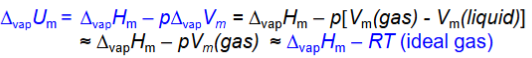
Hm-Hknotm= integral drom Tinitial to T (Cp,m)waterdT
Hm-Hmo=Cp,m(T-To)
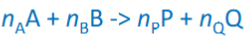
Heat effects chemical reactions
only PV work: deltarU=qv deltarH=qp → deltarH=deltarU+deltar(pV)
deltar(pV)=deltar(nRT)
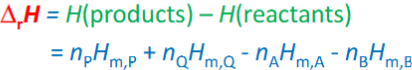
deltarU at constant V,T
deltarU<0, deltarH<0 at constant p, T: exothermic
deltarU>0, deltarH>0 at constant p, T: endothermic
deltarH is VoS, so can be calculated where qp not measured
T dependence deltarH
Known at T1, needed T2,
sum of the equations = overall equations
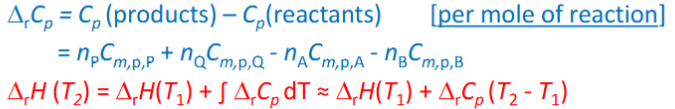
Standard enthalpies of formation Hknot
only differences of enthalpies of rxn of interest
absolute not determined, use molar standrd enthalpes from elements in their most stable allotrope in std state
for elements in most stable Ho=0J/mol
for rxn at 25C: deltarHo=npHop+nqHoq-nAHoA -nBHoB (no enthalpy changes arising from reactant mixing)
Estimation of reaction enthalpu differences from bond dissociation energies, quantum chemical calculations
Entropy
VoS, “S” ds=deltaqrev/T and dS>qirrev/T
change in s = reversible exchange amt of heat/temperature, only reversible path allows to measure, “carnot cycle”
s=(kB)(lnW), absolute values S require stat tehrmodynamics and 3rd law
proportional to logarithm microstates W; kB represents proportionality constant, R=kBNA
Carnot cycle
Idealized/cyclic heat enging, heat to work consisting of 4 reversible steps of ideal gas in closed system without friction
1) Isothermal expansion +q1 → -w1 at Thot
2) Adiabtatic expansion sys cools due to Tcold at -w2
3) Isothermal compression: +w3 → -q3 at Tcold
4) Adiabatic compression sys heats due to Thot at +w4
steps summed to get -wcycle for total amount work in 1 cycle
Discovery of S with the carnot cycle, qrev1/Thot +qrev3/Tcold=0
1) efficiency of Carnot cycle
as q1/thot + q3/tcold = 0 → q1/thot = -q3/tcold
effiency= 1 - tcold/thot = (thot-tcold)/thot = deltaT/Thot
efficency solely of ratio of temp in heat reservoir
2) carnot cycle can be run in reverse as a heat pump
3) proof by contradiction/indirect proof: coupling carnot heat engine and heat pump with lower efficiency results in construction of “impossible machine” heat flow spontaneously low → high, violates 2nd law
Reversing heat enging → heat pump (wcycle > 0)
COP heat pump: heat or cooling: wcycle>0 COPheat=COPcool+1

Isothermal
Heat exchanged reversibly at constant temperature
cyclic revrsible path: TdS=deltaqrev
pV vs TS diagram: Carnot cycle
deltaU cycle=0, -wcycle,rev=qcycle,rev
deltaqrev=TdS=-pdV → cyclic path integral represents any conceivable reversible cyclic path of closed system
p+V and T+S pairs of conjugate variables
qin,rev exchanged isothermally, deltaS scales with 1/T
deltaShot=qin,rev/Thot<deltaScold=qin,rev/Tcold
2nd Law thermodynamics
entropy system and surroundings = entropy universe = - if isolated, = not ratop if reversible
delta s for expansion ideal gas: reversible vs irrev: 1 mole irreversible against pex=0, no pV work, qirrev=0, but s(surr)=0 and s(sus)>0 (value of s(sys) unknown)
Reversible paths enable measurement of S by…
measuring amount of reversibly exchanged heat, reversiblity implies pex=p throughout expansion
wrev=-RTln(V2/V1)=-RTln2 → qrev=-wrev=RTln2 → s(sys)=qrev/T=Rln2=-s(surr) → suniv=0
s(sys)=integral final-initial delta qrev/T > integral final-initial delta qirrev/T
Reversible process…
process can be reversed by means of infinitesimal change of system property without increasing the entropy of the universe, system in thermodynamic equilibrium with surroundings
given p and T deltarS=?
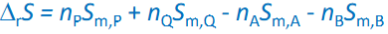
Sm,x
Molar entropy of compound x, reference 1 bar usually 25C
VoS, deltarS not depend on reaction path, cycles can be constructed
absolute entropies can be gotten with 3rd law
3rd law thermodynamics
Entropy pure perfect crystal at 0K=0, only microstate for macrostate of A
S=kBlnW=kVln(1)=0
Temperature dependence S
dS=deltaqrev/T, deltaqrev=CpdT at constant p
dSp/dT=Cp/T
isobaric: constant p = Cpln(T2/T1)
isochoric: constant V = Cvln(T2/T1)
Units S and C: J/K
C always positive → increase T = increase S
Entropy change 1 mol ideal gas heated at constant p
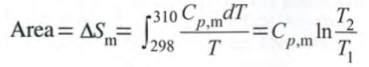
Entropy changes associated with phase transitions
determination of absolute 3rd law entropies
driben reversibly at constant ptrssure associated with deltaphiH=qrev → deltaphiS=deltaphiH/Tphi
Third law entropy Som liquid at 24C, Cp=infinity
For gas: 0→T: solid Tm→Tb: liquid Tb→infinity gas