IMED1003 - Inborn Errors of Metabolism 1 and 2 (L27 and 28)
1/33
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No study sessions yet.
34 Terms
IEM
- Sir Archibald Garrod pioneered this field
- Inborn errors not just enzyme defects. Might be transport proteins, receptors or structural components
- Mechanisms of disease: accumulation of a toxin; energy deficiency; deficient production of essential metabolite/srtuctural component
Inherited Disorders
- All feature a genetic defect (often a result of single base substitution or deletion), reduced synthesis or absence of particular protein or change in primary structure
- More than 4000 disorders involving single genes
- Most inherited disorders are autosomal recessive, heterozygotes usually normal. E.g sickle cell anaemia - haemoglobin. some autosomal dominant - hypercholesterolaemia
- many diseases have defective enzyme that cause metabolic disorder
Inherited disorders may be detected at different stages of life:
- heterozygote carriers, often found during screening family members of affected indivdual
- prenatal diagnosis e.g cystic fibrosis
- neonatal screening. e.g phenylketonuria
- neonate developing symptoms in first few weeks
- adult onset. e.g huntingtons disease
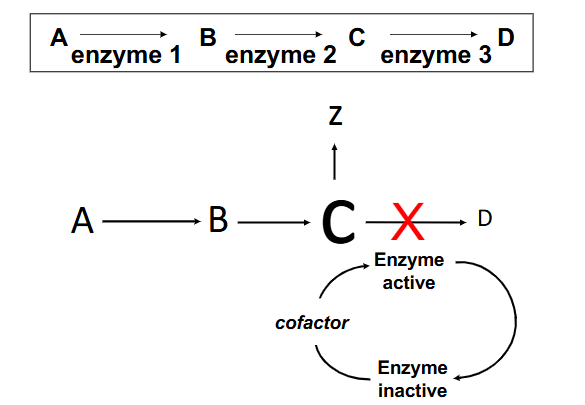
Enzyme Deficiencies
- some enzymes require cofactors for activity, and often require modification before use
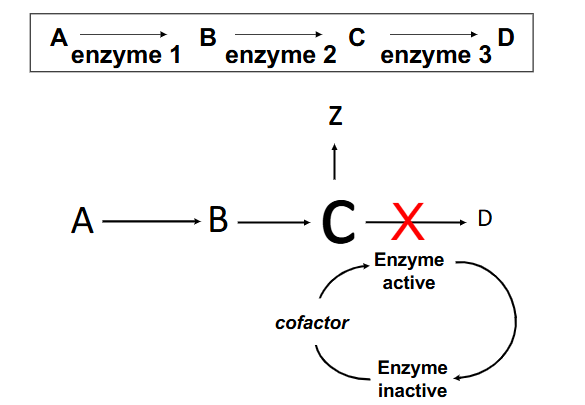
Classical inborn errors of metabolism ("metabolic disorders")
- involve a missing or defective enzyme, causing block in metabolic pathway and often production of toxic metabolites
.
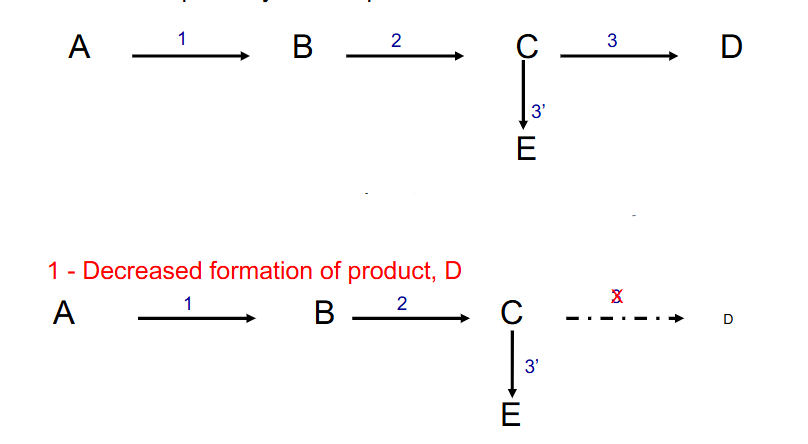
- metabolic imbalance secondary to enzyme deficiency. Three distinct sequelae of a lack of enzyme 3
1. decreased formation of product, D
DIAGRAM ON SLIDE 7
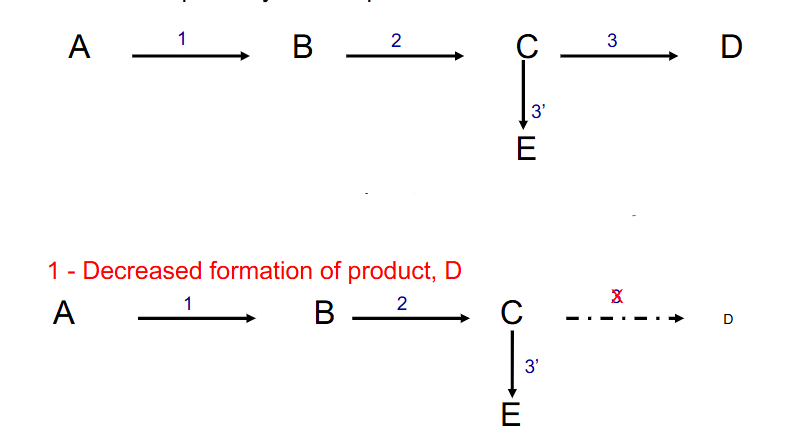
1. decreased formation of product, D
DIAGRAM ON SLIDE 7
DIAGRAM ON SLIDE 7
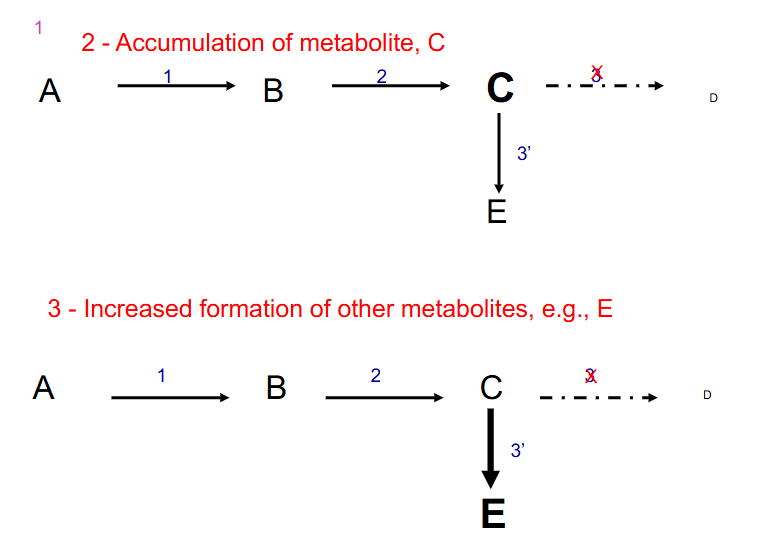
2. Accumulation of metabolite, C
DIAGRAM ON SLIDE 8
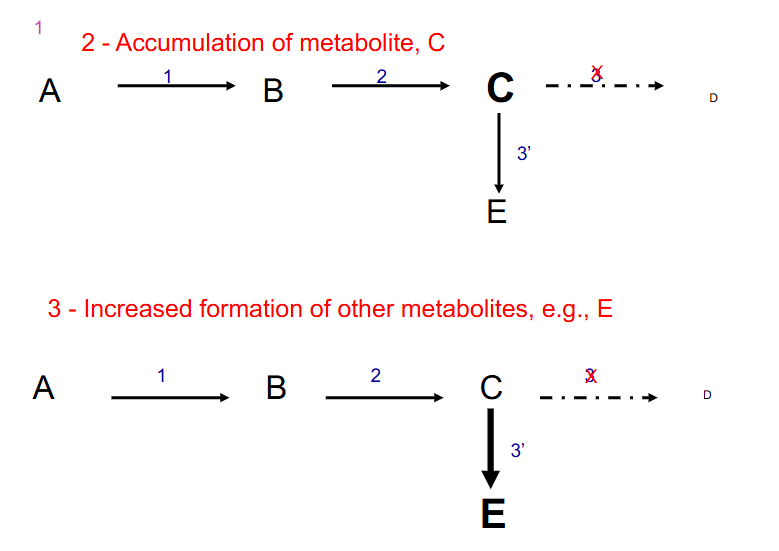
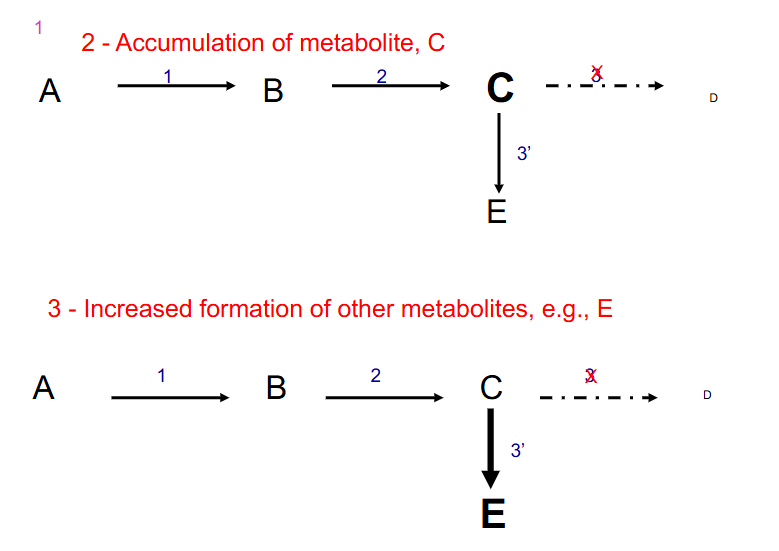
3. Increased formation of other metabolites, e.g E
- other things that can happen if enzyme 3 is defective: if there is feedback inhibition it is interuptted
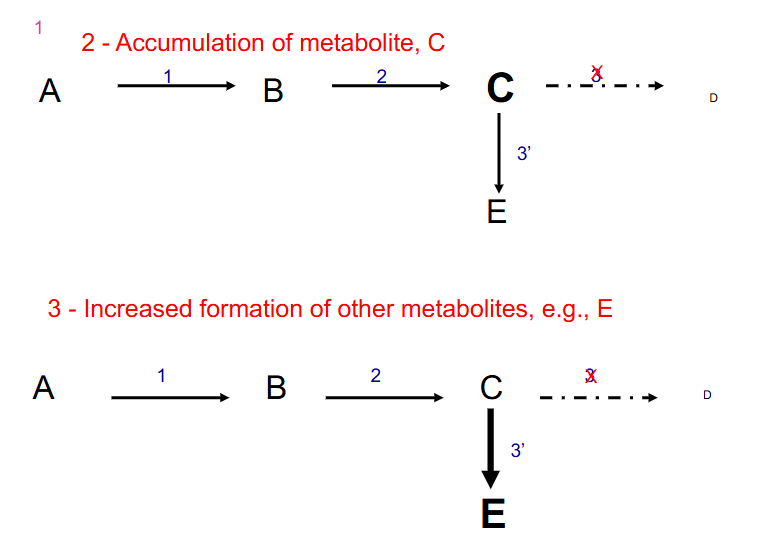
Inherited disorders diagnosed in first weeks of life often involve central metabolic pathways, AA and CH2O metabolism and storage, urea cycle ect.
- Frequently display non-specific symptoms: vomiting, lethargy, failure to thrive, feeding difficulties, respiratory distress, hypotonia, seizures, hypothermia (especially neonates)
- other telling signs: ocular findings, hepatosplenomegaly, cardiomyopathy, abnormal odour, colour, unusual skin and/or hair
Recognising Metabolic Disorders
- IEM are mainly autosomal recessive
- Some are X-linked recessive
- few autosomal dominant e.g hyperlipidaemia
- DNA analysis useful
.
Test for: glucose, electrolytes, gas, ketones, blood urea, nitrogen, creatinine, lactate, ammonia, bilirubin, amino acids, organic acids, and more
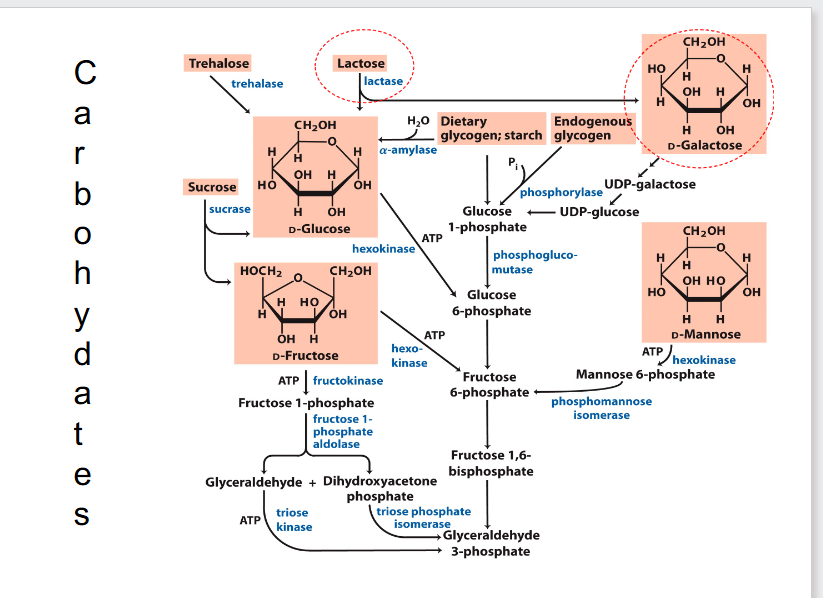
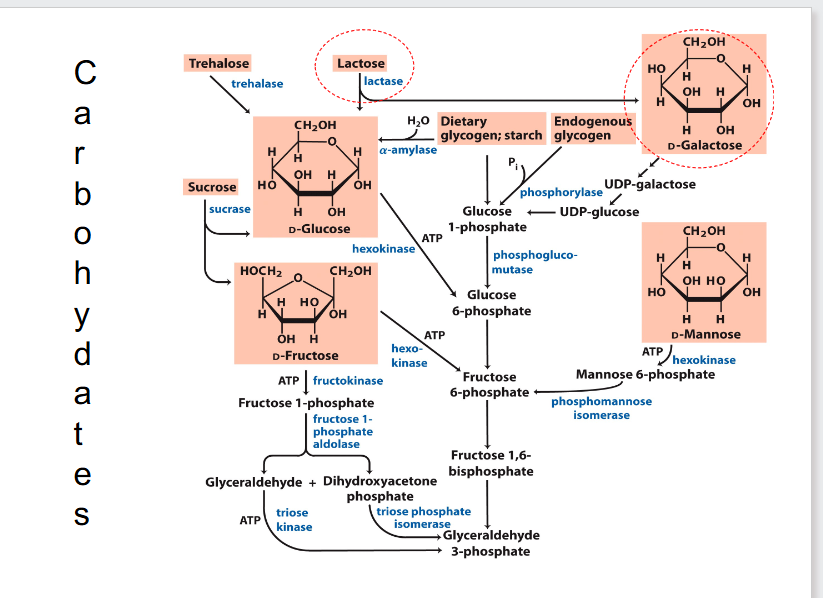
Carbohydrates
- most carbohydrates fit into the glycolytic pathway
- glycosidic bond is important
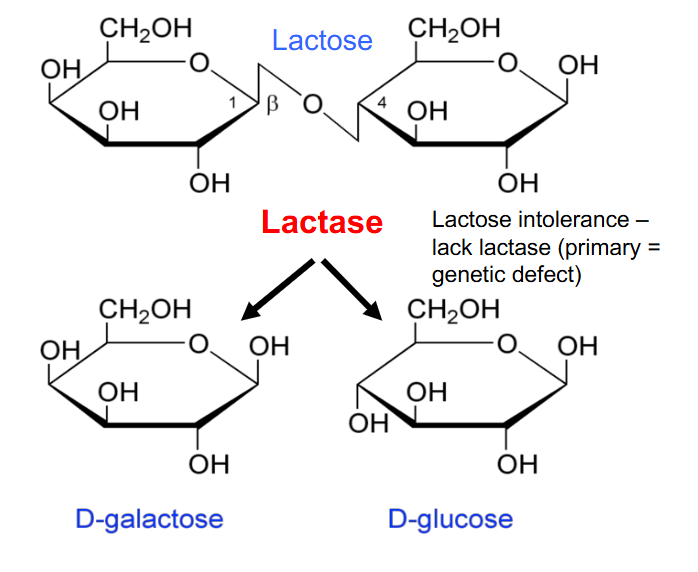
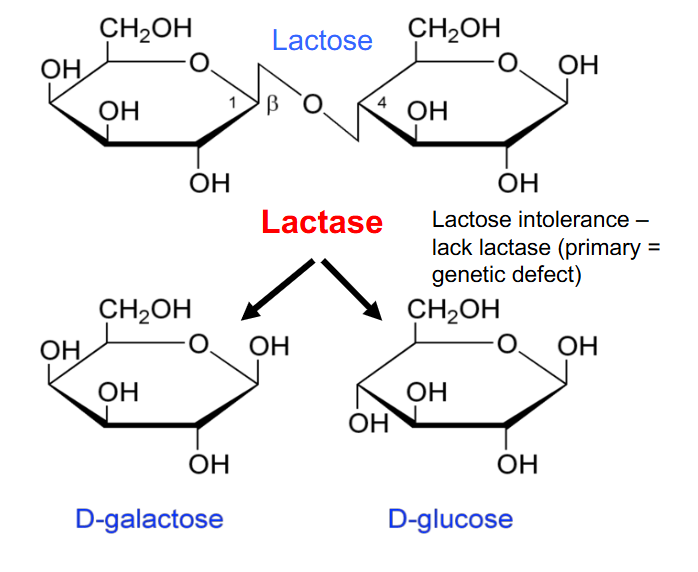
Lactose Intolerance
- lack lactase (primary genetic defect)
- primary genetic defect: enzyme they produce doesn't fold properly due to a mutation or isnt produced in adequate amounts due to some sort of mutation
- these people would have a consequence of not being able to break the glycosidic bond, which means lactose is accumulated
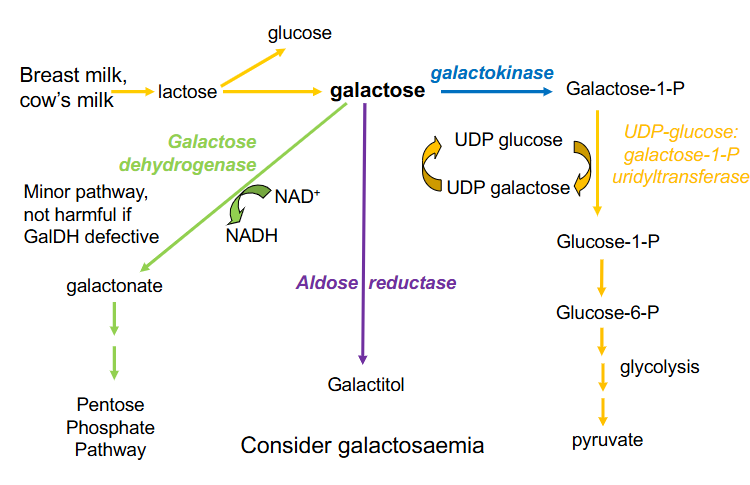
Three ways to metabolise galactose
1. Galactose Dehydrogenase:
- minor pathway, not harmful if GalDH defective
- oxidises galactose to galactonate (requires NAD+ to be reduced to NADH and galactonate can enter PPP
.
2. Aldose reductase
- forms galactitol
- when it accumulates it causes cataracts
.
3. Galactokinase:
- forms galactose-1-Phosphate
- that can enter glycolytic pathway if we can process it through to glucose-1-phosphate and then glucose-6-phosphate
- UDP-glucose: galactose-1-P uridyltransferase
- this enzyme transfers from a UDP glucose onto galactose-1-P, the UDP, the uridyl thats attached to UDP glucose
- we hence produce glucose-1-phosphate and we add galactose to the UDP forming UDP galactose
- this needs to be recycled so we can continue the reaction
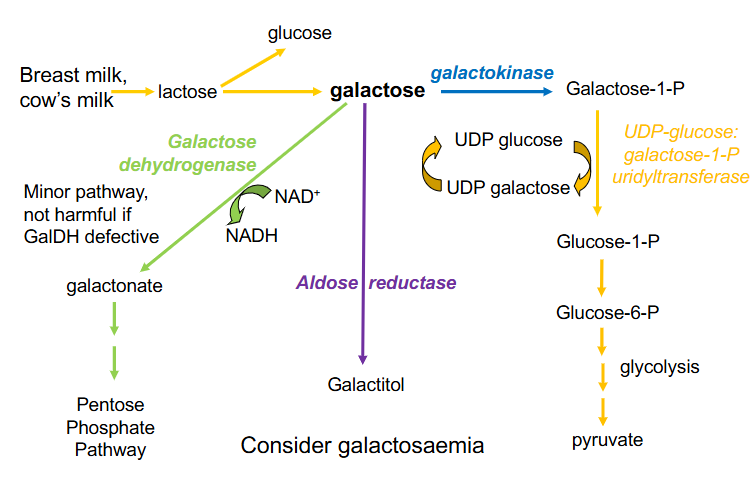
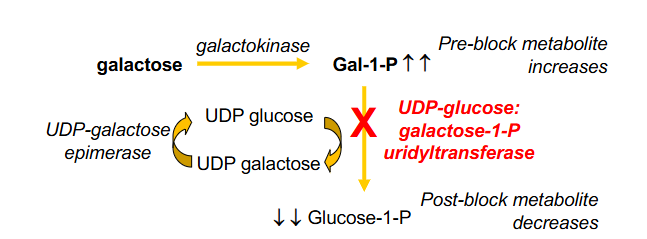
Classical Galactosaemia
- Inborn Error Carbohydrate metabolism, lack of hepatic enzyme
- UDP-glucose: galactose 1-P uridyltransferase
- Galactose-1-P accumulation leads to intellectual disability, jaundice, hepatomegaly, cirrhosis, infantile cataracts, hepatic failure and death
- treatment: galactose-free diet, ophthamology and developmental follow up
.
- Consequences: pre-block metabolite increases, post-block metabolite decreases
- if Galactose-1-P builds up it can feed back to galactose which can accumulate
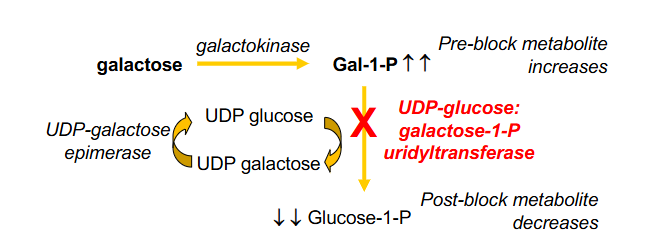

Galactokinase Deficiency Galactosaemia
- Aldose reductase is physiologically unimportant, but excess galactose
- with galactokinase stopped the galactose takes an alternative pathway through aldose reductase, which causes build up of galactitol, which causes galactosuria and galactosaemia. Elevated galactitol can cause cataracts
.
Consequences:
- increase in pre-block metabolite and decrease in post-block metabolite
.
- classical galactosaemia is miuch more com,mon then galactokinase deficiency

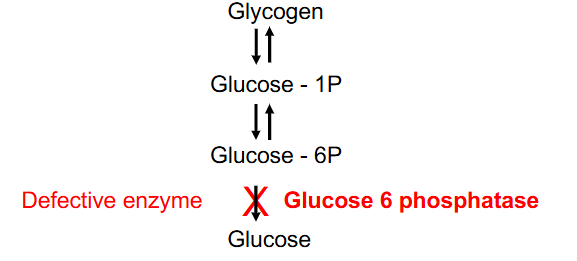
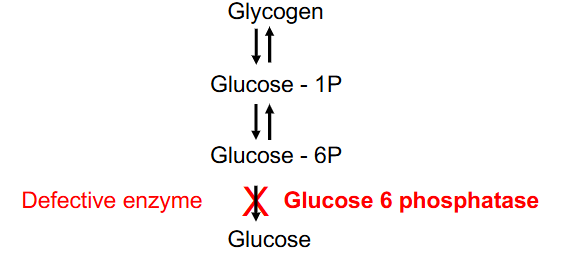
Type Ia Glycogen-Storage Disease
- most common glycogen storage disorder, 25% of total, causes excessive glycogen and fat accumulation (liver, kidney)
- initial symptoms = neonatal hypoglycaemia
- other symptoms include: hepatomegaly, tremors, irritability, seizures, apnoea, coma
- Glucose-6P converted to glucose (glucose 6 phosphotase defective)
.
- glucose 6 phosphotase - liver, kidney
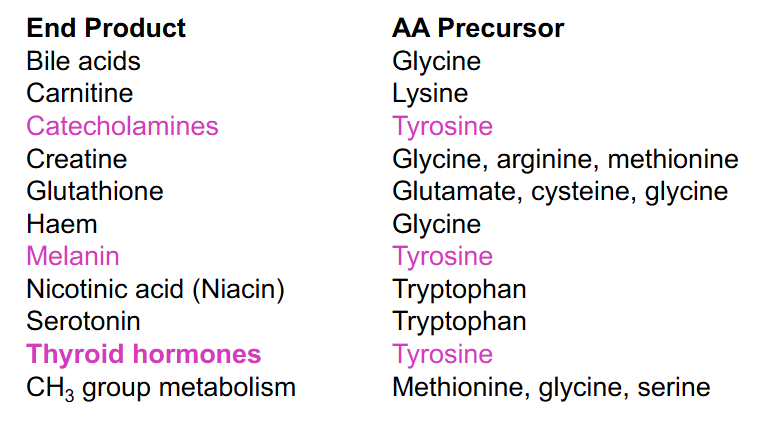
Amino Acid as Precursors
DIAGRAM ON SLIDE 24
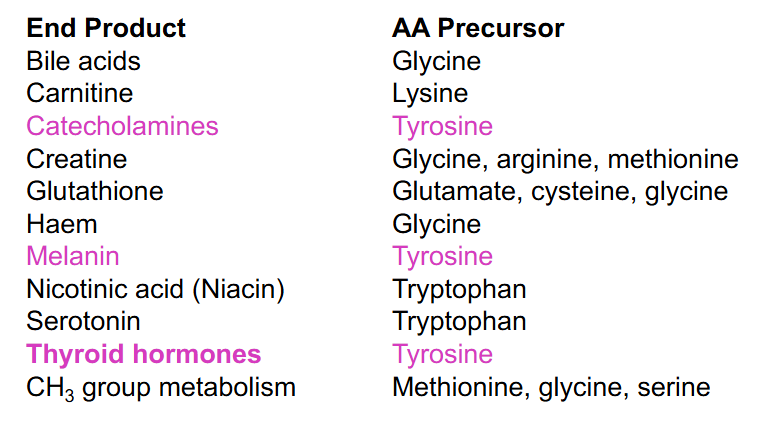
Tyrosine Related Disorders (NO LEARNING OUTCOME)
DIAGRAM ON SLIDE 25
- it is ketogenic and glucogenic (both aromatic aa are ketogenic and glucogenic)
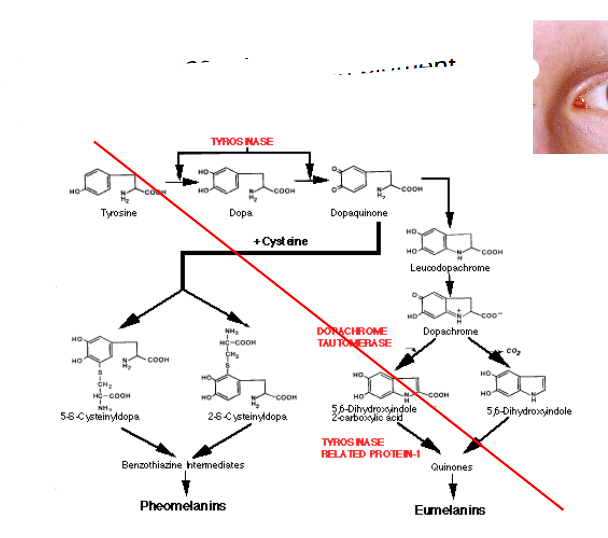
Albinism
- absence of melanin pigment
- tyrosine is a melanin precursor
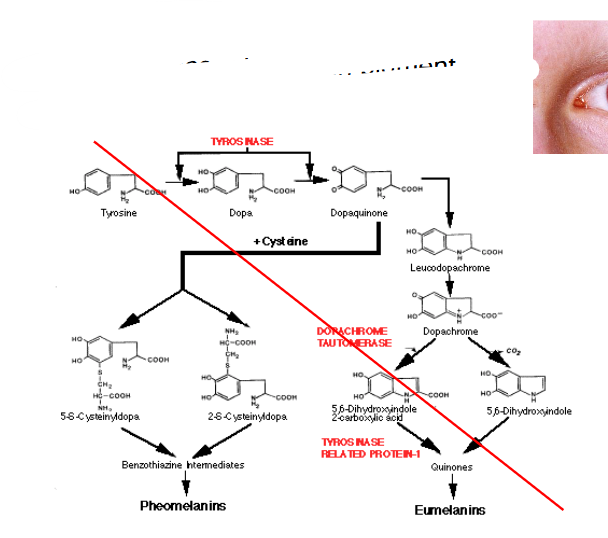
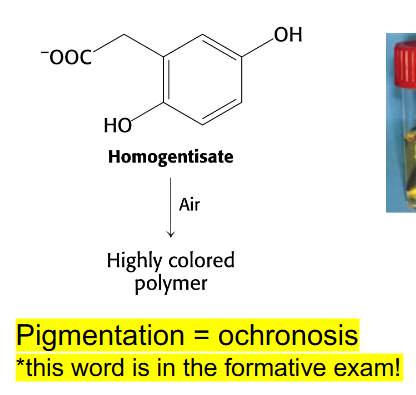
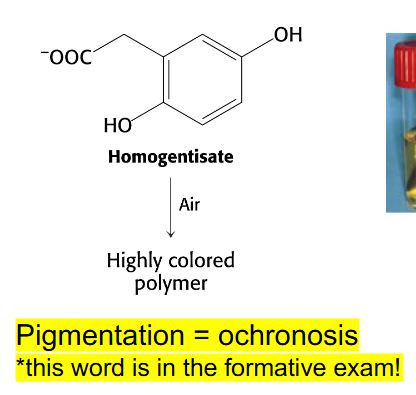
Alkaptonuria
- absence of homogentisate oxidase activity
- homogentisate accumulates - can damage cartilage (osteoarthritis), heart valves, precipitate in kidney (kidney stones), usually after age 30
- pigmentation = ochronosis (this word is in the formative exam)
- any enzyme in the pathway can be affected
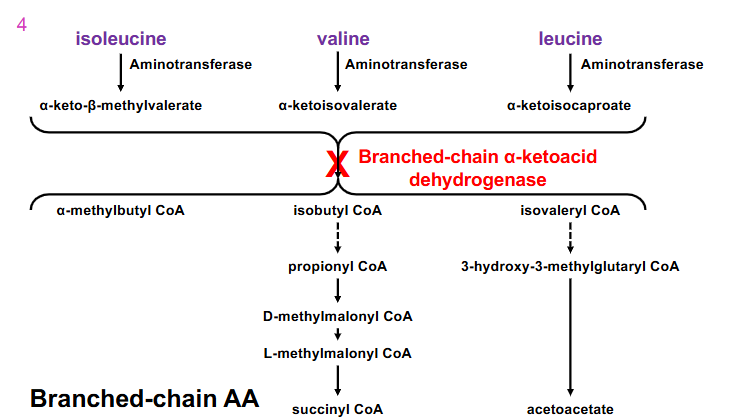
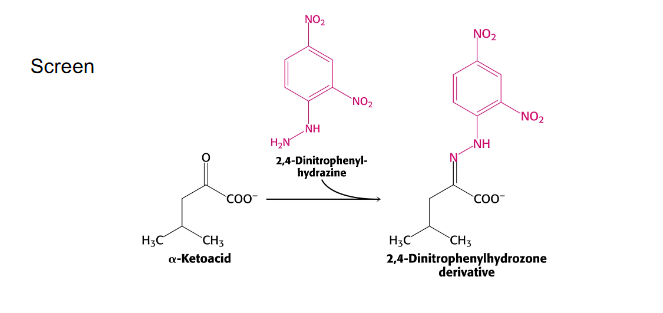
Maple Syrup Urine Disease
- From early infancy, symptoms include poor feeding, vomiting, dehydration, lethargy, hypotonia, seizures, hypoglycaemia, ketoacidosis, pancreatitis, coma and neurological decline
- branched-chain alpha-ketoacid dehydrogenase is the enzymes thats defective in this disease
.
- liver does not have the enzymes to process branched chain amino acids and the liver cannot use ketone bodies as fuel source
-
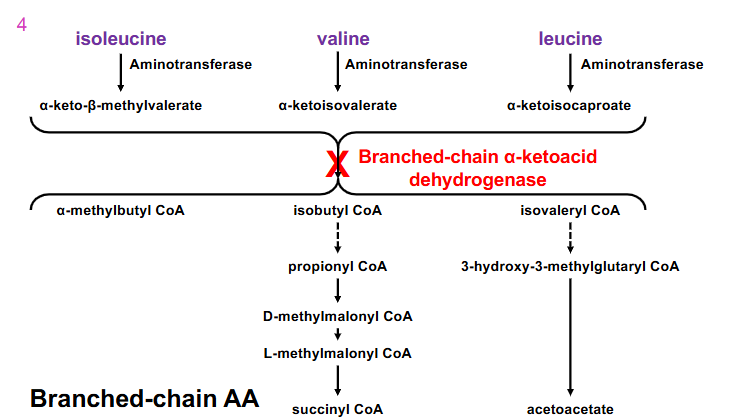
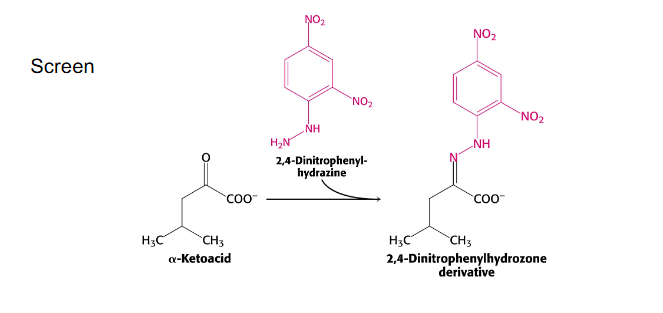
Maple Syrup urine Disease other info
- lack of branch-chain dehydrogenase activity
- leads to elevation of branched-chain amino acids and alpha-keto acids in urine. Urine smells like maple syrup
- neurological symptoms - unless very early intervention via restricting dietary intake of valine, isoleucine, leucine
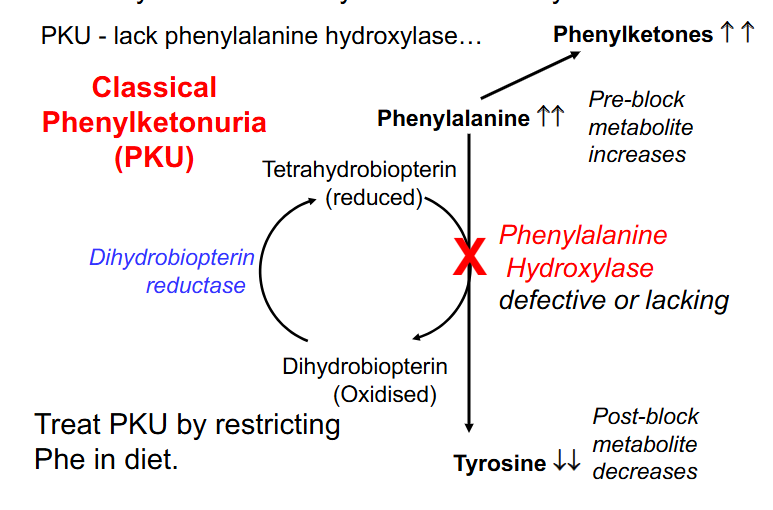
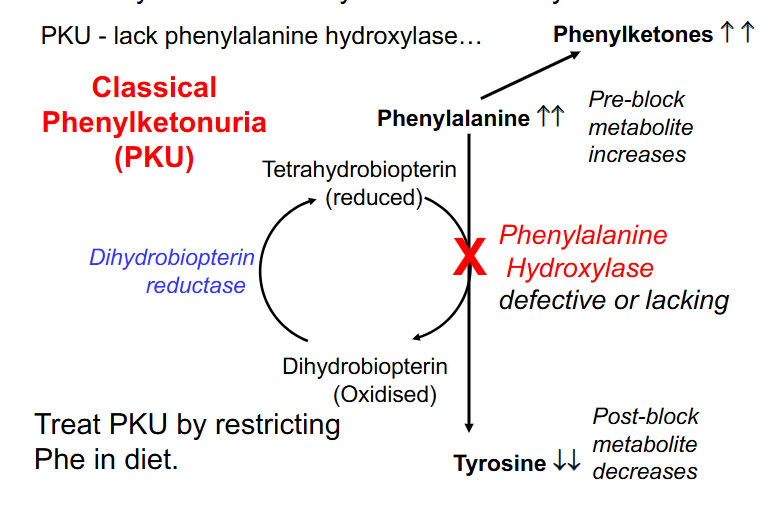
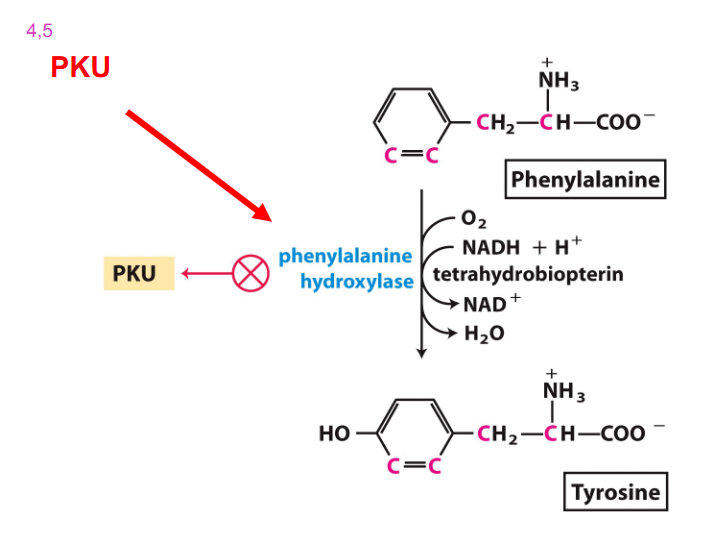
Phenylalanine Usually converted to Tyrosine
- PKU - lack phenylalanine hydroxylase
- Classical Phenylketonuria (PKU)
- treat PKU by restricting Phe in diet
- Consequences: increase in preblock metabolites (Phenylalanine) and decrease in post block metabolites (Tyrosine) due to the defective enzyme
- phenylalanine cant be excreted and so is excreted to phenylketones
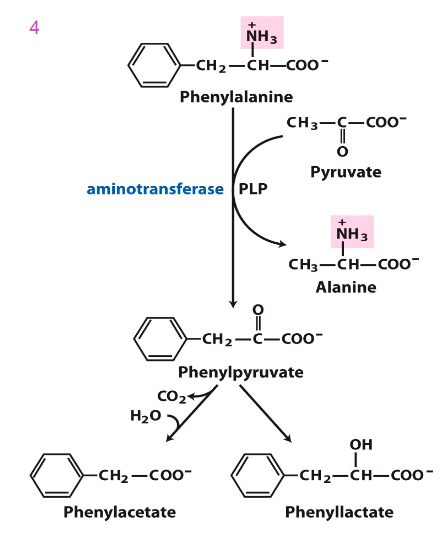
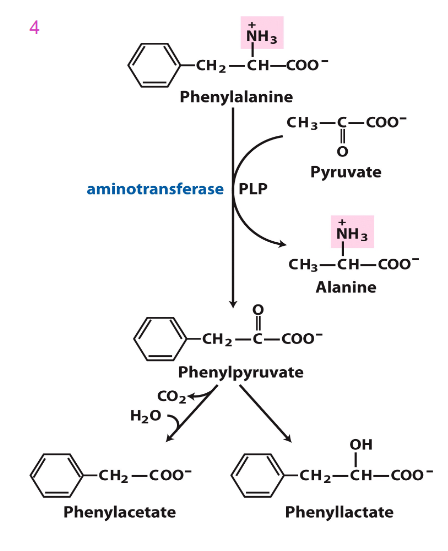
Accumulated Phe is converted into
phenylpyruvate, then phenylacetate and phenyllactate, which are phenylketone derivatives
- these are excreted in urine, hence the name phenylketonuria
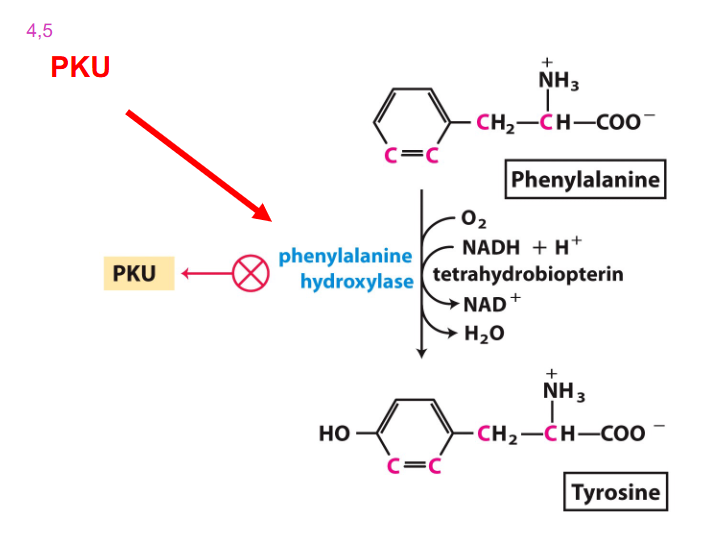
PKU Diagram at the main recation
DIAGRAM ON SLIDE 32
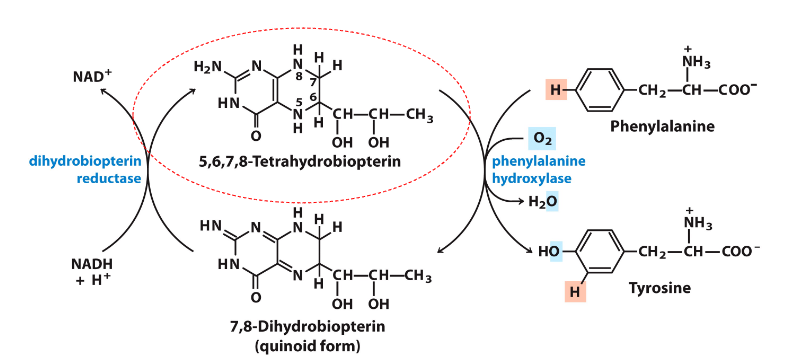
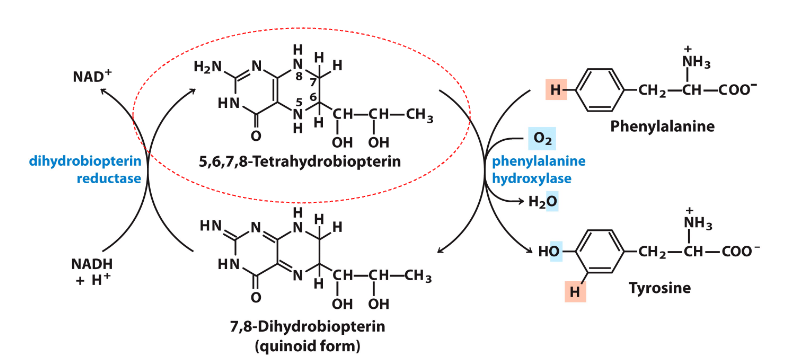
Phenylalanine Hydroxylase Reaction
DIAGRAM ON SLIDE 33
- need to be able to recognise and draw phenylalanine and tyrosine
- make sure you know this reaction well
- should be able to tell the enzyme that converts phenylalanine to tyrosine (it is hydroxylating the phenylalanine, hence its oxidising)
- thats why tetrahydrobiopterin is being reduced to dihydrobiopterin
- NADH is converted to NAD+
Disorders of Carnitine Metabolism
- Carnitine transports long chain FA into mitochondria
- Carnitine deficiency - primary or secondary
.
PRIMARY:
- caused by abnormal transport of carnitine into cells = carnitine uptake disorder, AKA systemic carnitine deficiency
- Variably associated with hepatomegaly, liver disease, hypertrophic cardiomyopathy and potential arrhythmia
- autosomal recessive
Urea Cycle Disorders
- Appear unaffected at birth
- within days - vomiting, respiratory distress, coma
- symptoms mimic other illnesses
- untreated: results in death
.
non-functioning enzyme results in:
- infant: progressive loss of motor and cognitive skills
- pre-school: non-responsive state
- adolesence: death
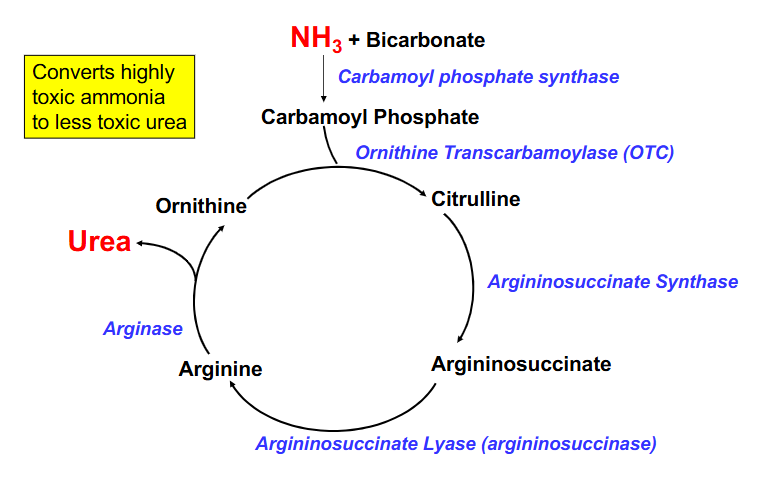
The Urea Cycle
DIAGRAM ON SLIDE 36
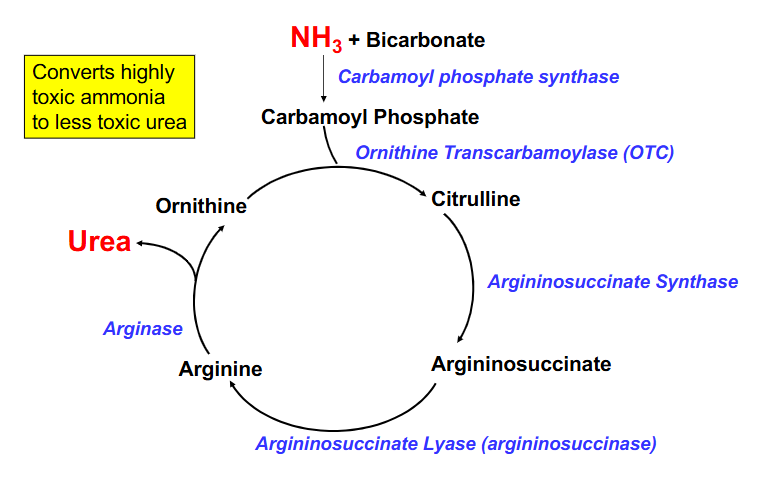
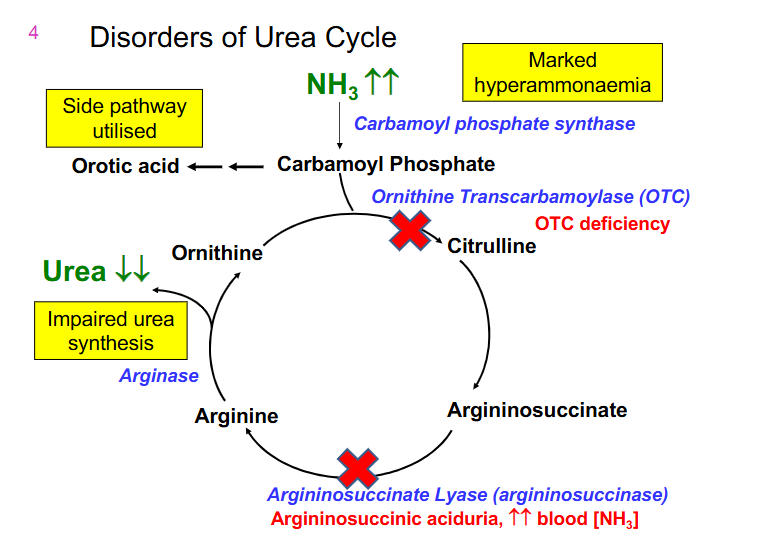
Disorders of Urea Cycle DAIGRAM
- Ornithine Transcarbamoylase (OTC) is the most common deficiency
- next most common is argininosuccinaste lyase (argininosuccinase) (accumulates argininosuccinate)
- results in increased NH3 and decreased urea output
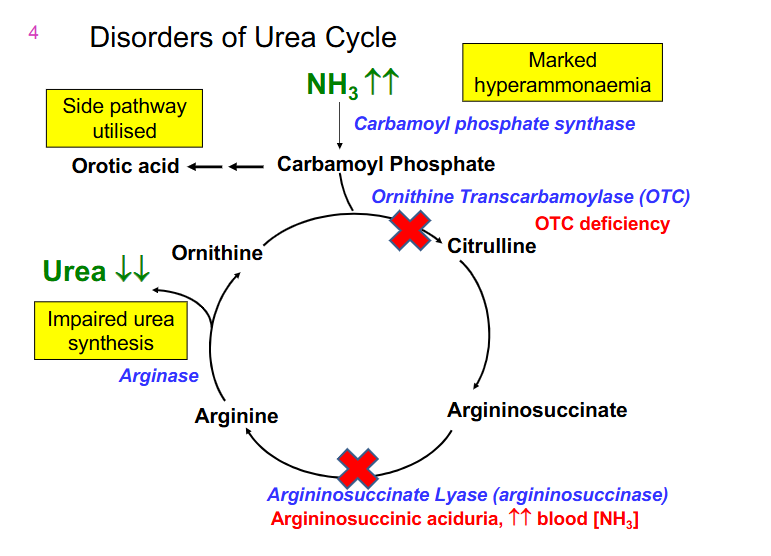
OTC deficiency
- X-linked
- Males (and females homozygotes) severely affected
- females heterozygotes are variably affected due to random X inactivation
.
- 12-72 hours
- lethargy, poor feeding, abnormal respiration, vomiting, seizures, decreasing conscious level
- if untreated - death
.
- management: early intervention. Restrict dietary intake of proteins, liver transplant, medication (benzoate/phenylacetate) can assist by removing excess ammonia (bypassing urea cycle)
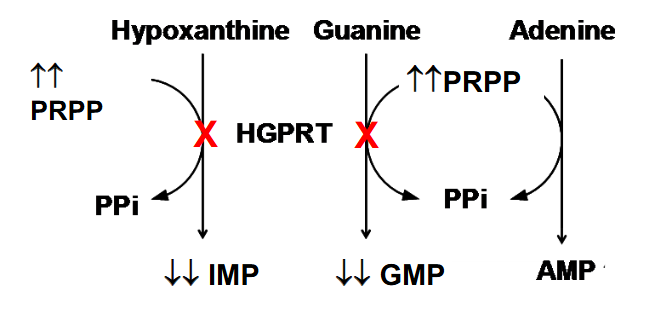
Disorders of Nucleotide Metabolism
Lesch-Nyhan Syndrome: X-linked, lack HGPRT
- impaired kidney function, acute gouty arthritis, self-mutilating behaviour such as lip and finger biting, and or head banging, involuntary muscle movements, neurological impairment
.
- results in elevated levles of PRPP if we cant salvage our purine and pyrimidne bases
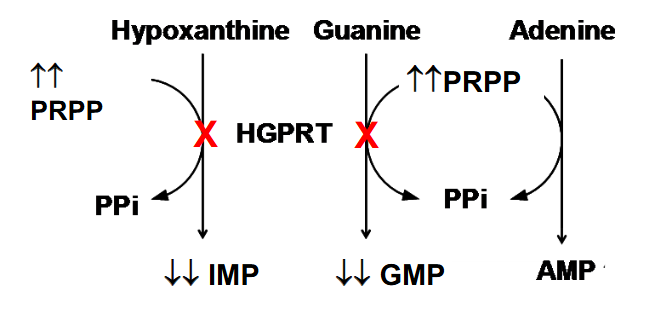
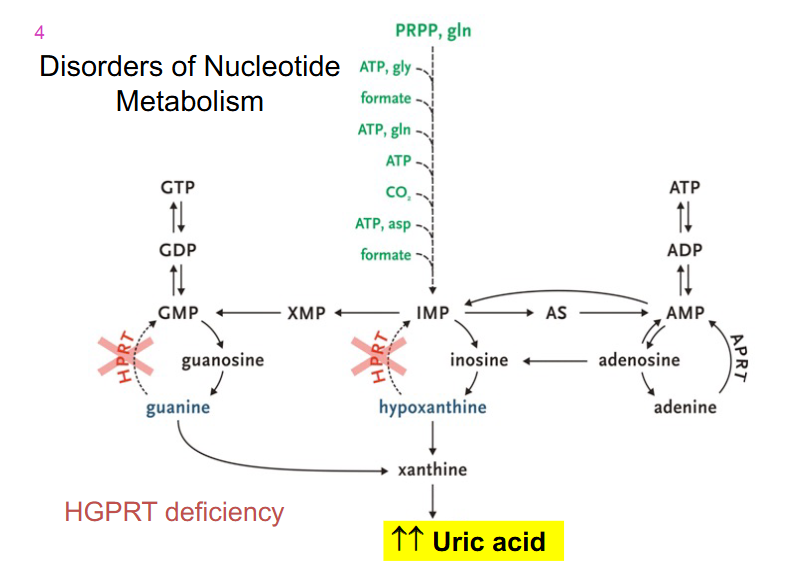
Disorders of Nucleotide metabolism
- HGPRT deficiency
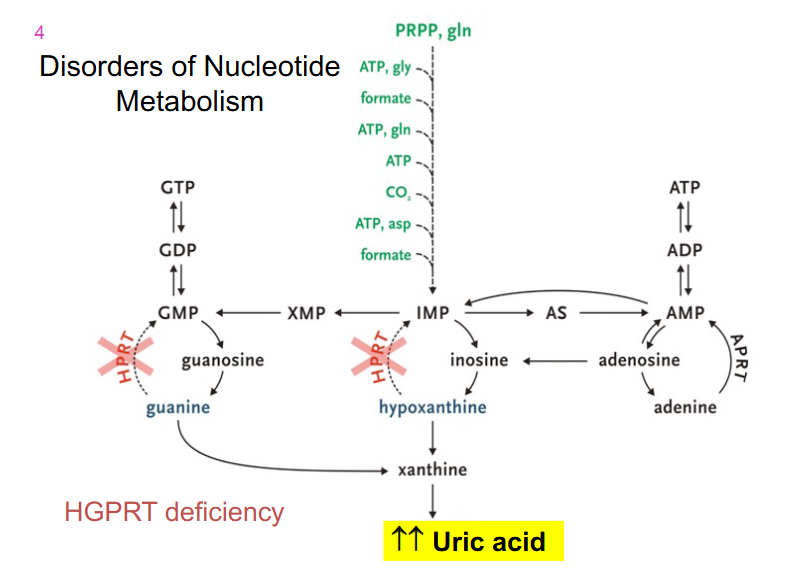

Summary
DIAGRAM ON SLIDE 41 and 42
