Immunology Unit 4- Immunodeficiencies
Primary Immunodeficiency Disorders are caused by what?
Defects in components of Cellular Immunity or Humoral Immunity.
Cellular Immunity includes what two things?
T Cells and Phagocytes.
1/38
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
39 Terms
Primary Immunodeficiency Disorders are caused by what?
Defects in components of Cellular Immunity or Humoral Immunity.
Cellular Immunity includes what two things?
T Cells and Phagocytes.
Humoral Immunity includes what two things?
Complement and Antibodies (B Cells).
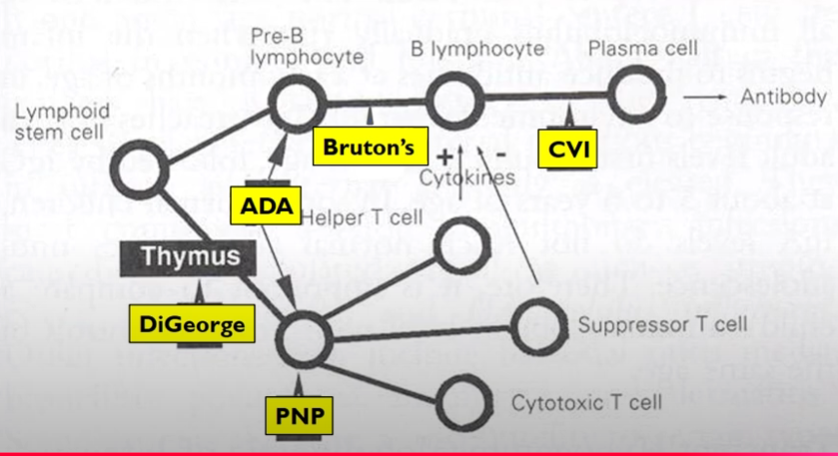
Agammaglobulinemia:
Primary deficiency of B cells. Humoral Immunity defect.
Cause pyogenic bacterial infections- especially in upper and lower RT. Viral infections ail them about the same as normal individual.
May be due to genetic defects in B cell maturation or defective T and B cell interactions.
True or false. IgM, IgG and IgA levels increase with age.
True.
Transient Hypogammaglobulinemia of Infancy:
Unknown cause.
Prolonged low Ig levels.
Problems with recurring infections.
Ig levels usually normalize by 9 to 15 months.
Mechanism of disease is not known.
Have normal B cells. May be due to delayed maturation of T helper cells.
X-Linked Bruton’s Agammaglobulinemia:
Linked to chromosome X.
Seen almost exclusively in males.
Development blocked at pro-B-cell stage to the pre-B-cell stage- lack all classes of Ig.
T cells are normal.
Cause: Deficiency of enzyme- Bruton’s tyrosine kinase.
Results in failure of Vh gene rearrangement.
Manifestations: Recurrent otitis and areas of cellulitis in the diaper area. Pseudomonas aeruginosa and Staphylococcus aureus isolated from the skin lesions.
How can you differentiate X-Linked Bruton’s not Transient Hypogammaglobulinemia?
Persistence beyond 2 years of age would tell us it’s X-Linked Bruton’s. As well as an abnormal histology of lymphoid tissue.
What is the treatment for X-Linked Bruton’s?
Regular injections of Ig and antibiotics as needed.
IgA Deficiency:
Most common primary immunodeficiency.
Most patients are asymptomatic.
Anti-IgA antibodies are produced by some patients important cause of anaphylaxis following blood transfusion.
Treatment: Antibiotics as needed.
How do you treat an IgA deficiency?
Antibiotics.
Why can you not treat IgA deficiency with IgA therapy?
IgA has a half-life of seven days, would have to get shots every week for your entire life. Not cost effective.
Typical IgA antibodies found in mucosal regions of body, so if we received them intravenously they would never make it to where they need to go.
Common Variable Immunodeficiency (CVI):
Normal number of mature B cells but can’t differentiate into plasma cells- for variety of reasons.
Patients usually in 20s and 30s.
Manifestations: Malabsorption and diarrhea; malignancies, and autoimmune disorders.
How is Common Variable Immunodeficiency diagnosed?
Low serum IgG in patient with recurrent bacterial infections.
What is the treatment for Common Variable Immunodeficiency?
Injection of IV Ig preparations; antibiotics as needed.
Transient Hypogammaglobulinemia Definition:
Slow development of immune system.
X-Linked Bruton’s Agammaglobulinemia Definition:
Development arrested at pro to pre-B cell; deficiency in Bruton’s tyrosine kinase.
IgA deficiency Definition:
Most common; blood transfusion anaphylaxis.
Common Variable Immunodeficiency Definition:
Development arrested at mature B cell; hypogammaglobulinemia.
Cellular Immunity Deficiencies:
Manifestations: Recurrent infections with intracellular pathogens: Viruses, fungi (Candida), and intracellular bacteria.
Increase of malignancy.
Defects can occur at many different stages of T cell development.
Can affect humoral immunity as well.
What is the treatment for Cellular Immunity deficiencies?
Bone marrow transplant.
True or false. Cellular Immunity deficiencies are more difficult to manage than Humoral Immunity deficiencies.
True.
Graft Versus Host Disease (GVH):
Donor cells attack recipient’s tissues.
Prevention: Irradiation of marrow transplant or blood products.
DiGeorge Anomaly:
Most missing portion of chromosome 22.
Embryonic development defect- affects thymus and variety or organs.
Partial DiGeorge: Near normal immune system.
Insufficient number of mature T Cells.
Treatment: Fetal thymus and bone marrow transplant; thymic hormones.
Purine-Nucleoside Phosphorylase Deficiency (PNP):
Due to defect in enzyme involved in purine metabolism.
Number of T cells decreases as a toxic purine metabolite accumulates.
Humoral Immunity is okay.
Name two T-cell deficiencies:
DiGeorge Syndrome: Thymus defective.
Purine-nucleoside phosphorylase (PNP) deficiency: Toxic purine products cause decrease in T cells.
Combined Cellular and Humoral Deficiencies:
Defect in development of both types of T and B lymphocytes or defective interaction between T and B lymphocytes.
Severe Combined Immunodeficiency (SCID):
Most severe of the congenital immune deficiency.
Variety of defects.
X-linked SCID most common- 46% in US.
Variety of defects grouped under SCID.
Autosomal recessive SCID- child inherits two defective copies.
Formerly known as Swiss-type agammaglobulinemia.
A higher prevalence is found in areas and culture among which there is a higher rate of consanguineous mating.
Adenosine deaminase (ADA) deficiency.
Wiskott-Aldrich Syndrome (WAS):
Manifestations: The triad of immunodeficiency, eczema and thrombocytopenia.
Usually lethal in childhood due to infection, malignancy or hemorrhage.
Cause is in X-linked protein defect.
Condition with variable expression, but commonly includes IgM deficiency.
Severe deficiency of the naturally occurring antibodies to blood group antigens (isohemagglutinins).
Ataxia-Telangiectasia (AT):
Ataxia: Muscle incoordination.
Telangiectasis: Capillary dilation.
Defect in gene recombination process (TCR and Ig)
Autosomal recessive.
What are the three B and T cell deficiencies?
SCID- Severe combined immunodeficiency.
Wiskott-Aldrich Syndrome (WAS)- X-linked protein defect.
Ataxia-telangiectasis- Defect in Ig and TCR gene recombination process.
Defective Neutrophils:
1st line of defense. Involved in antibody mediated killing.
Effective ingesting/killing.
Defects allow recurrent pyogenic bacterial infections.
Chronic Granulomatous Disease (CGD):
Several defects: Each affects production of reactive oxygen forms.
Defective microbial killing results in recurrent infections.
Death usually in childhood.
Treatment: Granulocyte transfusions, cytokine administration and bone marrow transplant.
Myeloperoxidase Deficiency (MPO):
Genetic abnormalities.
Most common neutrophil abnormality.
Generally does not cause disease- occasional disseminated candidiasis in diabetics.
No treatment in most cases.
Leukocyte Adhesion Molecule Deficiency:
Defect in adhesion receptors: PMN, monocytes, and T cells.
Results in abnormal adhesion, motility, chemotaxis and endocytosis.
Manifestations: Delayed wound healing and chronic skin infections.
What are three Neutrophil Defects?
Chronic Granulomatous Disease (CGD)- Inability to kill ingested organisms.
Myeloperoxidase deficiency- Candida infections.
Leukocyte adhesion molecule deficiency- Defect in adhesion receptors.
Secondary Immune Deficiencies:
Abnormality due to another disorder or treatment.
Malignancies- Hodgkin’s disease and lymphoma, leukemia, myeloma, macroglobulinemia.
Infections- Bacterial, viral, protozoan, helminthic and fungal.
Example: Hodgkin’s disease, Herpes Zoster.
Laboratory Evaluation of Deficiencies:
Controls critical- Age matched controls.
Screening tests- CBC and diff.
Humoral Immunity:
Measures serum: IgG, M and A, G subclasses.
Isohemagglutinins: ABO antibodies by 2 years.
IgG response to protein/CHO antigens.
Cell Mediated Immunity:
Delayed type hypersensitivity skin tests (CA and diph/tetanus after immunization)
Phagocytes defects.
Complement- Total C activity and components.
Confirmatory Tests:
Flow Cytometry to enumerate: B cells, T cells and types of T cells and NK Cells.