Hit to lead 2
1/21
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
22 Terms
explain the optimisation cycle and the pros and cons of different strategies
improve hits drug like characteristics, synthesis of analogues, test for structure activity relantionship SAR ( potency, bioavailability, stability, selectivity), repeat multiple times develop a pharmacophore
explain lipinsky rule of 5 and its role in orally bioavailable drugs
Mw< 500 Da, < 5H-bond donors, <10 H-bond acceptors, LogP <5
explain key safety considerations during lead optimisation
mechanism based pharmacology, formation of reactive metabolites, activation of receptors like hERG, interaction with other receptors, idiosyncratic toxicity
a d2-5Ht2 antagonist drug was designed with napthalene ring ans it was potent in vitro, but had poor efficiacy in vivo due to poor solubility, explain how to improve solubility
introduce polar groups e.g. replace napthalene ring with heteroaromatic system containing N or S like pyridine ring
what are the considerations for good lead
log P, Mw, potency, polar interactions, heteroaromatic groups, bioisosters, structural simplification, chemical tractability
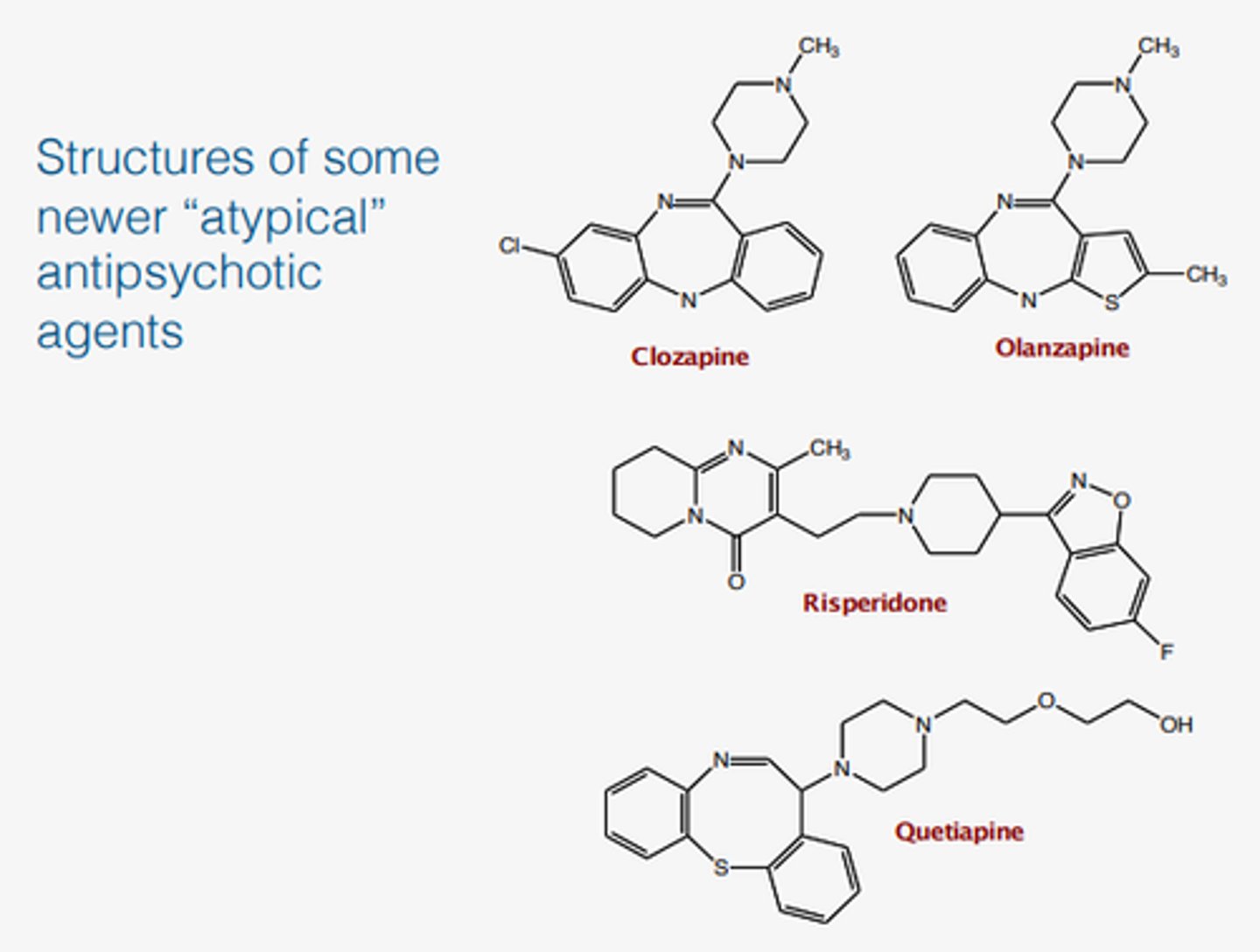
Chlozapine is poorly water soluble drug and 55% bioavailability, so it was made to Olanzapine which has 100% bioavailability. Explain how they did it?
they added polar groups to the ring and reduced LogP, which made the drug more bioavailable
what are the assumptions made before human studies
In vitro/in vivo* correlations: The assumption that effects observed in vitro will predict effects in vivo. o Animal-to-human extrapolation: The assumption that toxicity observed in animals will be relevant to humans. o Use of high doses in animals: The assumption that effects seen at high doses in animals can be used to predict effects at therapeutic doses in humans.
patient overdoses on salbutamol, which he used for his asthma and had a heart attack. Explain the mechanism behind toxicity?
salbutamol is a beta agonist, which causes airways to dilate. If the drug is overdosed then the levels in the systemic circulation arise and the drug binds to the beta recptors in th heart causing palpitations. This is an example of mechanism based pharmacology, where activation of the target in another tissue can cause side effects. This is avoided by inhaled salbutamol.
the patient took 10g of paracetamol and presented to the hospital with liver damage. Explain the mechanism behind toxicity and why it happened?
in normal 1 g dose phase 2 metabolism sulphate metabolite and glucoronide metabolite are added to the same position, which maked the drug highly water soluble and it's excreted via urine. alternative pathway is where the OH group is oxidised to form a reactive metabolite NAPQI, which cause toxicity. In normal dose glutathione acts as a scavenger of electrophiles, it reacts with the reactive metabolites and removes them via excretion. The patient overdosed, so there was not enough glutathione to react with all the NAPQI, so it bound to liver proteins and iron causing liver damage.
How to avoid reactive metabolite toxicity
avoid functional groups known to form reactive metabolites
look for reactive groups or their products such as protein binding or glutathione with mass spectometry
do an ames test to detect mutagenicity that can be caused reactive metabolites damaging DNA
explain aimes test
use a genetically modified Salmonella typhimurium bacterium which cannot synthesise histidine & cannot grow in its absence, mutant grown on histidine-containing media, drug and a liver microsomal enzyme preparation (to test for reactive metabolites) added to bacteria, histidine becomes depleted and only back-mutants can grow, if the drug can cause mutations, the genetic modification can be reversed and the bacteria will grow, mutation rate measured
the patient overdosed on terfendaine antihistamine and had a heart attack, explain the mechanism behind that.
the drug caused an off-target toxicity, because it conatins a hERG pharmacophore, which allows the drug to bind to the K channel in the heart and cause hyperpolarisation, so QT interval is prolonged, lengthening the time required to repolarise the cell, which leads to fatal arrythmias
Farnesyltransferase inhibitor conatins an aromatic ring, alkyl chain and nitrogenous based and can cause heart toxicity, explain how you could change the structure to get rid of toxicity?
introduce a heteroaromatic ring instead of phenylring, add an OH group, this changes polarity, which reduces LogP, so drug cannot diffuse to the heart and hERG IC50 reduces
Patient was taking terfenadine with erythomycin and ketokonazole and died of heart attack, explain the mechanism?
erythomycin and ketoconazole inhibit CYP P450, which meant that Terfenadine was not metabolised, so it reached toxic levels and was able to bind to hERG causing heart attack.
what is the criteria from lead optimisation to preclinical candidate
Preferred crystalline form identified.
Compound sufficiently stable to allow a shelf life of >2 years.
• Scale-up of lead compound to 100g demonstrated.
• Full PK and metabolite profiling in two species e.g. (rodent and monkey)
• Predicted human half-life and dose ( low dose and frequency means high potency, less side effects)
• No toxicity in extended animal study
explain pharmacokinetic screening during lead optimisation
advanced lead compounds are screened in animals like rodents before efficacy and safety studies
measure Cmax, Tmax, Half-life, volume of distribution, clearance, bioavailability,
help to determine dose, frequency and drug half-life
What is hERG pharmacophore and how to avoid it
A strong lipophilic base, often a tertiary amine. An aromatic ring close to the basic group. A 2-5 atom chain connecting these groups.
avoid hERG groups
Checking for hERG activity using in silico (computer-based) structural alert predictions.
Screening compounds in vitro using a high-throughput hERG assay.
Ensuring a sufficient safety margin: hERG IC50 > 30.
What are the preclinical drug development tests that are done
In silico techniques DEREK, to predict potential toxicity.
in vitro tests for mutagenicity Ames test
In vitro cytogenetic evaluation of chromosome damage in response to a drug.
Carcinogenicity testing: Chronic drug dosing in animals to look for tumor development.
Reproductive (teratogenicity) testing: Dosing pregnant animals (from one rodent and one non-rodent species) with the drug at different stages of organogenesis and examining the offspring for birth defects.
Preliminary in vivo toxicity testing
Maximum non-toxic dose (given for 28 days to 2 species). Animals examined post-mortem and tissue damaged assessed
Lethal dose LD50 - the dose of drug which kills 50% of treated animals within a specified short amount of time
what is NOAEL
no observed adverse effects level)
LOAEL
lowest observed adverse effects level
Therapeutic index
(TI) is the ratio of the dose of a drug that produces an unwanted (toxic) effect to the dose that produces a wanted (therapeutic) effect LD50 / ED50