PHSI3009: Module 3 - Protein Structure & Function
1/104
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
105 Terms
Integral membrane proteins are responsible for…
Transport across the membrane
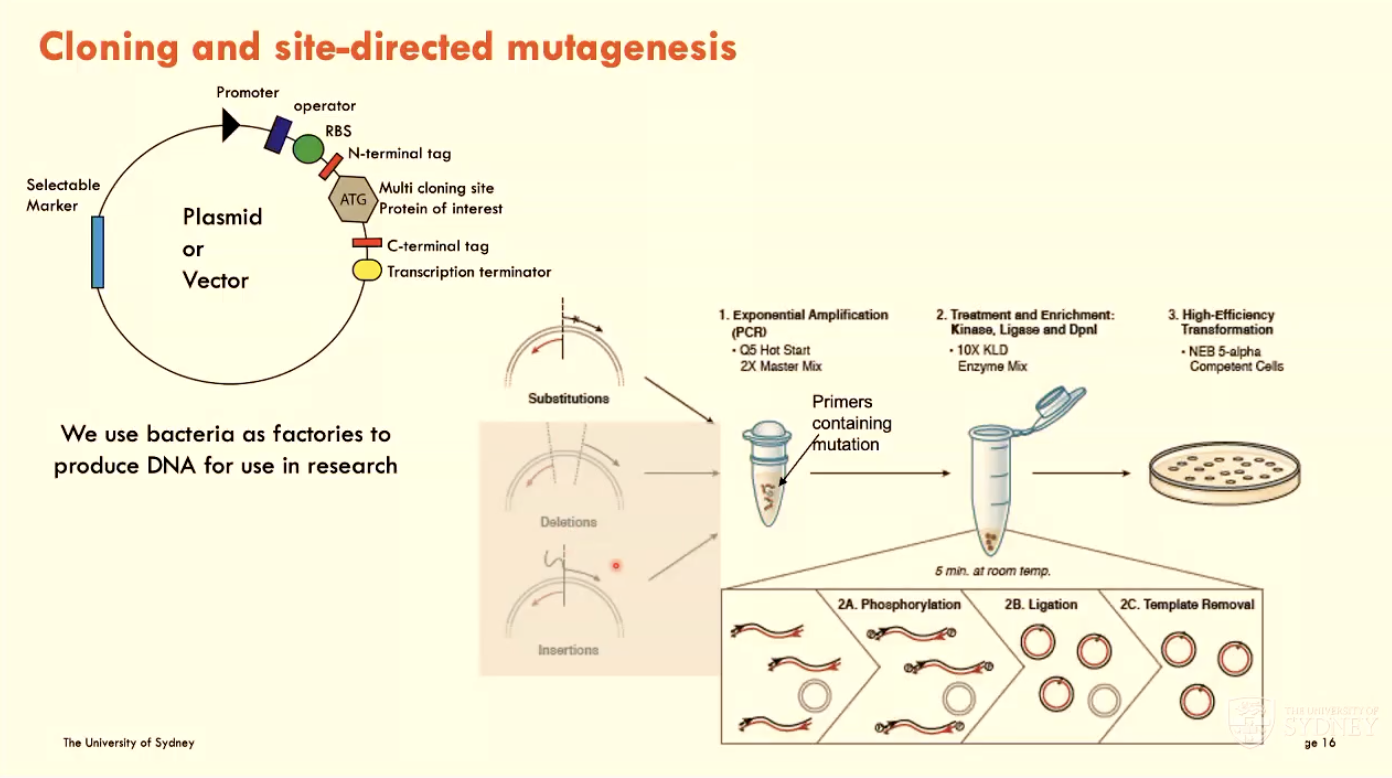
What is cloning and site-directed mutagenesis?
Using bacteria in the lab as factories to produce DNA for research use
done using a plasmid or vector
Use PCR to change AAs to desired protein of interest
Site-directed mutagenesis: introduce a single point mutation and investigate functional effect
start by designing primers that introduce mutation
confirm mutation using sequencing
express protein and check function of altered AA using chosen experiment
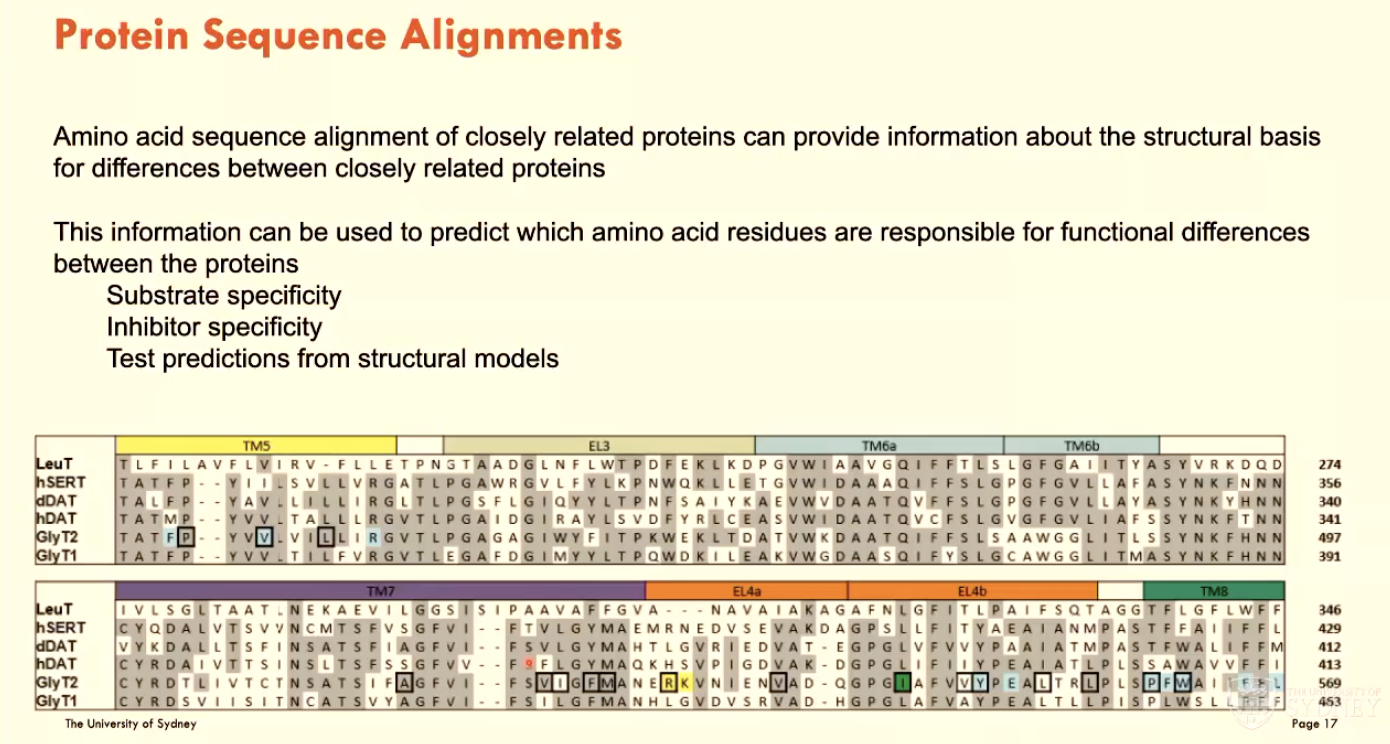
What are protein sequence alignments?
Aligning AAs between closely related proteins to predict which AA residues are responsible for functional differences between the proteins:
substrate specificity
inhibitor specificity
test predictions from structural models
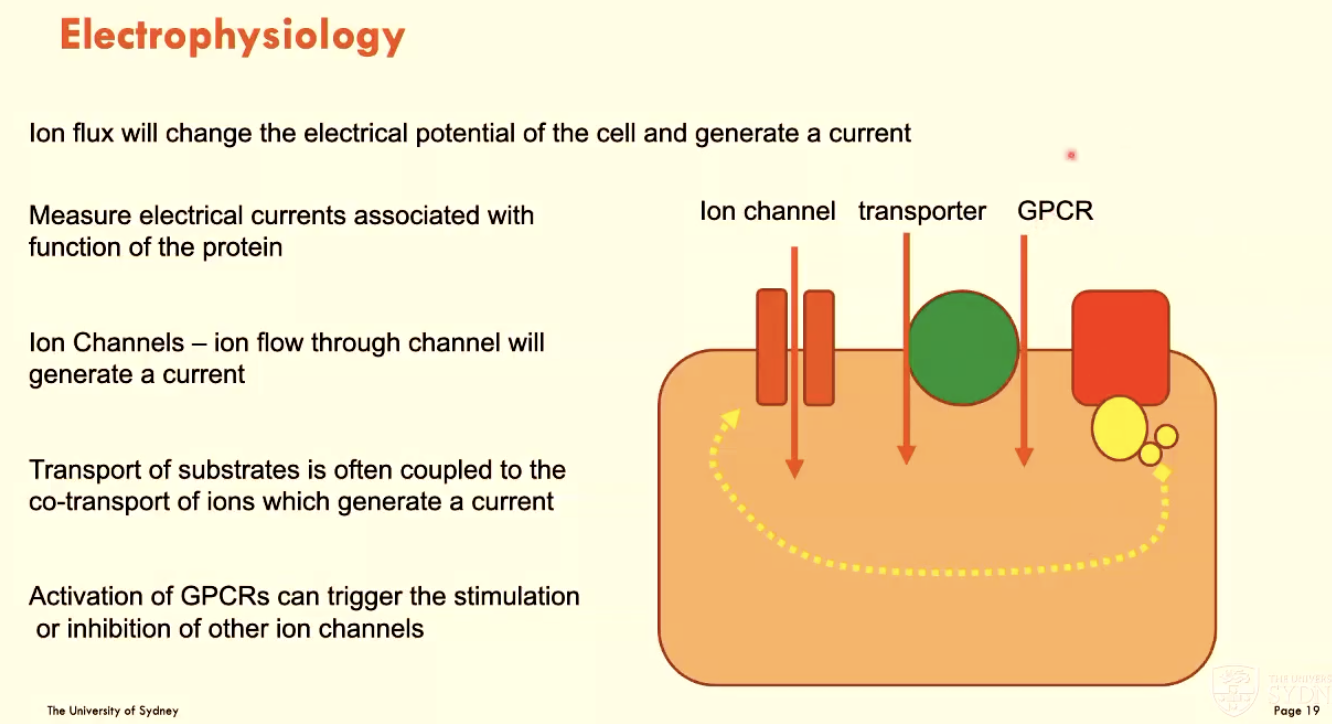
What is electrophysiology?
Using ion flux to change cell electrical potential and generate a current
measure electrical currents associated with protein function
activation of GPCRs can trigger ion channel stimulation/inhibition
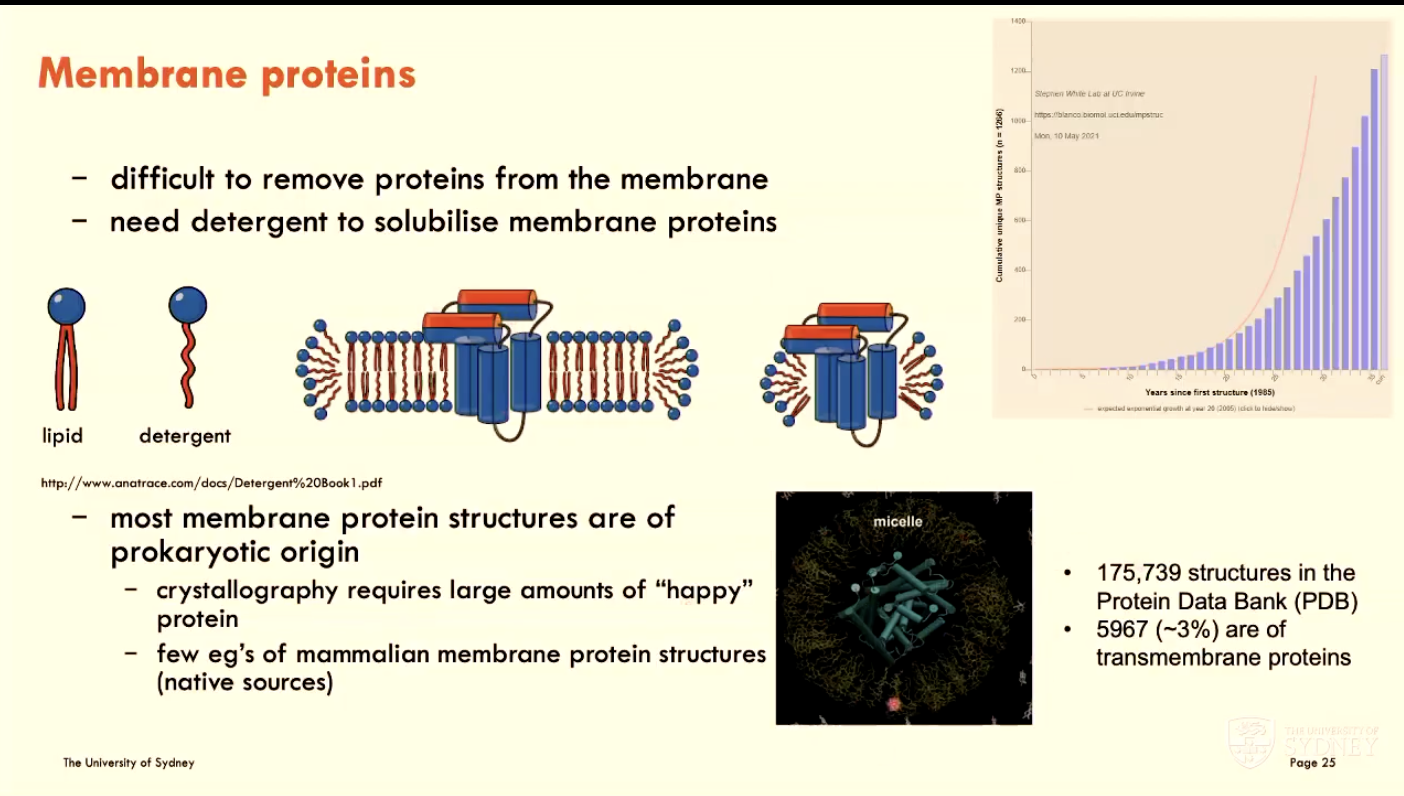
What is the challenge of working with membrane proteins?
They are difficult to remove from the membrane (natural environment), so we need detergent to solubilise membrane proteins
note: most membrane protein structures are of prokaryotic origin
How is protein purified?
For structural biology, protein needs to be pure (homogenous), stable, soluble, and of high quantities
Example procedure: Gel filtration (size exclusion chromatography)
Then, functional assays can be conducted
What are some methods used in structural biology?
Structural biology is the study of the molecular structure and dynamics of biological macromolecules, particularly proteins and nucleic acids. Some methods used to study this are:
X-ray crystallography: Provides detailed information on protein interactions with ligands, cofactors, and ions, but requires 3D crystals
Cryo-Electron Microscopy (cryo-EM): Analyses macromolecule structures in their native environment, advancing rapidly, especially for membrane proteins, but limited by molecule size.
Solution and Solid-state NMR: rapidly advancing, but sensitivity and resolution enhancements are still needed to make this a robust technology
CryoEM and (A - technique) can be used to visualise (B).
This helps us (C - 2 things)
A - protein crystallography
B - protein structures at very high resolution (atomic scale)
C - understand conformational changes in proteins AND develop drugs to specifically bind to proteins

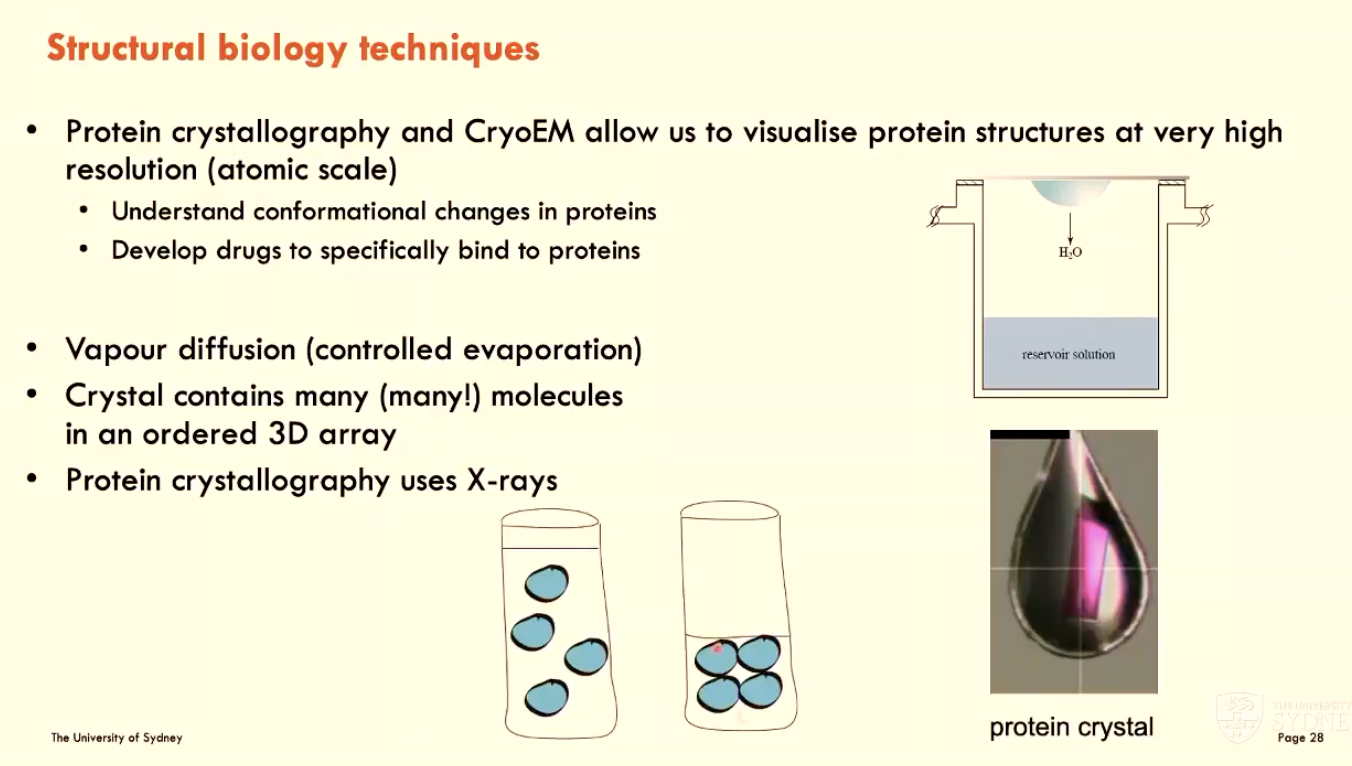
How does protein crystallography work?
Vapour diffusion (controlled evaporation)
crystal contains many molecules in an ordered 3D array
protein crystallography uses X-rays
What is a synchrotron?
A synchrotron produces light by accelerating electrons almost to the speed of light
infrared, UV, and X-rays are sent down pipes called beamlines to work areas
light is aimed at a very small sample → creates an image detecting molecular structure → sent to computer for analysis
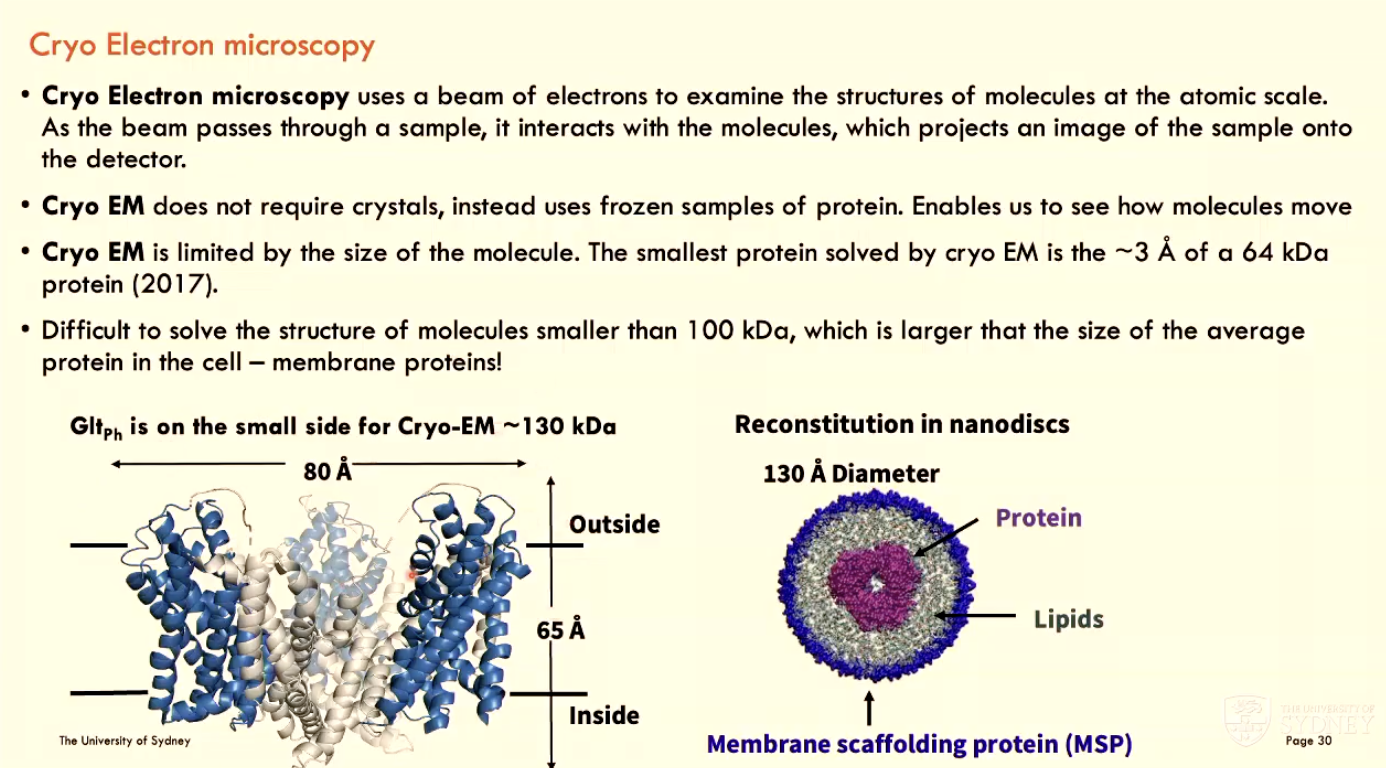
What is Cryo Electron Microscopy?
Cryo EM uses a beam of electrons to examine the structures of molecules at the atomic scale
as the beam passes through a sample, it interacts with molecules and projects an image of sample onto detector
Cryo EM does not require crystals, instead using frozen samples of protein (enables visualisation of molecule movement)
Cryo EM is limited by molecule size (difficult to solve structures of molecules smaller than 100 kDa, which is larger than the average protein size)
Identify the 5 classes of the Transport Classification System
Class 1: Channels and Pores
These are proteins that allow the relatively free flow of solutes across the membrane. They are divided into five subclasses.
Class 2: Electrochemical Potential-Driven Transporters (Carriers)
These proteins bind their solutes to form complexes before transporting them across the membrane via secondary active transport processes such as symport and antiport.
Class 3: Primary Active Transporters
These include ATPases and ATP-binding cassette (ABC) transporters. Other examples are transporters driven by redox reactions or by light (e.g., photosynthetic reaction centers).
Class 4: Group Translocators (not covered in module)
These transporters enable the phosphorylation of sugars during their transport into bacterial cells.
Class 5: Transmembrane Electron Transfer Carriers (not covered in module)
These include one-electron and two-electron carriers involved in electron transport across membranes. Redox proteins in this class are often not classified as transport proteins.

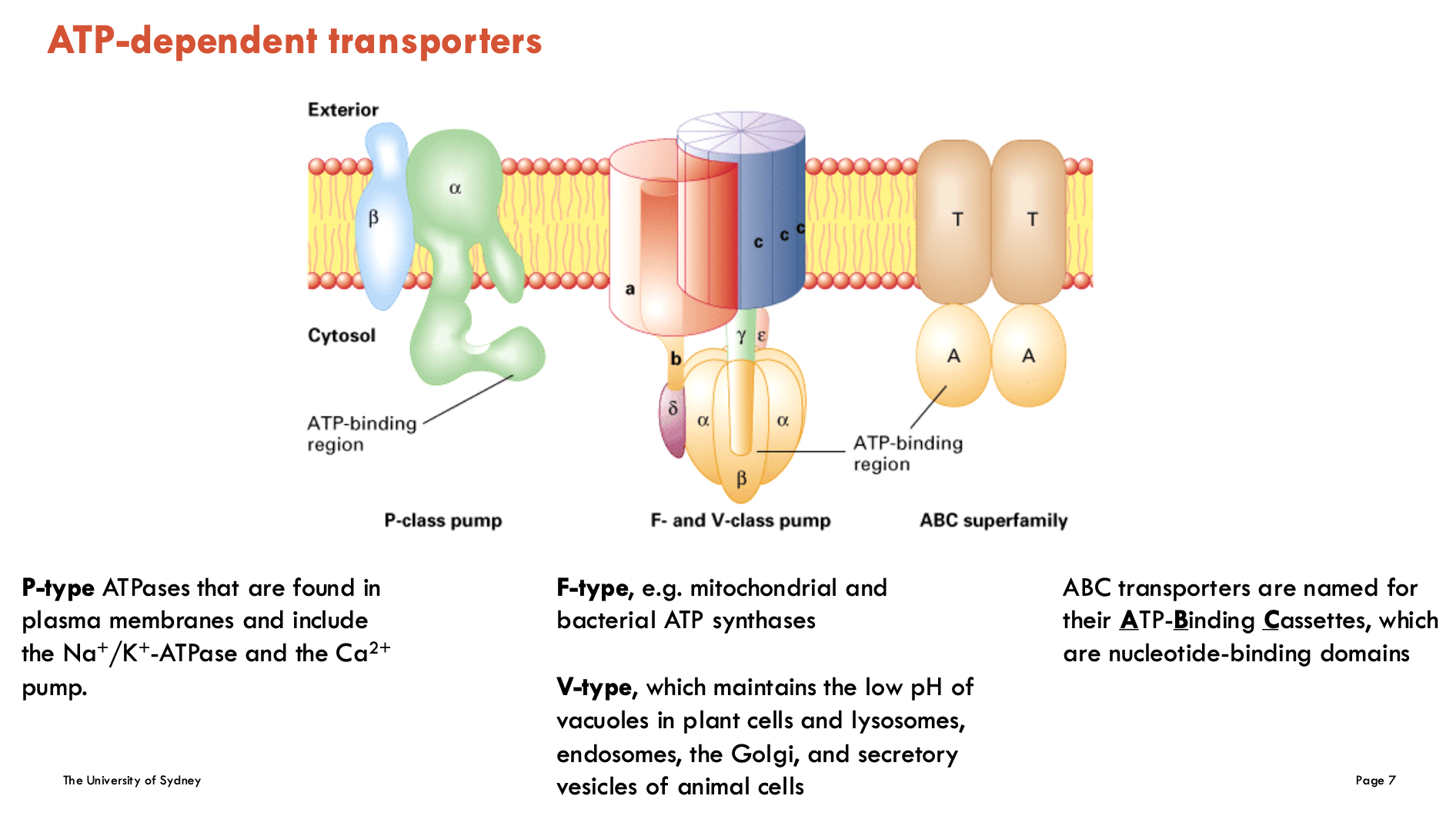
Identify ATP-dependent transporters (4)
P-type ATPases: found in plasma membrane and include the Na+/K+-ATPase and the Ca2+ pump (for regulating calcium in the muscle and heart)
F-type (e.g. mitochondrial and bacterial ATP synthases)
V-type: maintains the low pH in vacuoles in plant cells and lysosomes, endosomes, the Golgi, and secretory vesicles of animal cells
ABC (ATP-Binding Cassettes): nucleotide-binding domains
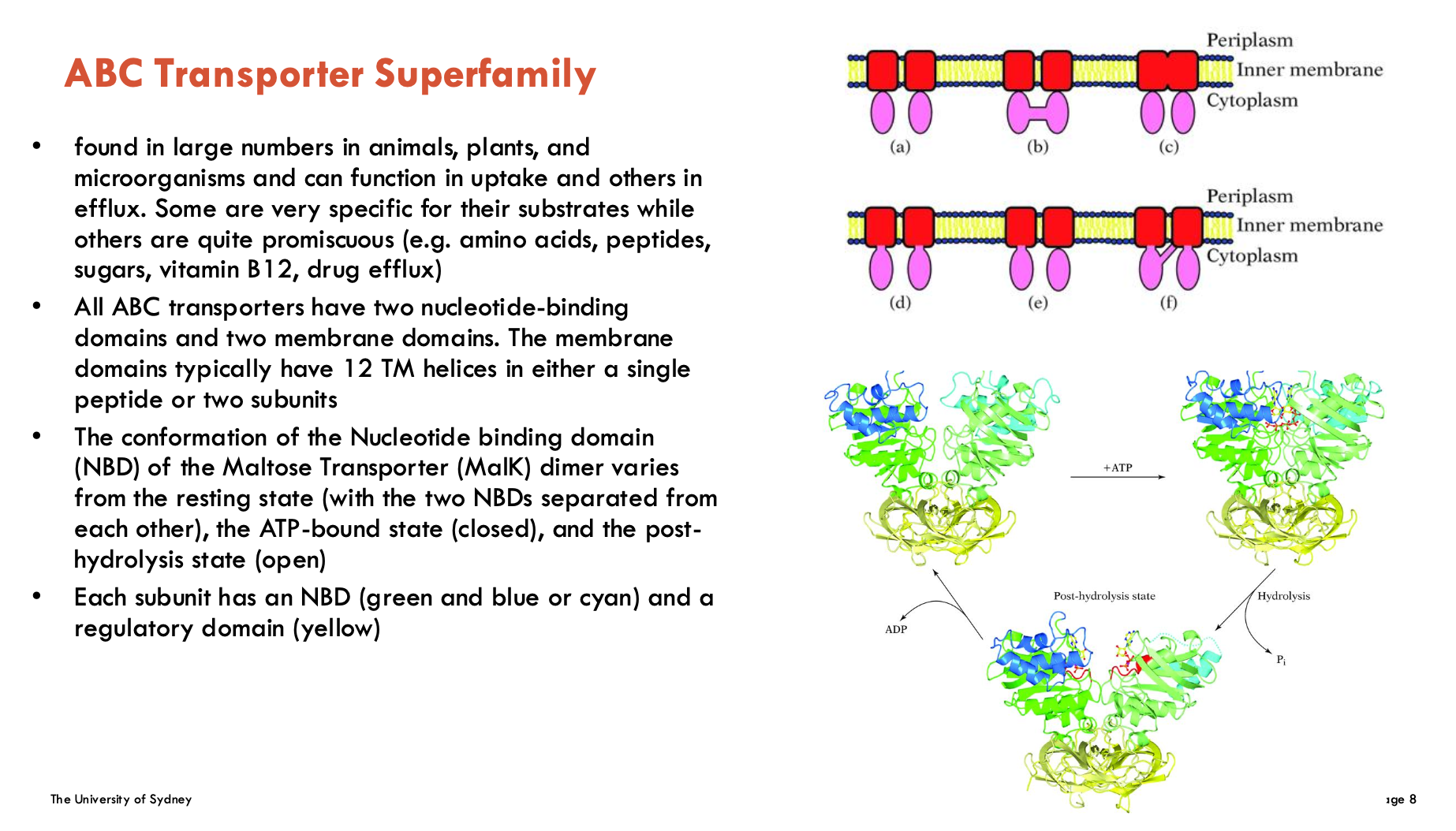
Features of ABC transporters (2)
Domain Structure
2 Nucleotide-Binding Domains (NBDs): Bind/hydrolyse ATP
2 Transmembrane Domains (TMDs): Form transport pathway (typically 12 helices total)
Conformational Changes
Resting state: NBDs apart
ATP-bound: NBDs dimerise (closed)
Post-hydrolysis: NBDs separate (open)
Has diverse functions and found in all organisms
Features of P-type ATPases (4)
driven by ATP (uses ~75% ATP in some cells)
forms a phosphorylated enzyme intermediate involving Asp residue in DKTGT motif
diverse substrates: ions and phospholipids
superfamily involving 300 known members, grouped into 5 subclasses
Na+/K+-ATPase is in class P2C
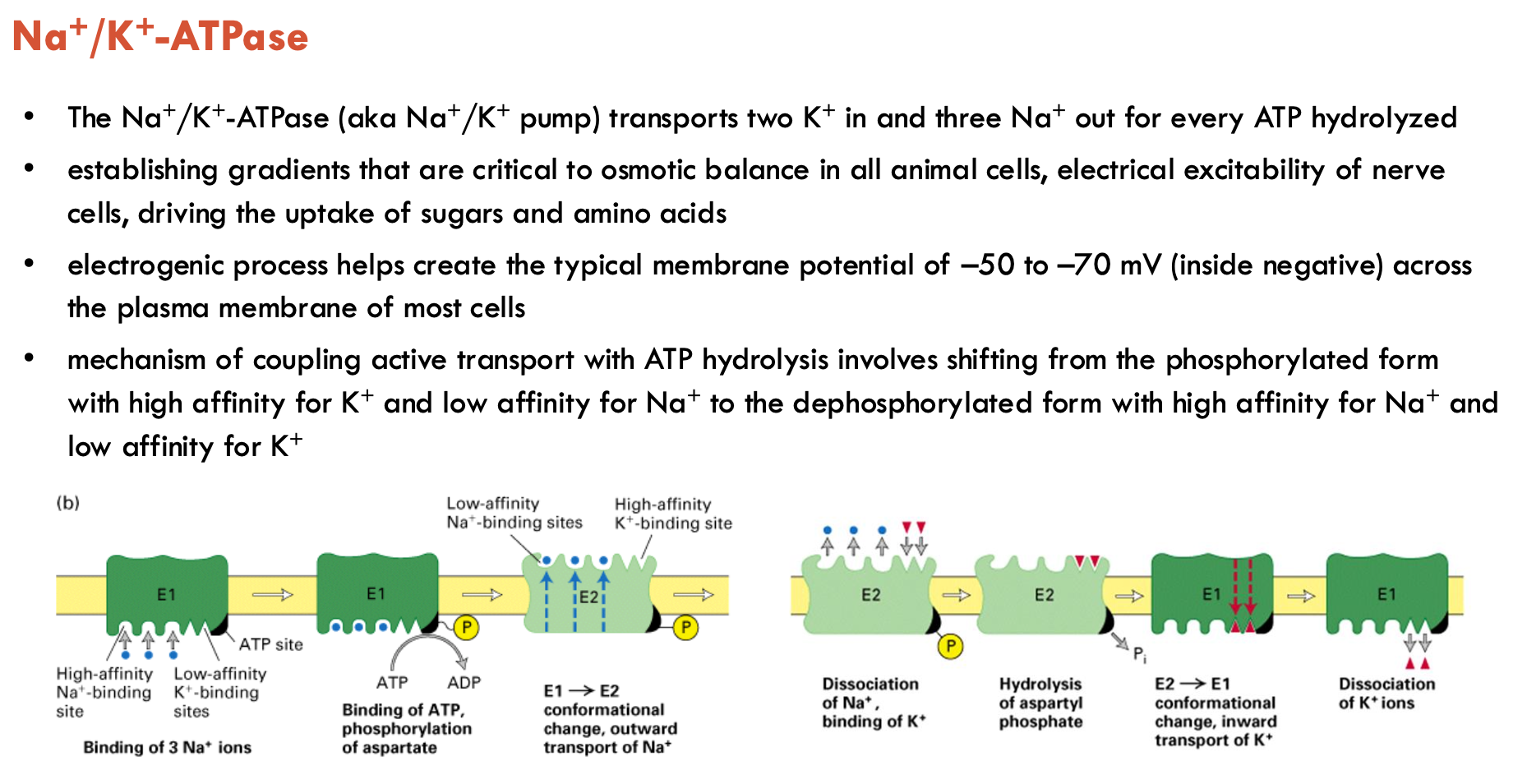
Features of Na+/K+-ATPase (3)
2 K+ in, 3 Na+ out per ATP hydrolysed
helps create typical membrane potential of -50 to -70 mV across plasma membrane of most cells
involves shifting from the phosphorylated form (high affinity (binds) for K+ and low affinity (releases) for Na+) to the dephosphorylated form (high affinity for Na+ and low affinity for K+)
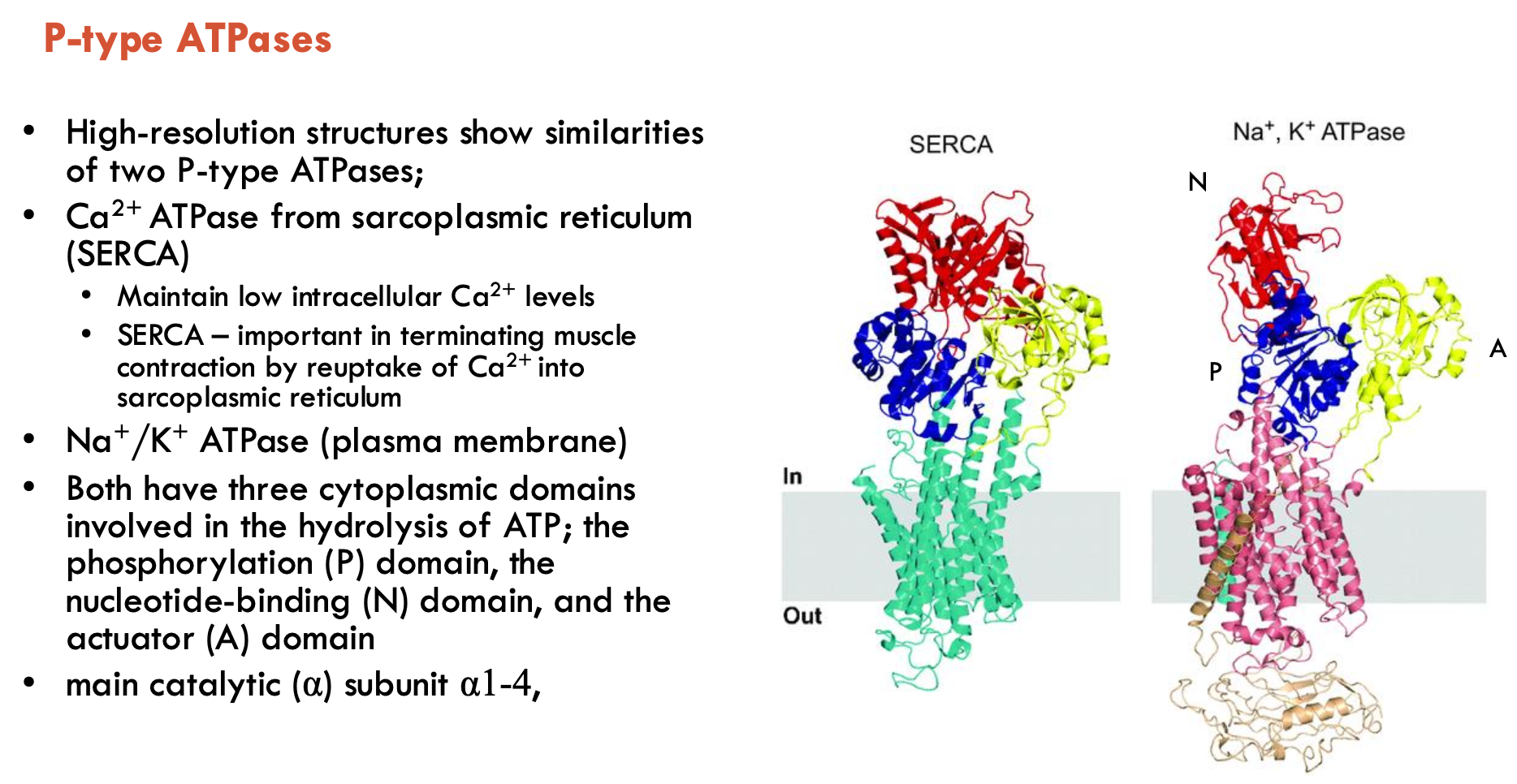
Which two P-type ATPases show similar structures?
Ca2+ ATPase from SR (SERCA) and Na+/K+ ATPase (plasma membrane)
both have 3 cytoplasmic domains involved in ATP hydrolysis:
phosphorylation (P), nucleotide-binding (N), and actuator (A) domains
main catalytic (α) subunit α1-4
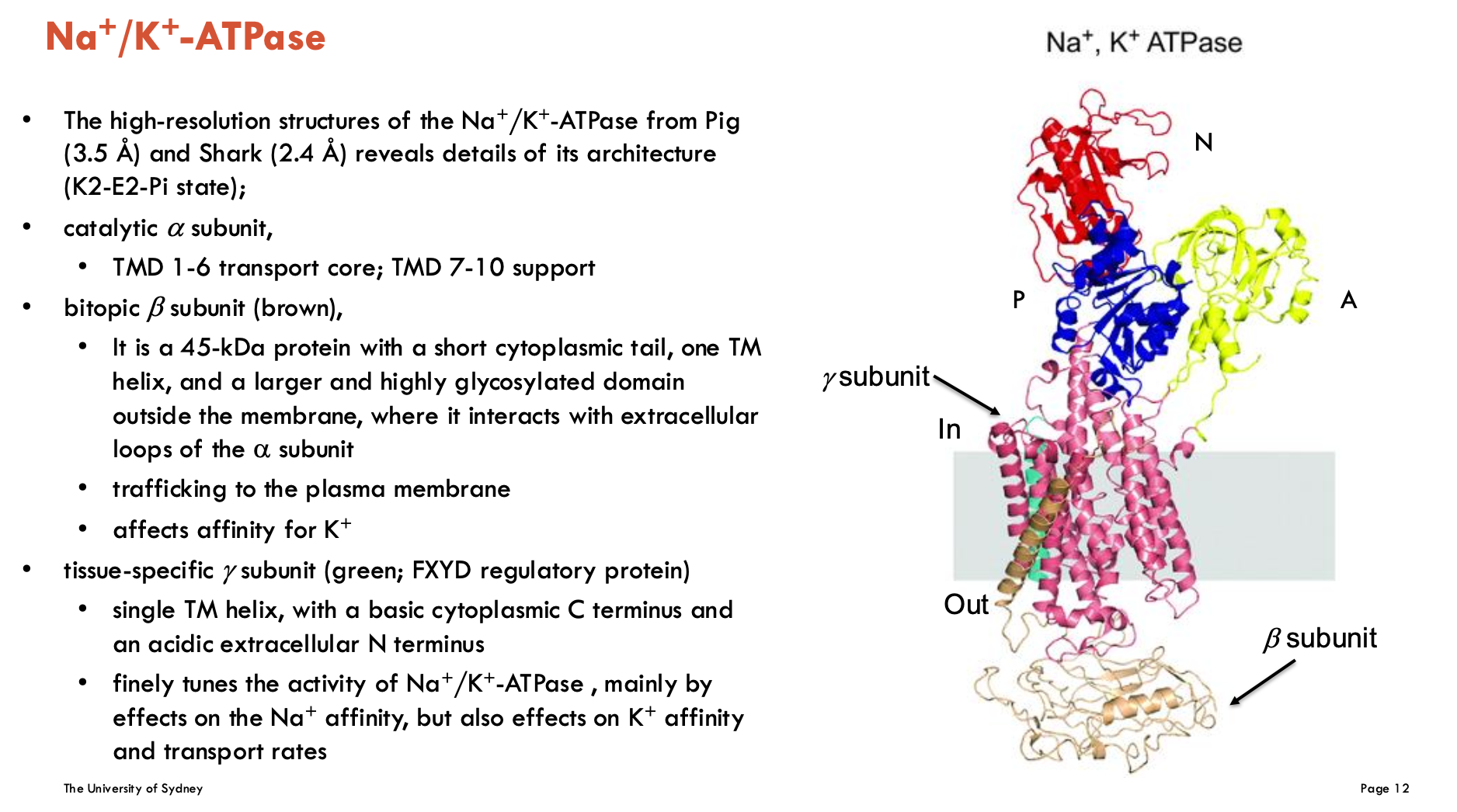
What subunits make up Na+/K+ ATPase?
catalytic α subunit
TMD 1-6 transport core, TMD 7-10 support
bitopic β subunit (brown)
is a 45-kDa protein with a short cytoplasmic tail, one TM helix, and a larger and highly glycosylated domain outside the membrane, where it interacts with extracellular loops of α subunit
trafficking to the plasma membrane
affects affinity for K+
tissue-specific γ subunit (green; FXFD regulatory protein)
single TM helix, with a basic cytoplasmic C terminus and an acidic extracellular N terminus
finely tunes the activity of Na+/K+-ATPase, mainly by effects on the Na+ affinity, but also effects on K+ affinity and transport rates
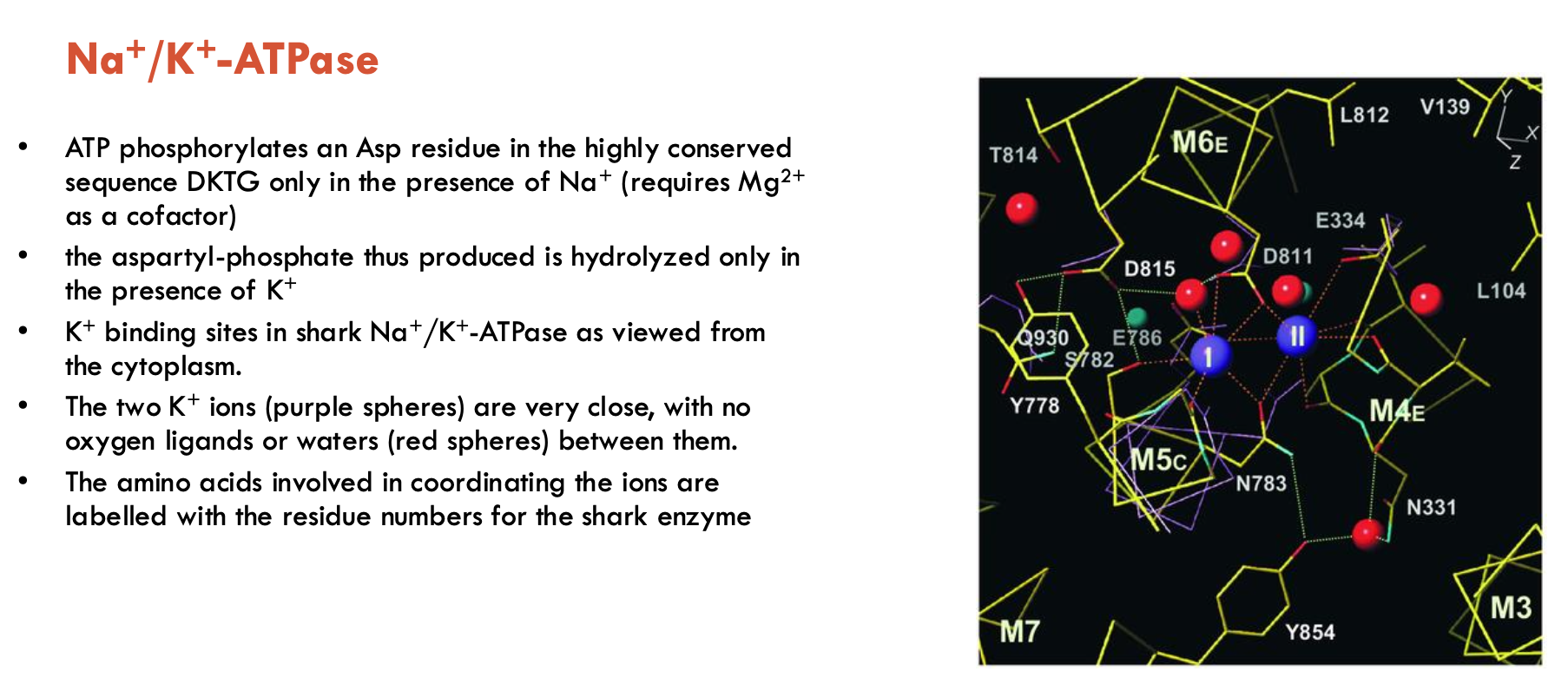
Describe the phosphorylation switch mechanism of Na+/K+ ATPase
Phosphorylation switch mechanism:
Na+-dependent step: In the presence of Na+, ATP phosphorylates Asp in DKTG motif (requires Mg2+ as a cofactor) → produces aspartyl-phosphate (drives conformational switch into phosphorylated form)
K+-triggered step: aspartyl-phosphate is only hydrolysed when 2 K+ ions are bound from the extracellular side (coordinated by AAs, no water or oxygen ligands separate them) → dephosphorylated form
This image shows K+ binding sites in shark Na+/K+ as viewed from cytoplasm
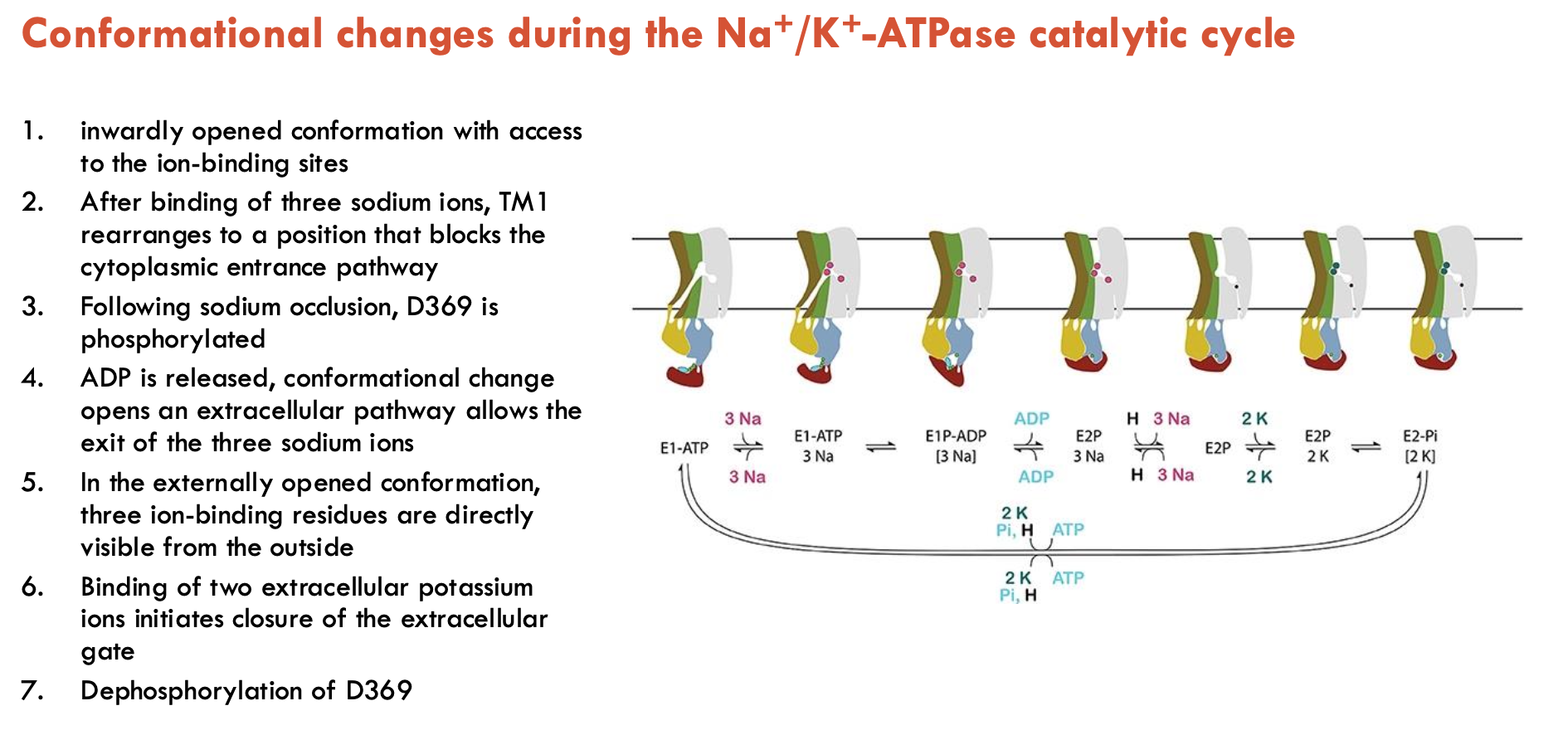
What conformational changes occur during the Na+/K+-ATPase catalytic cycle (7)?
Inward-open conformation → ion-binding sites accessible from cytoplasm
3 Na⁺ ions bind → TM1 shifts to block cytoplasmic entry
Na⁺ occlusion → ATP phosphorylates D369
ADP release → conformational shift to outward-open state
Na⁺ exit through extracellular pathway (3 binding sites exposed externally)
2 K⁺ ions bind from extracellular side → triggers extracellular gate closure
Dephosphorylation of D369 → prepares pump to return to inward-open state
How can Na+/K+-ATPase be manipulated in medical settings?
Cardiotonic steroids (e.g. ouabain) inhibit Na+/K+-ATPase
Decreases Na+ gradients → reduces Na+/Ca2+ exchange (less Na+ export) → more Ca2+ stored → allows stronger contractions (used in heart failure)
Endogenous cardiotonic steroids regulate blood pressure, kidney function, and cell growth/apoptosis
What medical conditions can be linked to Na+/K+-ATPase (5)?
FFXYD3 and 5 are upregulated in several cancers (functional effect not clear)
Na+/K+-ATPase malfunctions in cardiovascular, neurological, renal, and metabolic diseases
Hypertension; Adrenal overproduction of aldosterone is the cause of hypertension in up to 10% of hypertensive patients
Familial Hemiplegic Migraine (FHM) is an autosomally inherited form of migraine, experiences aura and weakness in one side
of the body during attacks
Rapid-onset Dystonia Parkinsonism (RDP), Alternating Hemiplegia of Childhood (AHC), CAPOS (cerebellar ataxia….)
Overall, a critical protein that affects many other proteins, and any mutation causing dysfunction may have large effects on health due to downstream effects

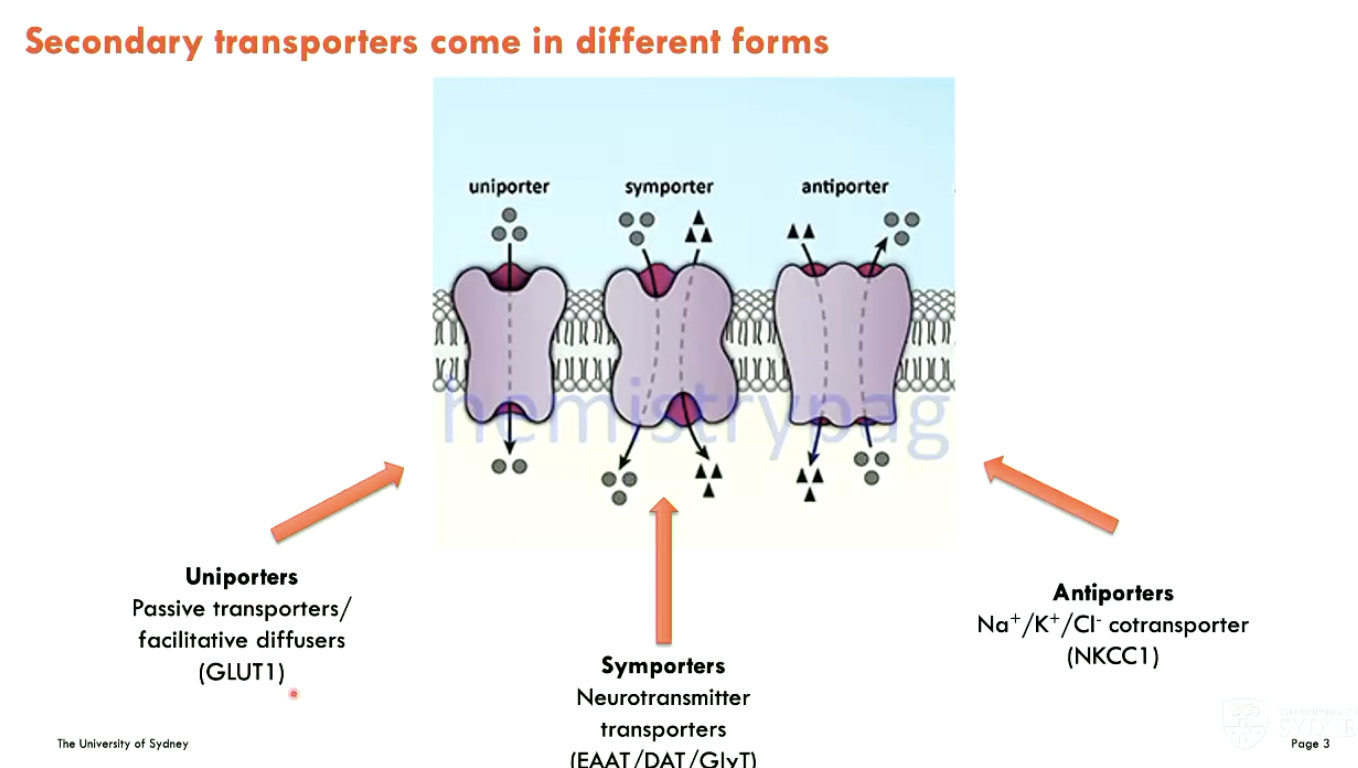
What different forms can secondary transporters be (3)?
Uniporters: passive transporters/facilitative diffusers (GLUT1)
Symporters: neurotransmitter transporters (EAAT/DAT/GlyT)
Antiporters: Na+/K+/Cl- cotransporter (NKCC1)
What are neurotransmitter transporters and what is their role?
All NT transporters are secondary active transporters that are coupled to pre-existing ion gradients
There are two main families found on post- and pre-synaptic and glial cells:
Glutamate transporter family: Glutamate/aspartate
Neurotransmitter Sodium Symporter (NSS) Family: GABA, glycine, dopamine, NA, serotonin
Bacterial homologues of both families have been crystallised and used as models - more recently, mammalian structures are available
Role: To clear NTs from synaptic cleft (important in brain)
What are EAAT transporters?
Excitatory Amino Acid Transporters (EAAT) are human plasma membrane glutamate transporters
transport glutamate and aspartate with similar affinities (2 - 20 µM)
5 human subtypes (EAAT1 - 5)
EAAT1, 2 are found on glial cells and are widely expressed
EAAT3 - widely expressed on neurons
EAAT4 - neurons in cerebellum
EAAT5 - neurons in retina
share ~50-60% amino acid identity
EAAT2 ~1% of total brain membrane protein

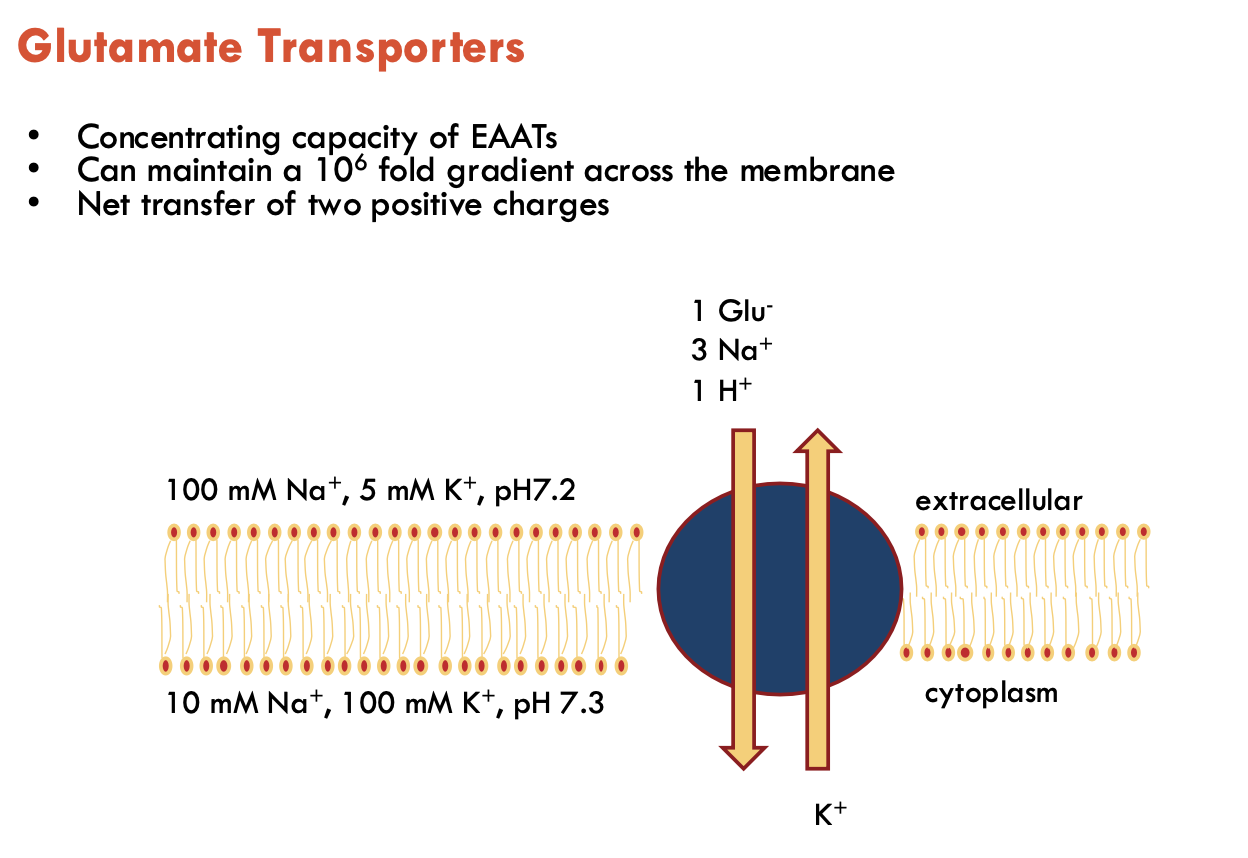
The concentrating capacity of EAATs is linked to (A).
It can maintain a (B - no.) gradient across the membrane and has a net transfer of (C).
A - stochiometry
B - 106 fold
C - two positive charges
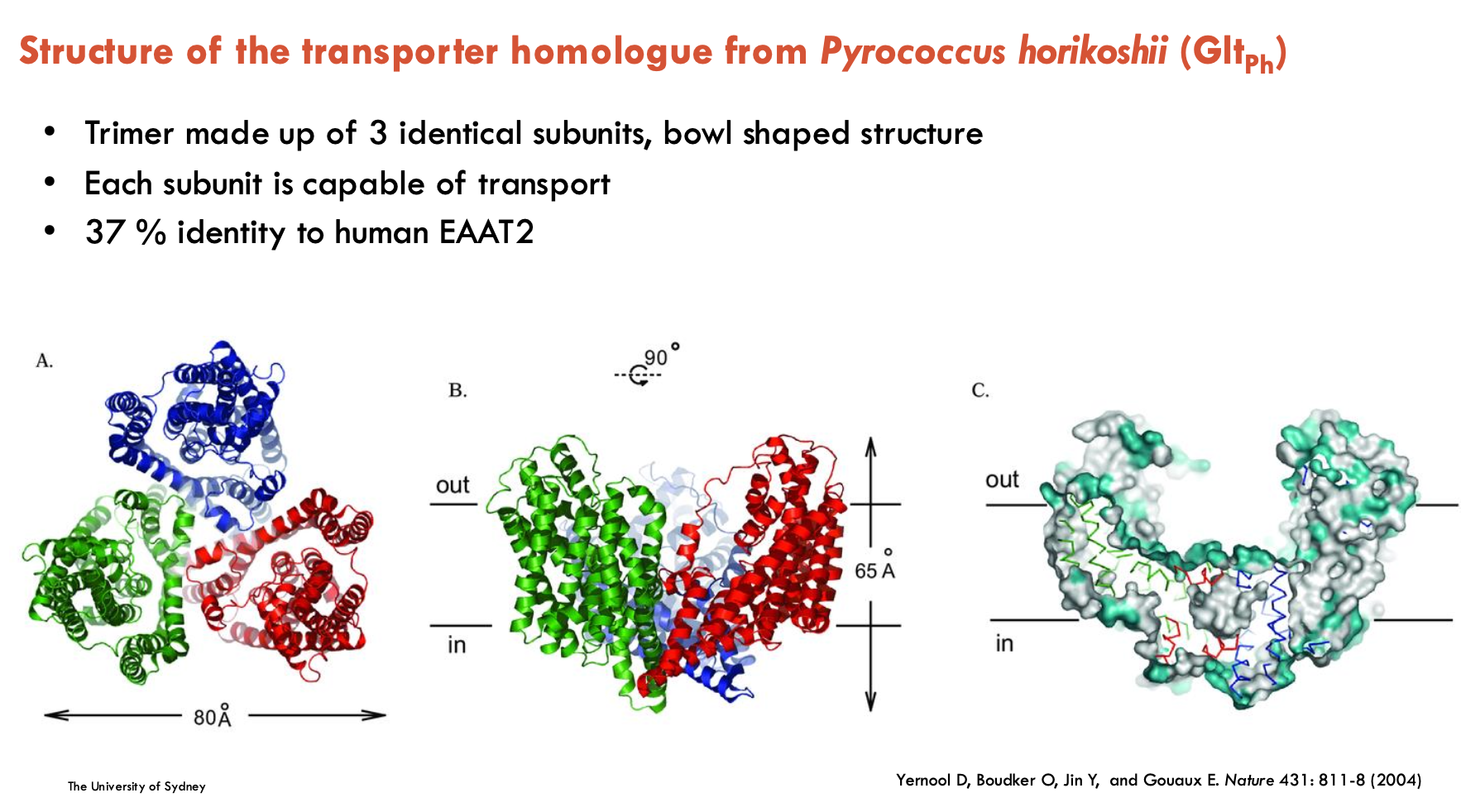
Describe the structure of the transporter homologue from Pyrococcus horikoshii (Gltph)
Trimer made up of 3 identical subunits, bowl-shaped structure
each subunit is capable of transport
37% identity to human EAAT2
is an Na+-dependent aspartate transporter
L-TBOA is a competitive inhibitor of…
glutamate
What are hairpins in transporter topology?
Hairpins are structural motifs where a single alpha-helix bends or breaks in the middle, forming a loop-like structure.
Instead of spanning the membrane completely, each half-helix enters and exits on the same side of the membrane.
often plays a critical role in forming the substrate and sodium binding sites within the transporter
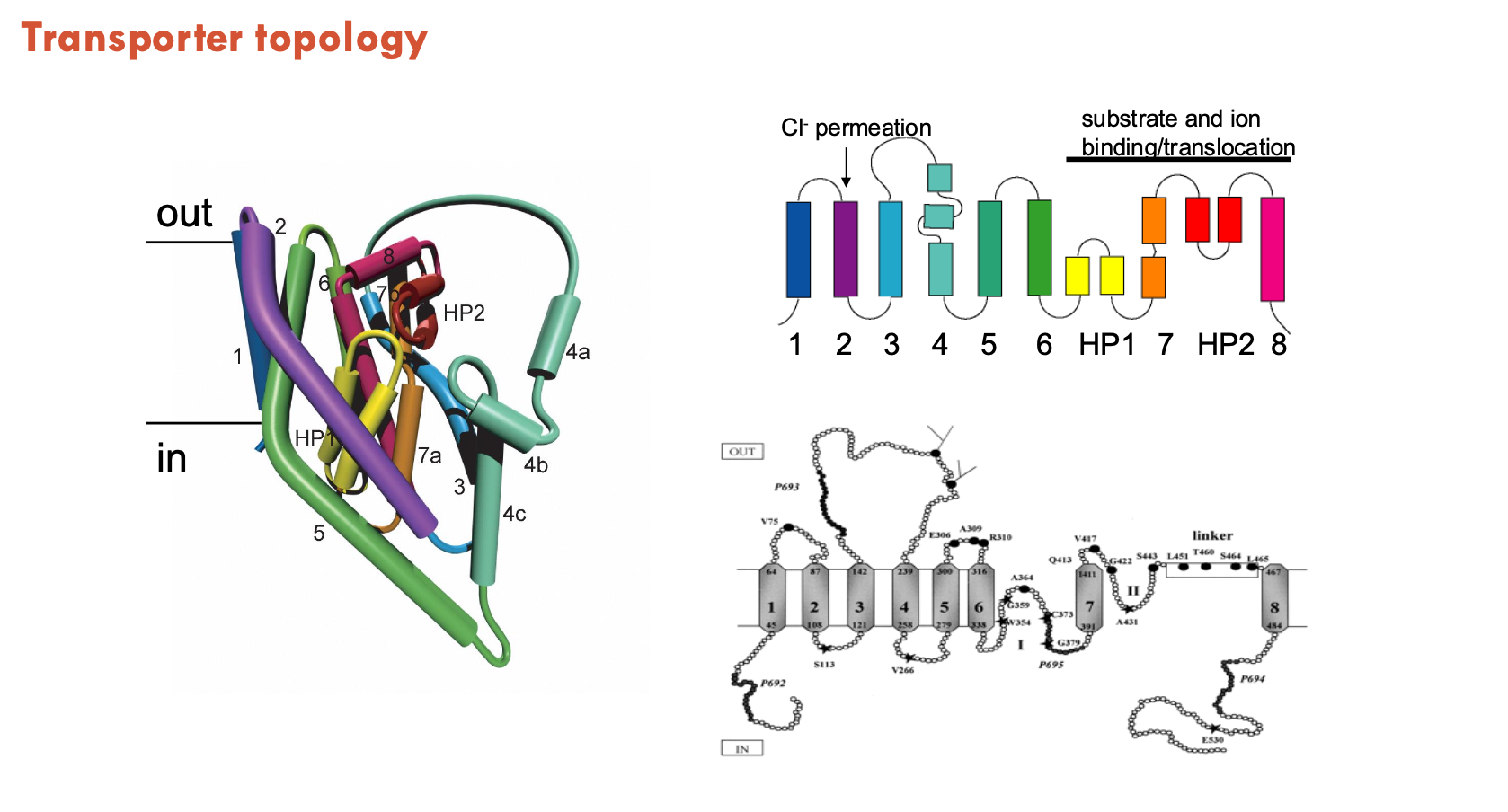
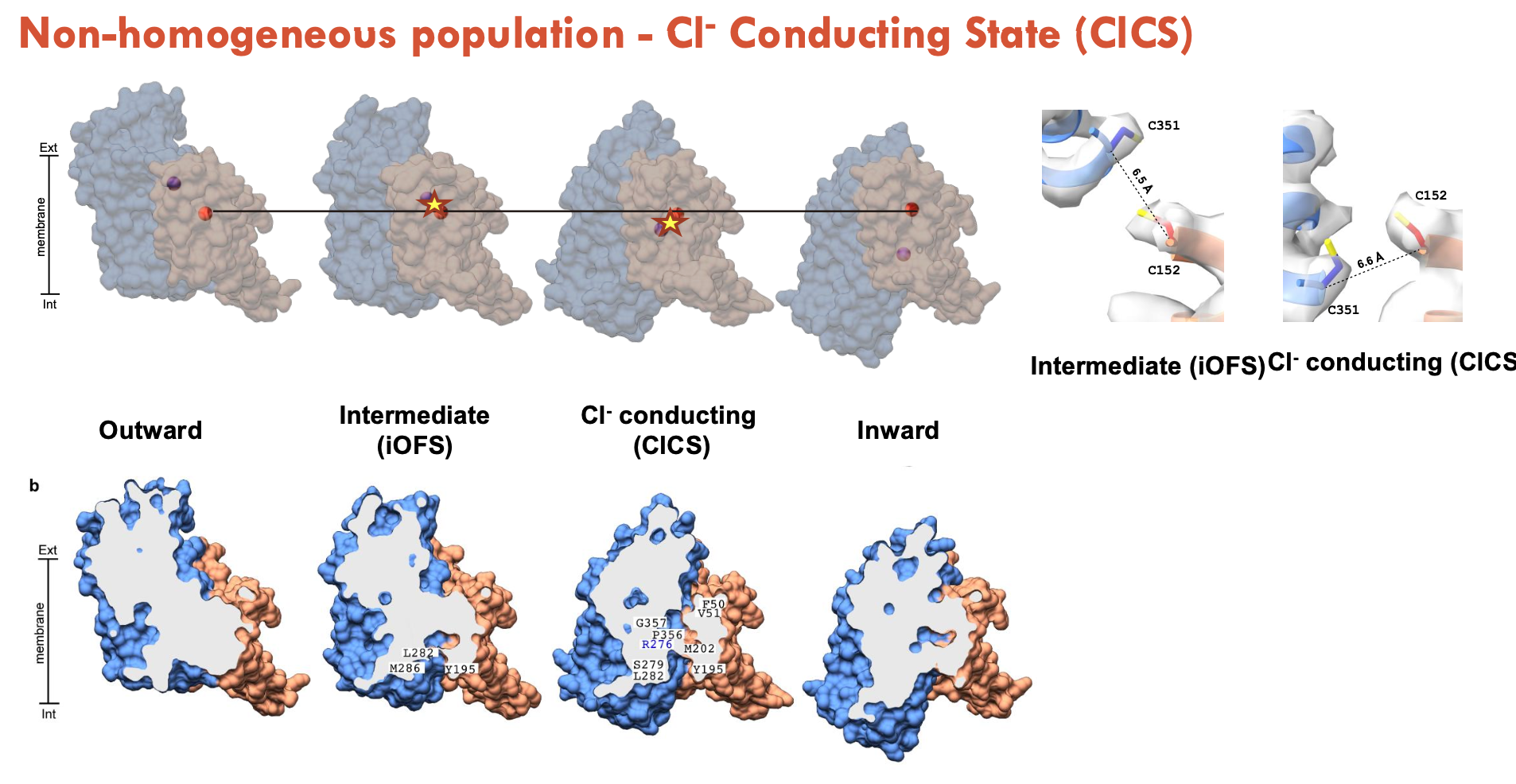
What did the cysteine proximity assay find in EAAT1 and Gltph?
The cysteine proximity assay tests if two cysteine residues are close enough (~6–7 Å) to form a disulfide bond, indicating spatial proximity.
In EAAT1, a double cysteine mutant showed:
Increased function with DTT (reduces disulphide bonds).
Decreased function with CuPh (forms disulphide bonds).
Suggests the two cysteines are near each other and that bonding DTT and CuPh affects transporter function
In GltPh (K55C/A634C mutation):
SDS-PAGE was used to detect disulphide bond formation
Confirms cysteine proximity biochemically
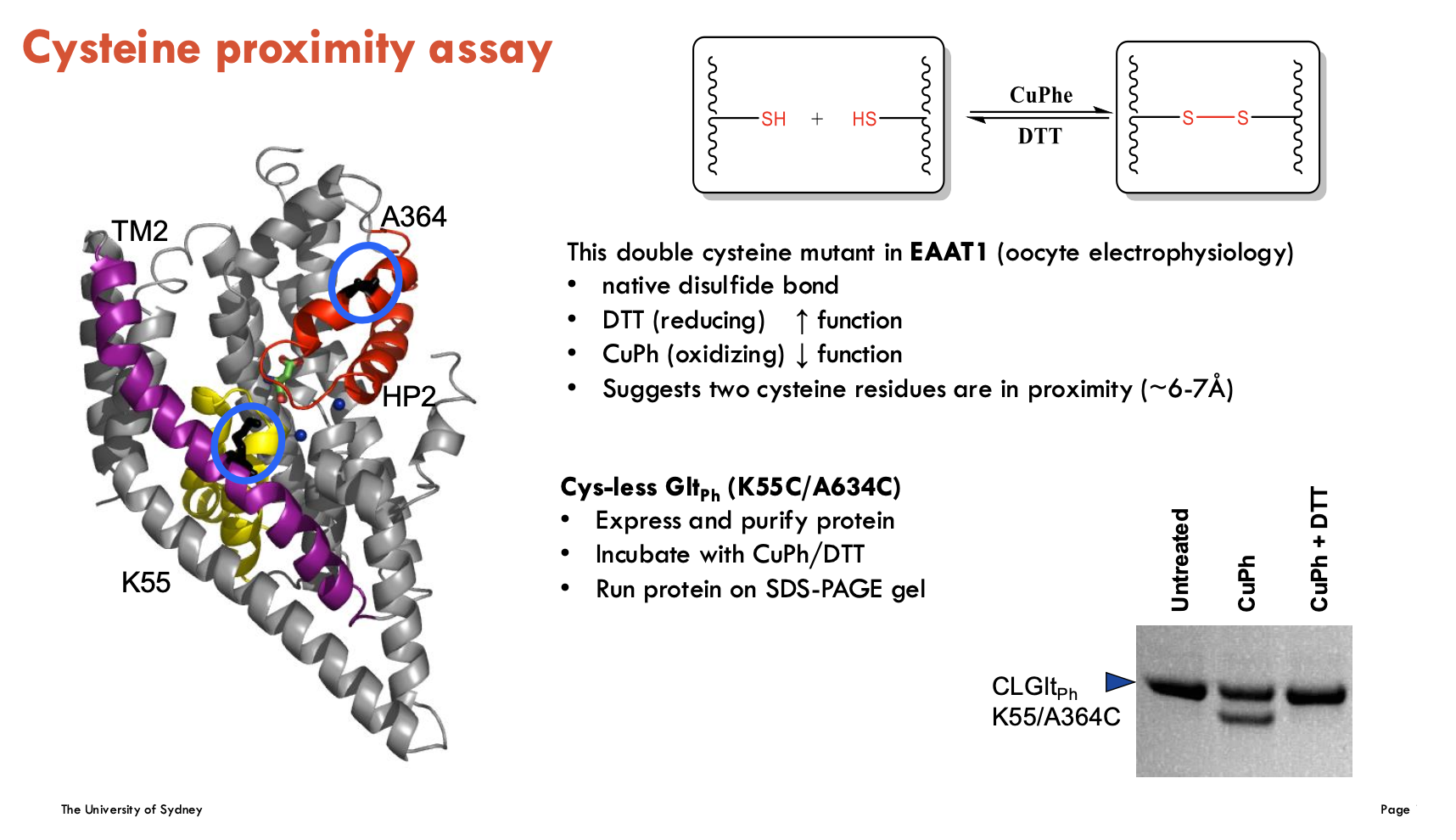
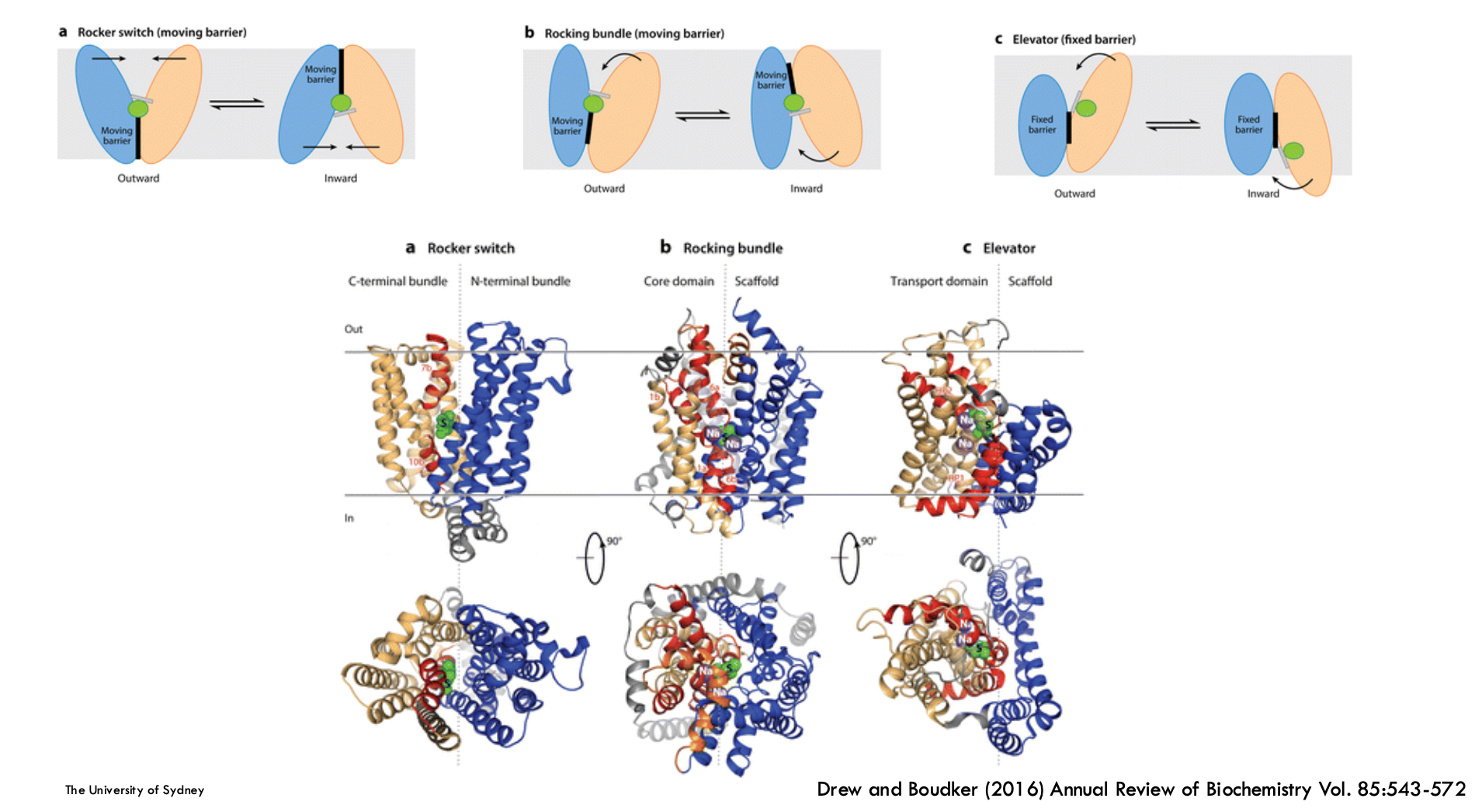
What is the elevator mechanism of transport?
The elevator mechanism of transport is when part of a transporter protein (the "transport domain") moves like an elevator through the membrane, carrying the substrate from one side to the other, while the rest of the protein (the "scaffold domain") stays still.
allows substrate to be moved across the membrane in a controlled, stepwise way
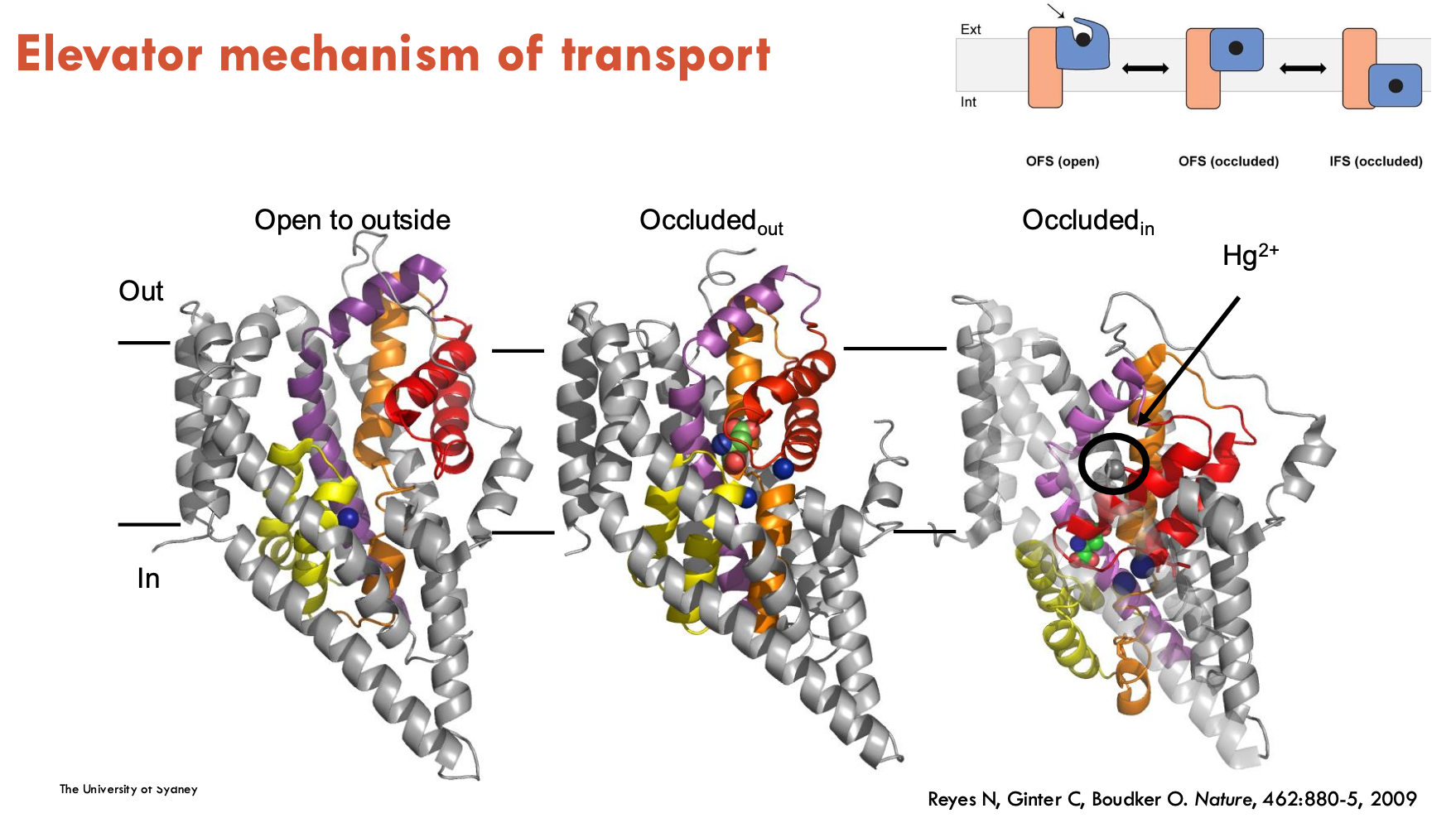
What is the mechanism of Gltph?
Gltph is the first elevator transporter
Binding: Sodium ions and substrate (like aspartate) bind to the transport domain on the outside of the membrane.
Elevator movement: The transport domain slides down through the membrane like an elevator, carrying the bound substrate to the inside.
Release: Substrate and sodium are released inside the cell.
Reset: The empty transport domain moves back up to the original position to start another cycle.
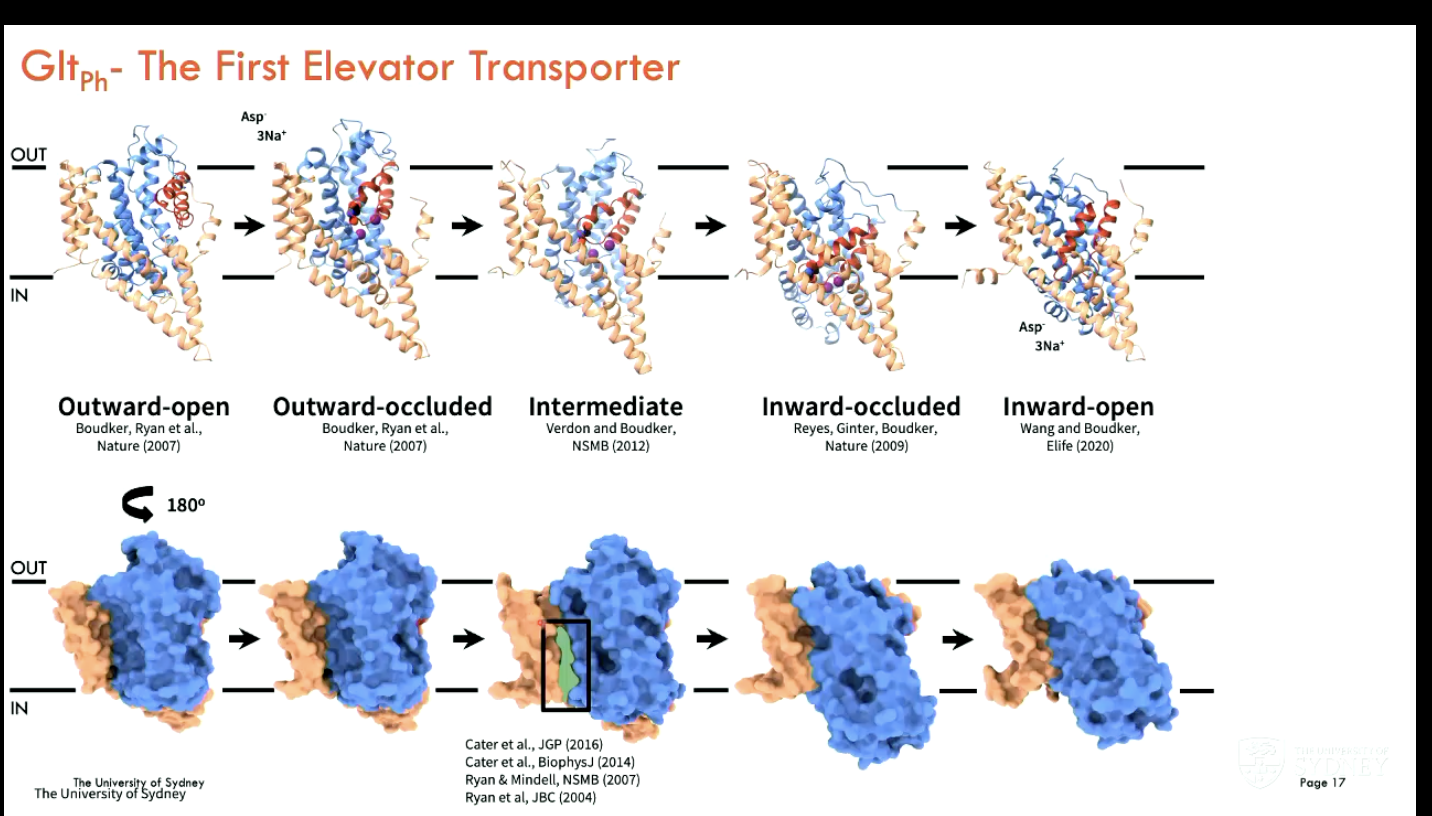
In which conformational states can Gltph exist (4)?
Outward-Facing State (OFS)
Transport domain open to extracellular space
glutamate + 3 Na⁺ ions bind
Occluded State
Substrate is trapped inside; binding site is closed off from both sides.
Prevents premature release or leak.
Inward-Facing State (IFS)
Transport domain shifts inward
Releases substrate/ions into cytoplasm
Cl⁻ Conducting State
Uncoupled from transport, forms a passive Cl⁻ channel
Triggered during intermediate conformations (likely between iOFS and IFS).
Role: charge balance and membrane potential regulation
Excitotoxicity (neuron damage in synaptic cleft due to excessive glutamate) is the link between (A) and (B)
A - Na+/K+-ATPase
B - EAATs
Glucose transporter GluT1 belong to (transporter family)
Major Facilitator Superfamily (MFS) of secondary active transportes
largest of all transporter families
can be uniporters, symporters, or antiporters
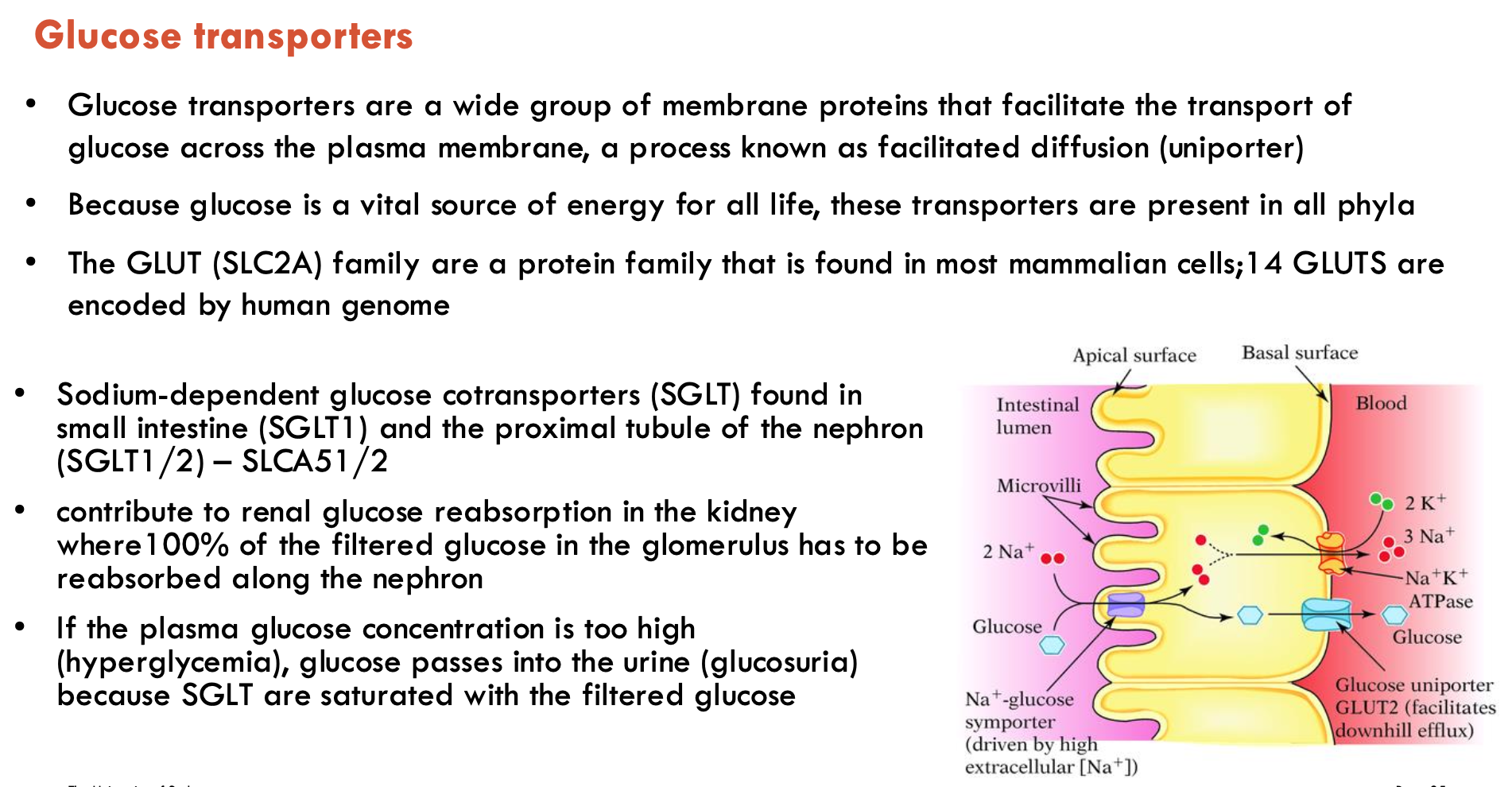
What are the two types of glucose transporters?
Two types:
GLUTs (facilitated diffusion) - 14 human isoforms
SGLTs (sodium-dependent) - 2 key subtypes (SGLT1 - small intestine, SGLT2 - proximal tubule)
If plasma glucose is too high, glucose passes into urine because SGLTs are saturated with filtered glucose → glucose in urine
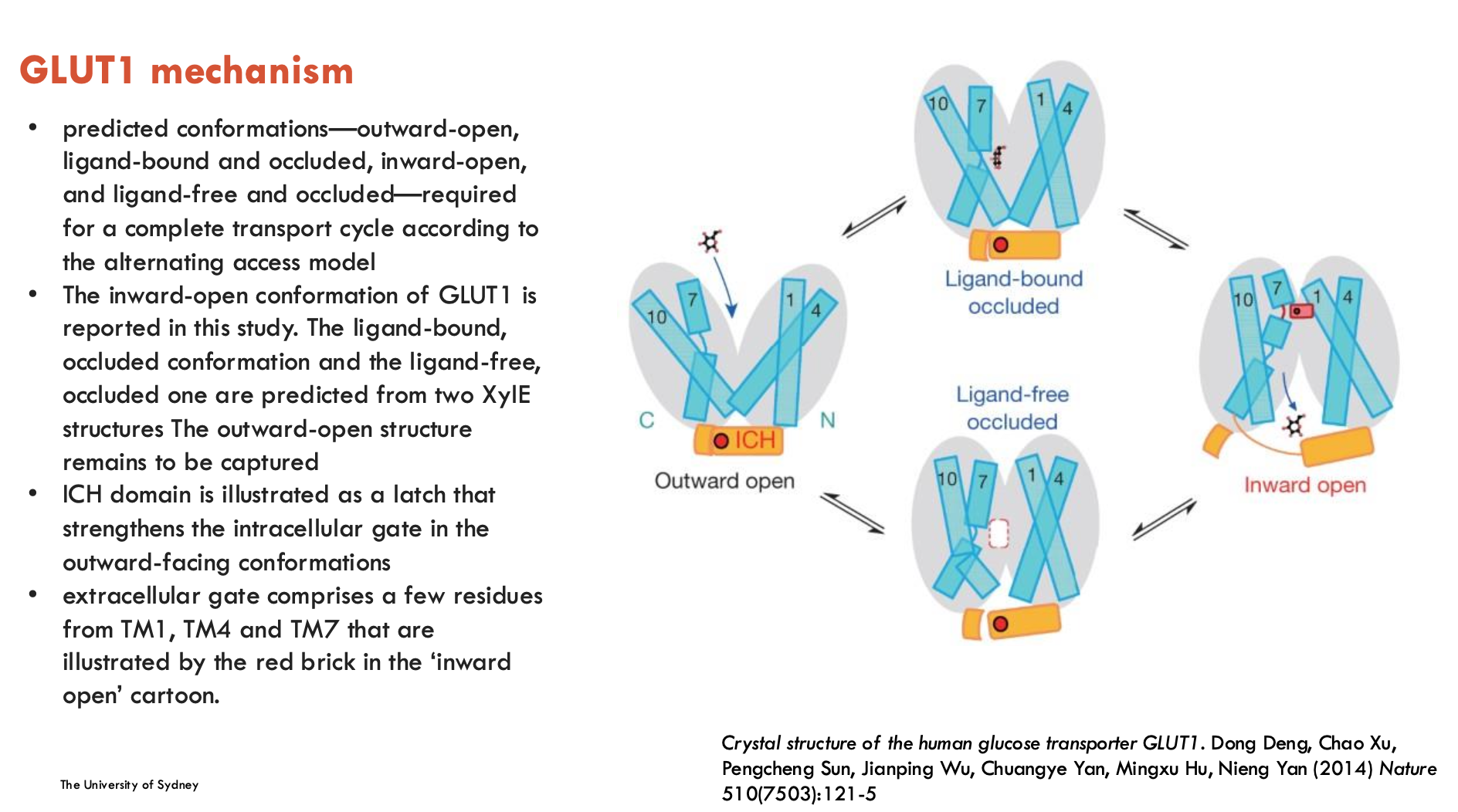
Describe the structure of GLUT1
12 TMD (2 × 6 bundle)
inward open conformation
N- and C- domains connected by intracellular helical bundle (ICH)
mostly unique to sugar transporters
latch that tightens the intracellular gate
Describe the structural mechanisms of GLUT1
Extracellular Gate: Formed by residues from TM1, TM4, TM7, controlling access to the binding site.
ICH (Intracellular helices) Domain: Latch that strengthens the intracellular gate in the outward-facing state.
Conformations:
Outward-open
Inward-open
Ligand-bound, occluded
Ligand-free, occluded
Alternating Access Model: Substrate-binding site switches between outside and inside of the membrane.
What is the neurotransmitter sodium symporter family?
NSS is a large family of NT transporters, includes:
glycine, GABA, monoamines (dopamine, NA, and serotonin)
All human subtypes are coupled to co-transport of Na+ and Cl-
They are targets for many therapies or drug abuse
bacterial homologue from LeuTAa has been crystallised and is a model of the structure of all transporters in this family
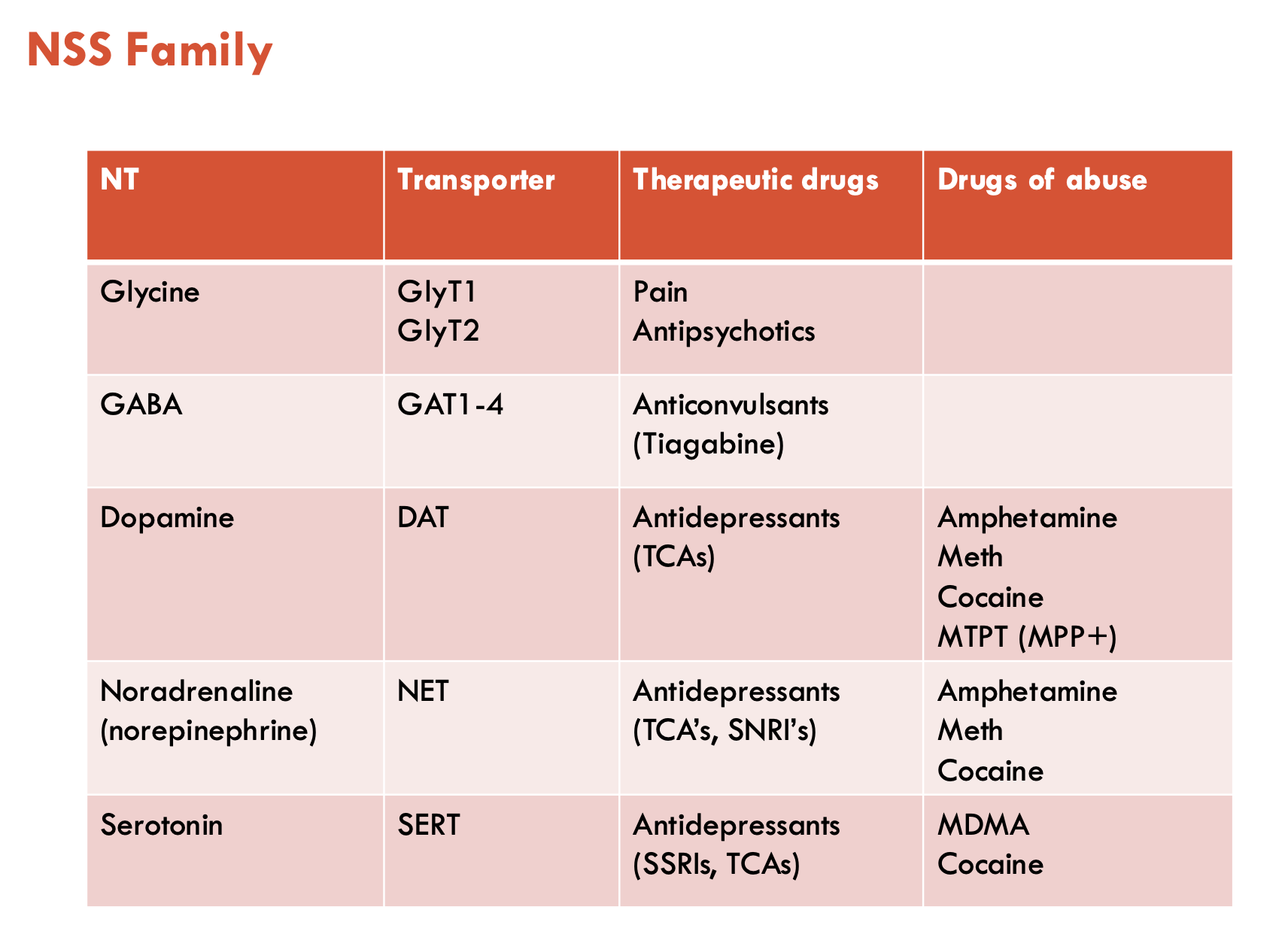
What are the roles of GlyT1 and GlyT2 in inhibitory and excitatory neurotransmission?
Inhibitory (GlyT2)
GlyT2 is found exclusively on inhibitory synapses as glycine is an inhibitory NT
Glycine receptors (GlyRs) are ligand-gated Cl- channels (not a transporter)
GlyT2 clears glycine from synapse and uptakes it into pre-synaptic neuron
Excitatory (GlyT1)
glutamate is released and act on NMDAR and AMPAR receptors
these receptors also require glycine to bind as a co-agonist
GlyT1 transports glycine from astrocytes for excitatory NT
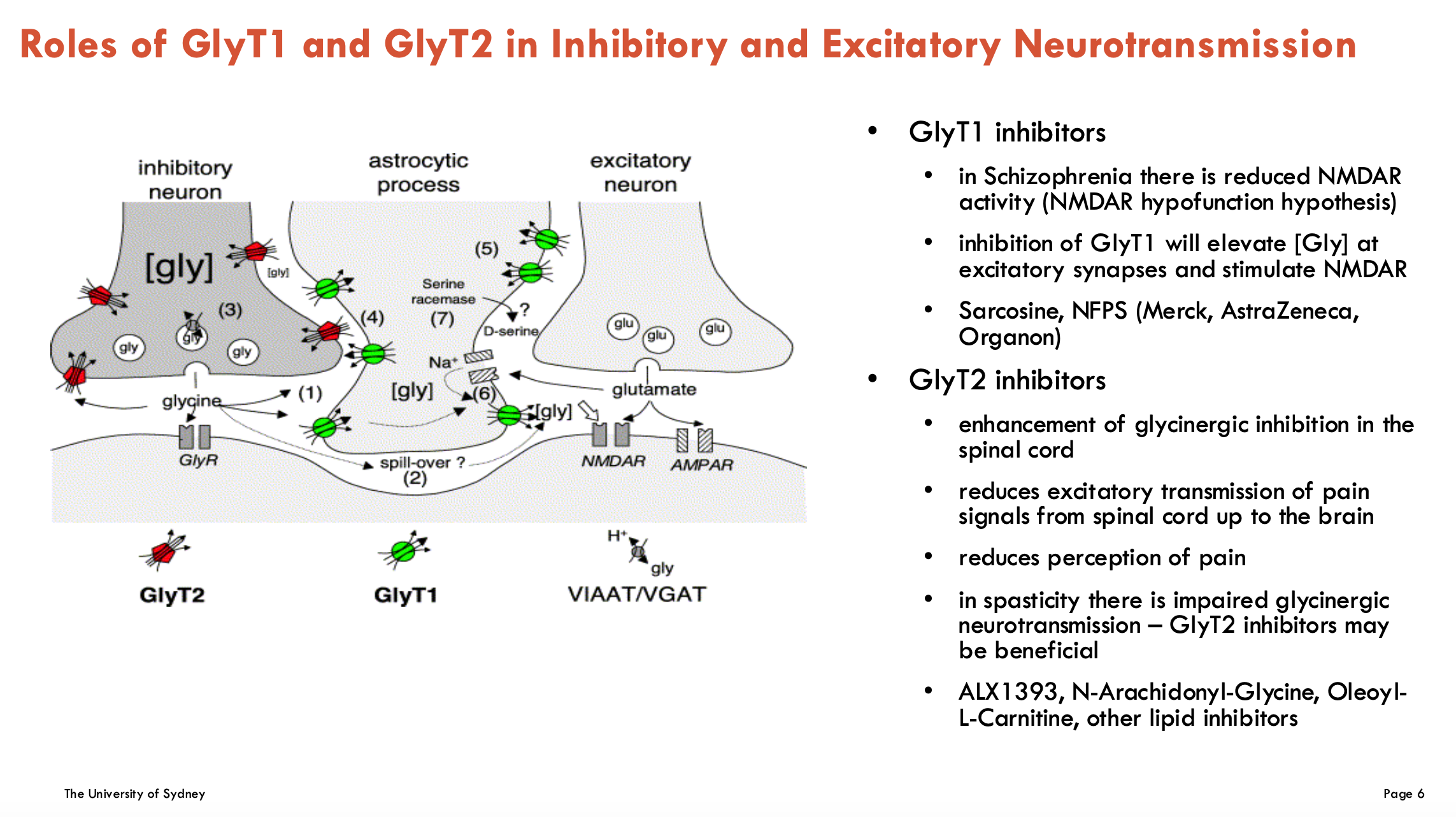
What could inhibition of GlyT1 and GlyT2 be used for?
GlyT1 inhibitors (↑ glycine → ↑ NMDA-R activity)
Target: Schizophrenia (NMDAR hypofunction)
Mechanism: ↑ Synaptic glycine → boosts NMDA-R signaling
Examples: sarcosine, NFPS (Merck, AstraZeneca, Organon)
GlyT2 inhibitors (↓ glycine reuptake → ↓ pain signalling)
Targets:
Chronic pain (spinal cord GlyT2 blockade → ↓ excitatory pain transmission)
Spasticity (restores glycinergic inhibition in motor pathways)
Examples: ALX1393, N-Arachidonyl-Glycine, Oleoyl-L-Carnitine, other lipid inhibitors

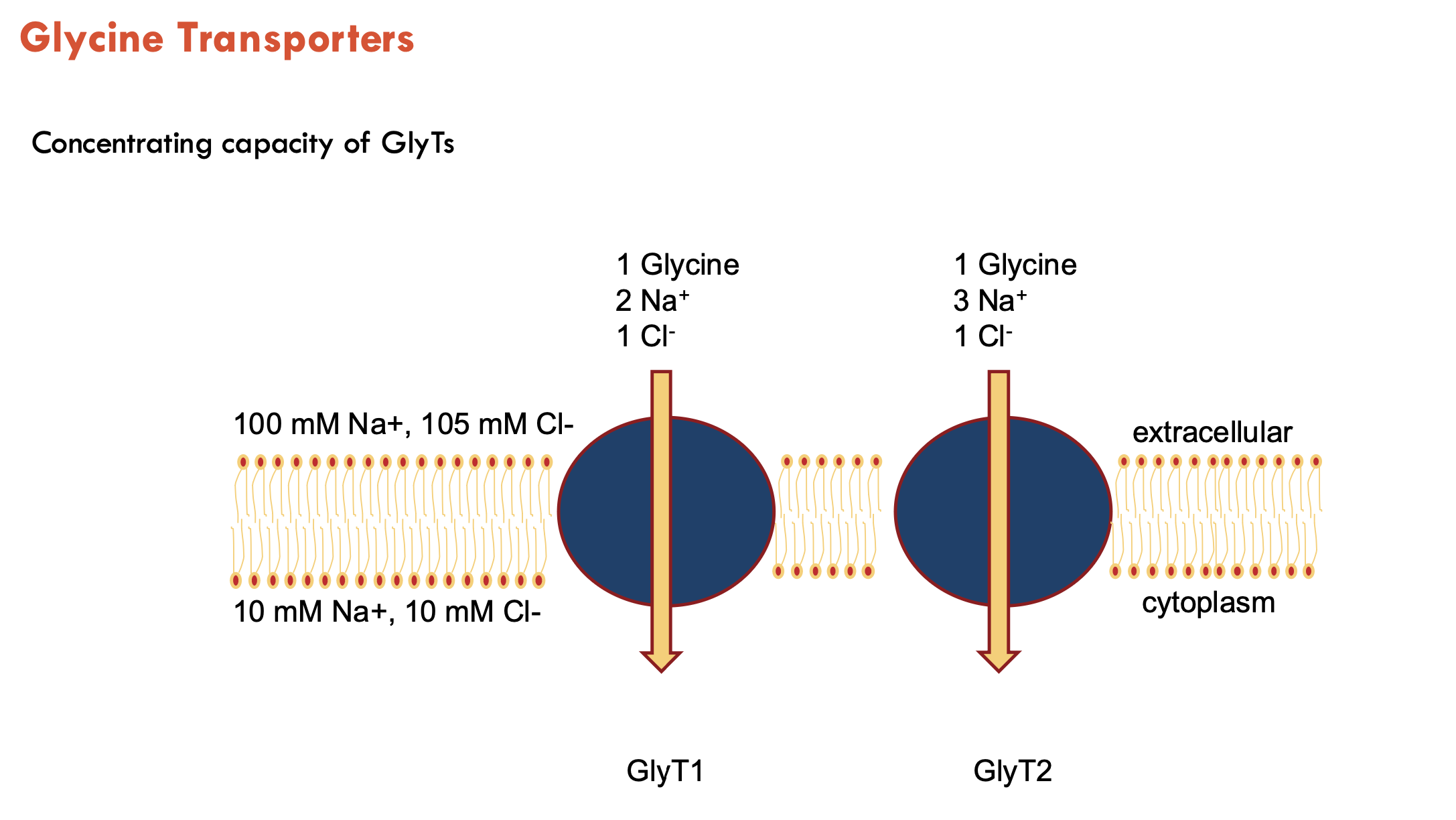
What are the two subtypes of glycine transporters?
GlyT1 and GlyT2
transports Na+ and Cl-
GlyT1 uses 2 Na+, GlyT2 uses 3 Na+ (stronger driving force → lower extracellular glycine)
What is the issue with current glycine transport inhibitors and how can it be addressed?
They are analgesic in chronic pain rodent models BUT have side effects - thought to be due to irreversible, competitive binding
→ without glycine, you can’t breathe, so you need some
Alternative: bioactive lipids are “atypical” GlyT2 inhibitors
comprised of AA head and a monounsaturated tail
both the head and tail group are required for inhibition
are non-competitive
some transport of glycine still remains even at high concentrations of inhibitor
has potential to be useful in reducing pain signals with no serious side effects
Describe the binding of bioactive lipids to GlyT2
Bioactive lipids do not bind in the vestibule allosteric site
act through extracellular allosteric site
spontaneously binds to extracellular allosteric site in molecular dynamic simulations
Describe the ion coupling of catecholamine and monoamine transporters
DAT, NET, and SERT are all slightly different (do not need to know in detail)
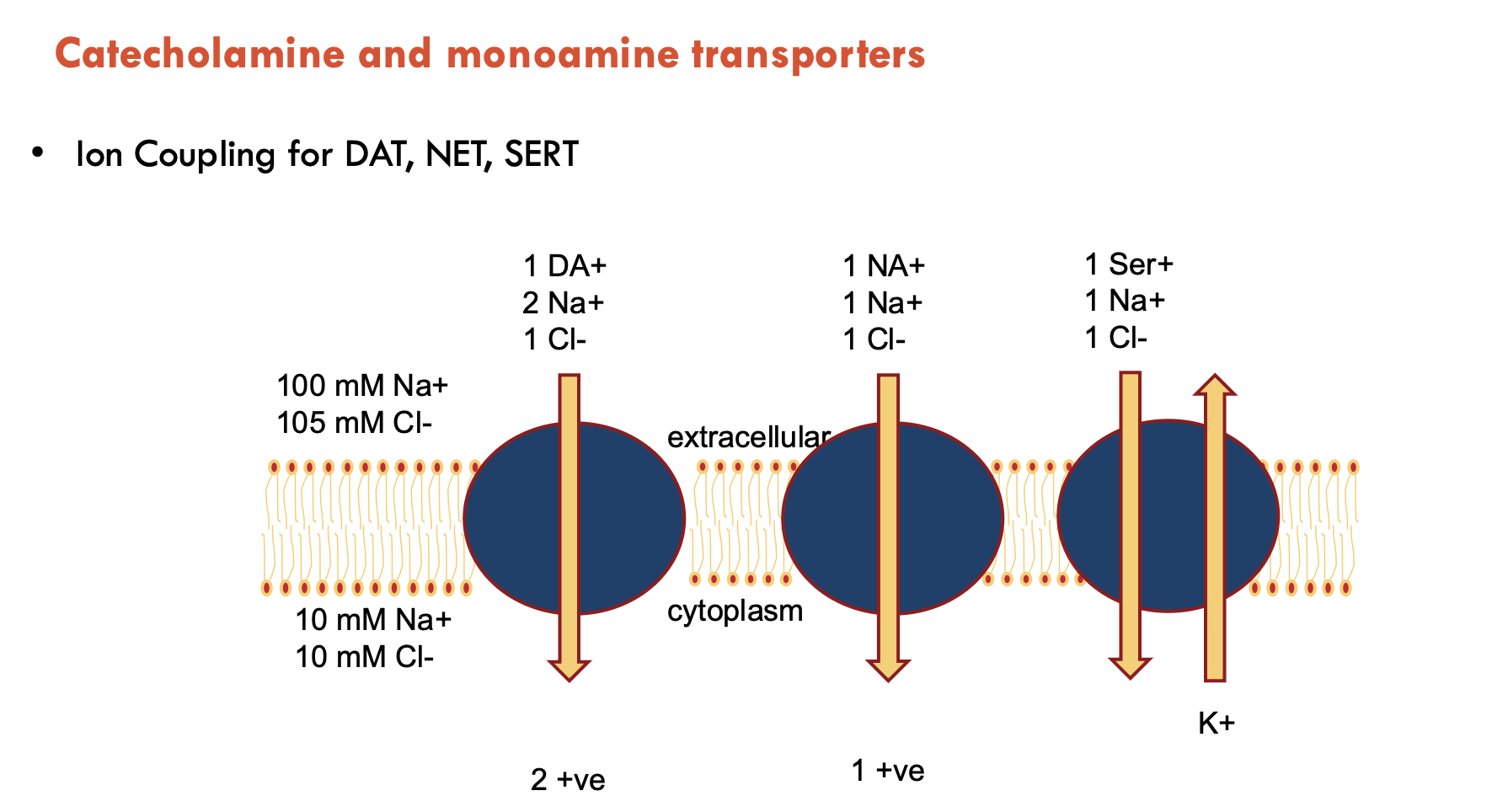
Features of dDAT
dDAT is the Drosophila melanogaster dopamine transporter (dDAT)
has 12 TM domains
shot glass shape
substrate buried
co-transports 2 Na+ and 1 Cl- ion
Substrate and Na+ sites are similar in all members of the NSS family
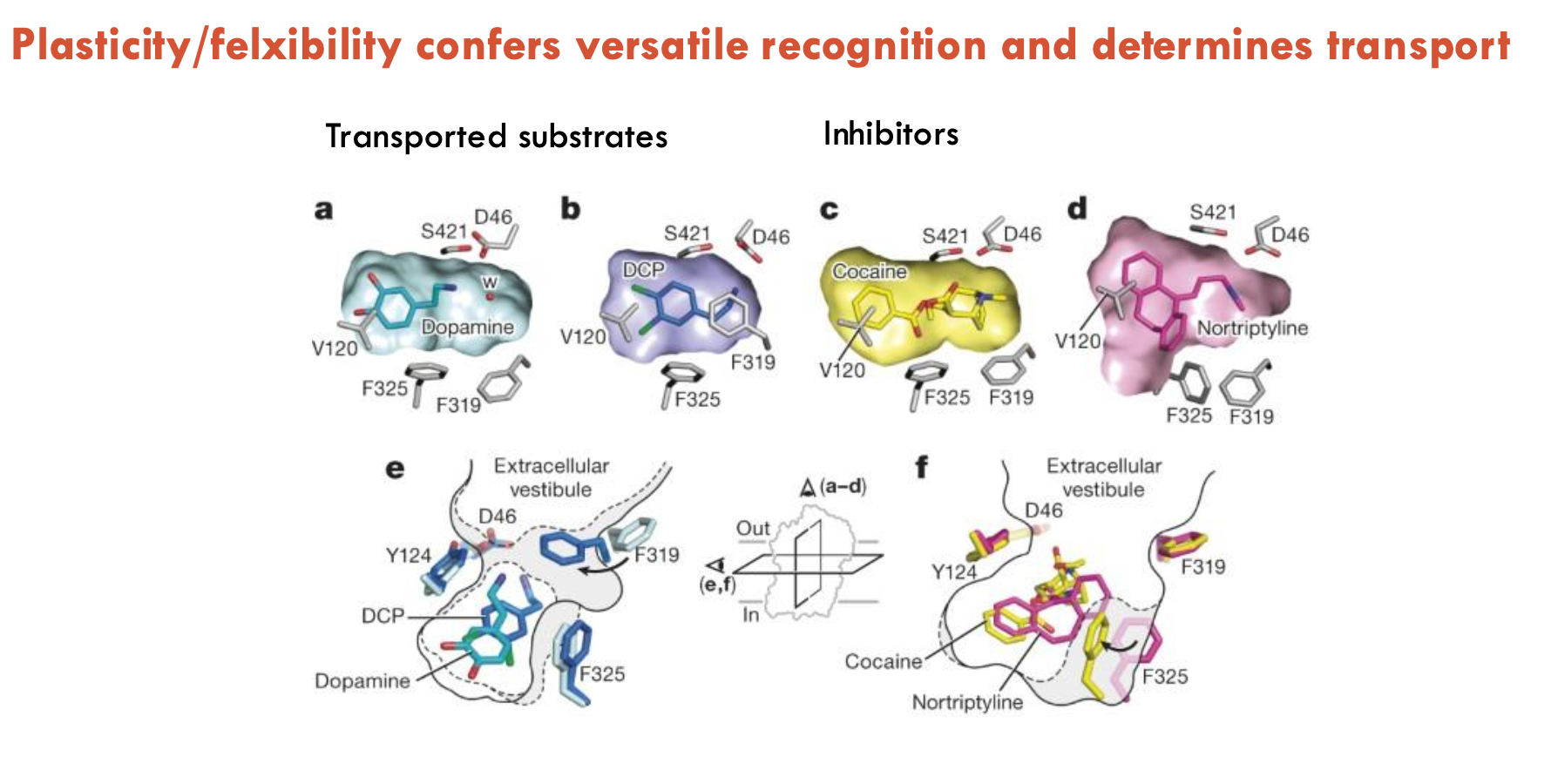
Plasticity/flexibility confers (A) and determines (B)
A - versatile recognition
B - transport
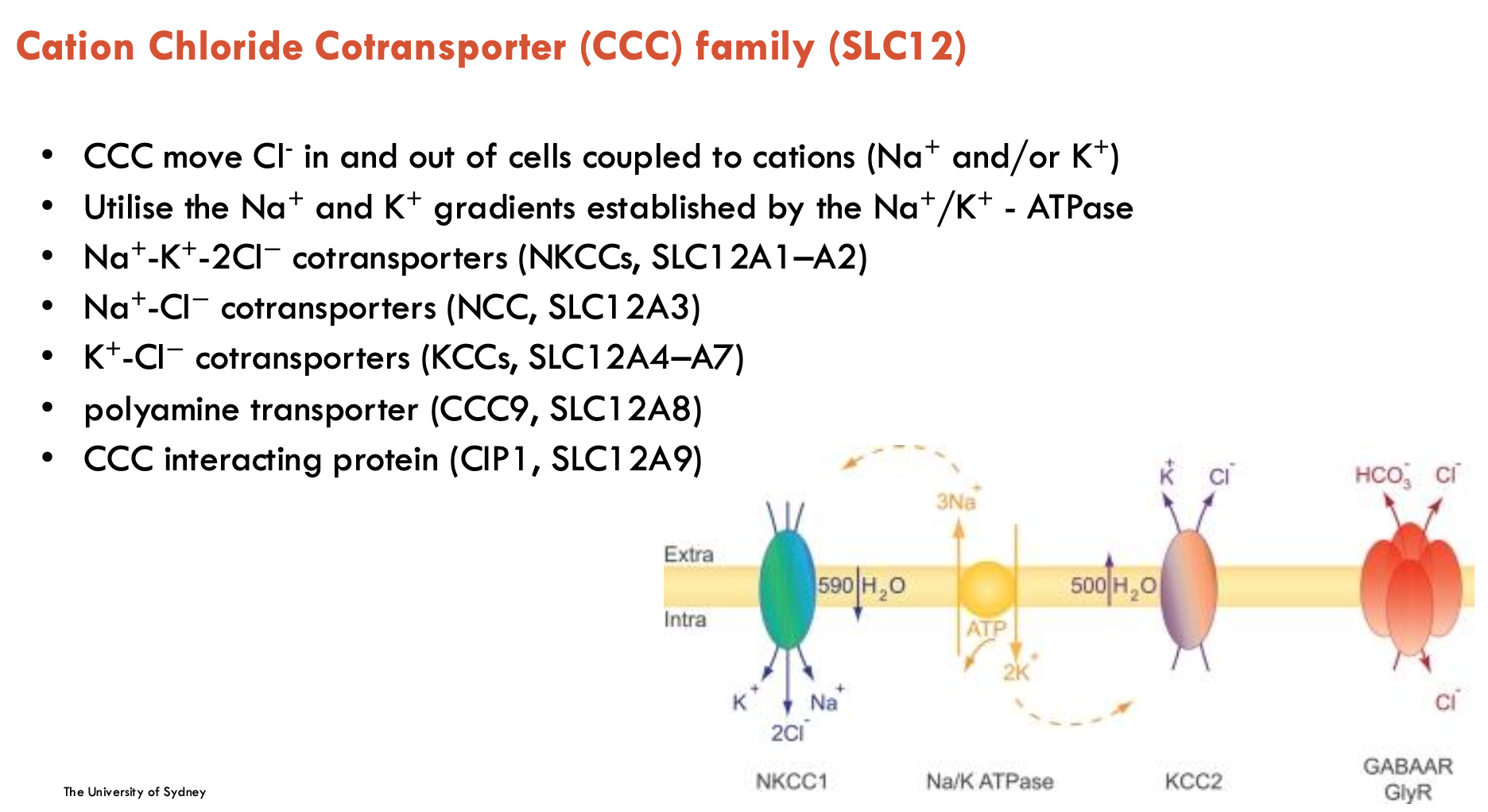
Describe the CCC family
CCC refers to the Cation Chloride Cotransporter family (SLC12)
moves Cl- in and out of cells coupled to cations (Na+ and/or K+)
uses Na+ and K+ gradients established by the Na+/K+-ATPase
Has many different types (only first two to know in detail):
Na+-K+-2Cl- co-transporters (NKCCs, SLC12A1-A2)
Na+-Cl- co-transporters (NCC, SLC12A3)
K+-Cl- co-transporters (KCCs, SLC12A4-A7)
polyamine transporter (CCC9, SLC12A8)
CCC interacting protein (CIP1, SLC12A9)
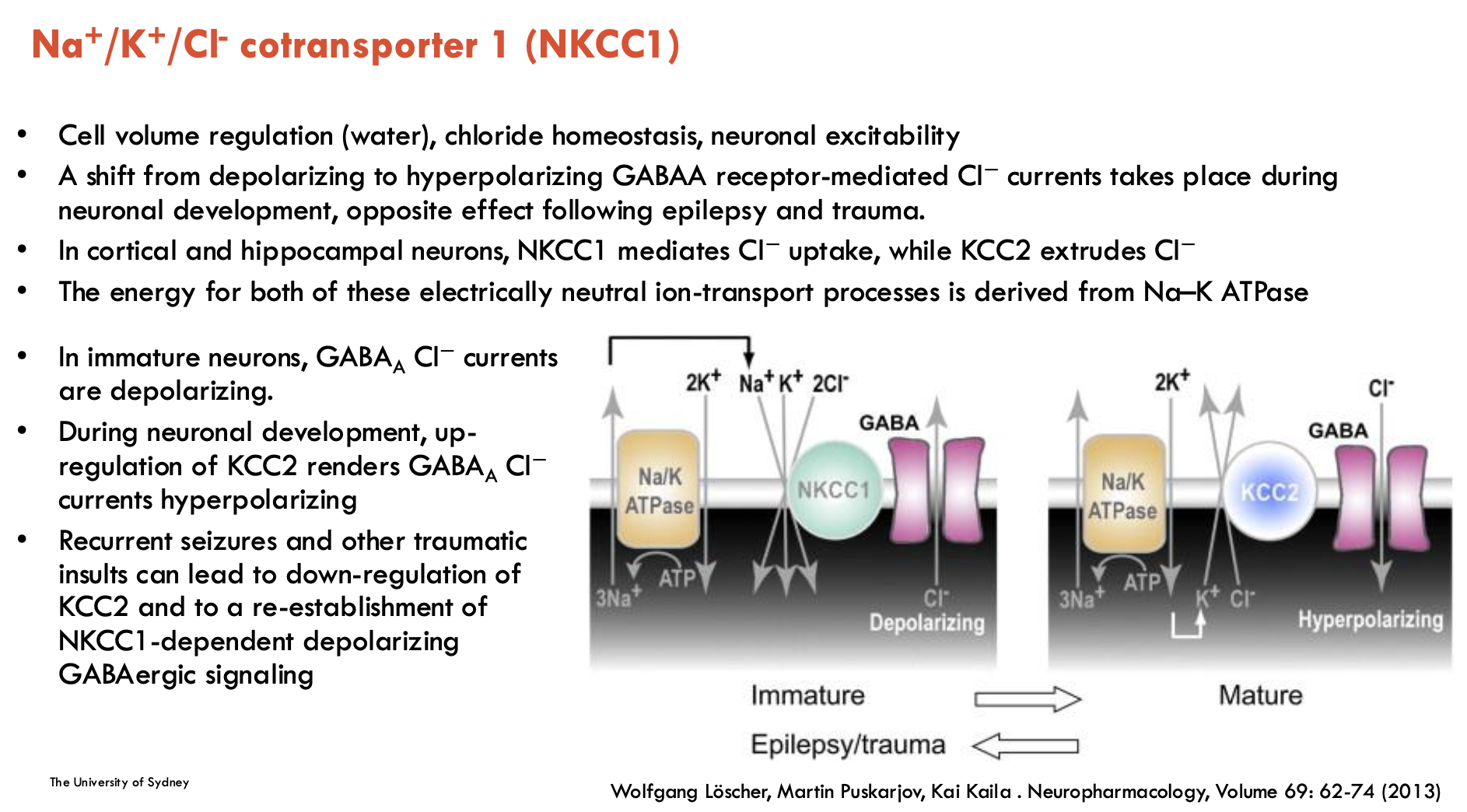
What is NKCC1 and its role?
NKCC1 is Na+/K+/Cl- co-transporter 1
responsible for cell volume regulation (water), Cl- homeostasis, GABAergic signalling
driven by Na+ gradient from Na+/K+-ATPase pump
Developmental Shifts
Immature Neurons: NKCC1 imports Cl⁻, making GABA depolarising (excitatory)
Mature Neurons: KCC2 replaces NKCC1 and exports Cl⁻, making GABA hyperpolarising (inhibitory)
Pathology: NKCC1 upregulated after epilepsy/trauma, which makes GABA excitatory again → contributes to seizures
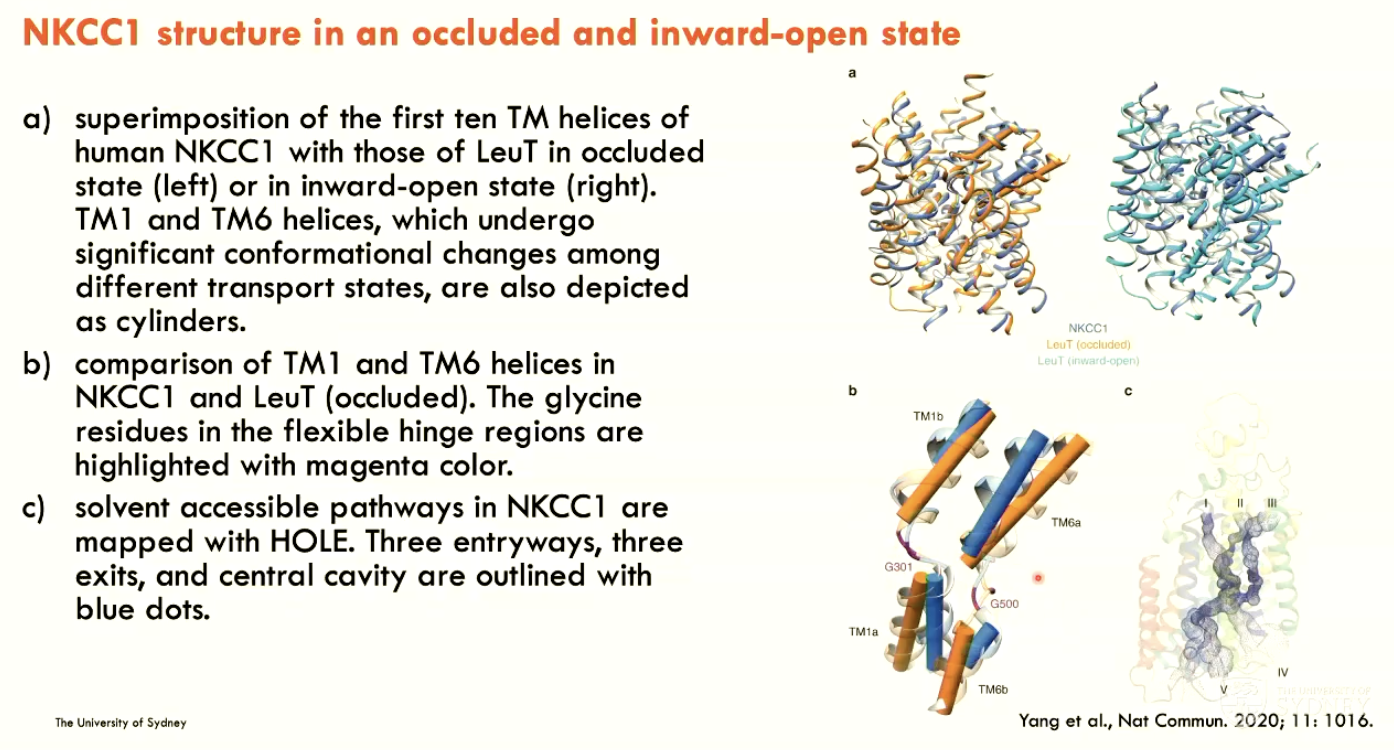
How was human NKCC1 structure determined?
By single-particle cryo-EM
Describe the structure of NKCC1
In an occluded and inward-open state
Membrane protein fold is highly linked to…
Mechanism (e.g. rocker switch, rocking bundle, elevator)
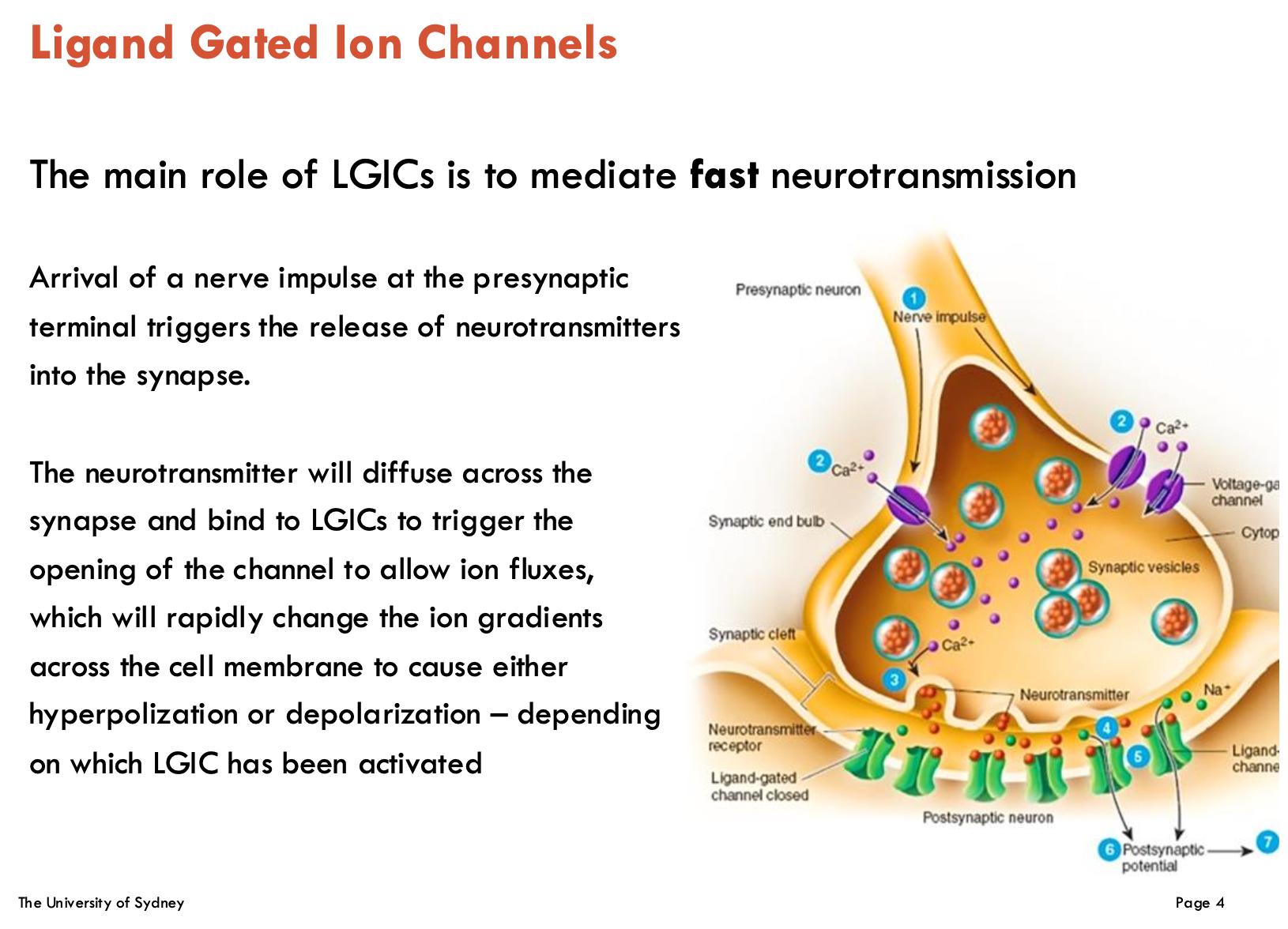
What is the main role of LGICs?
Fast neurotransmission
NTs diffuse across synapse and bind to LGICs to trigger channel openings → ion fluxes (change ion gradients across cell membrane)
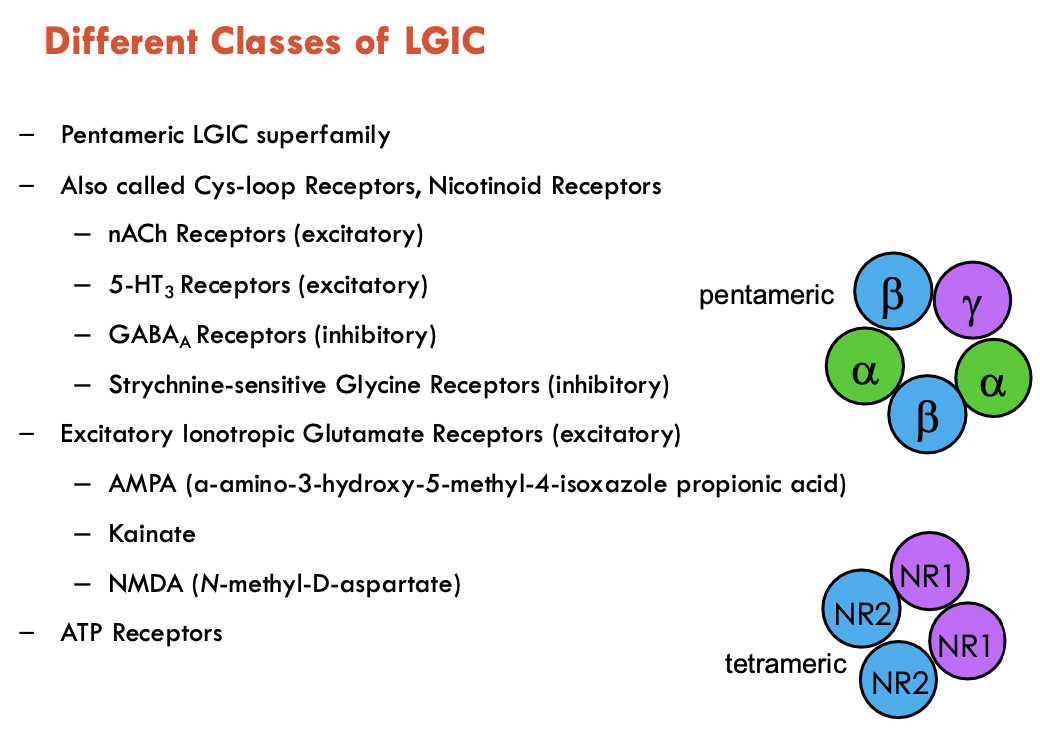
Identify the two main classes of LGICs
Pentemeric LGIC Superfamily (aka Cys-loop Receptors, Nicotinoid Receptors)
nACh receptors (excitatory)
5-HT3 Receptors (excitatory)
GABAA Receptors (inhibitory)
Strychnine-sensitive Glycine Receptors (inhibitory)
Tetrameric Excitatory Ionotropic Glutamate Receptors
AMPA (a-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid)
Kainate
NMDA (N-methyl-D-aspartate)
There are also ATP receptors (not covered in lecture)
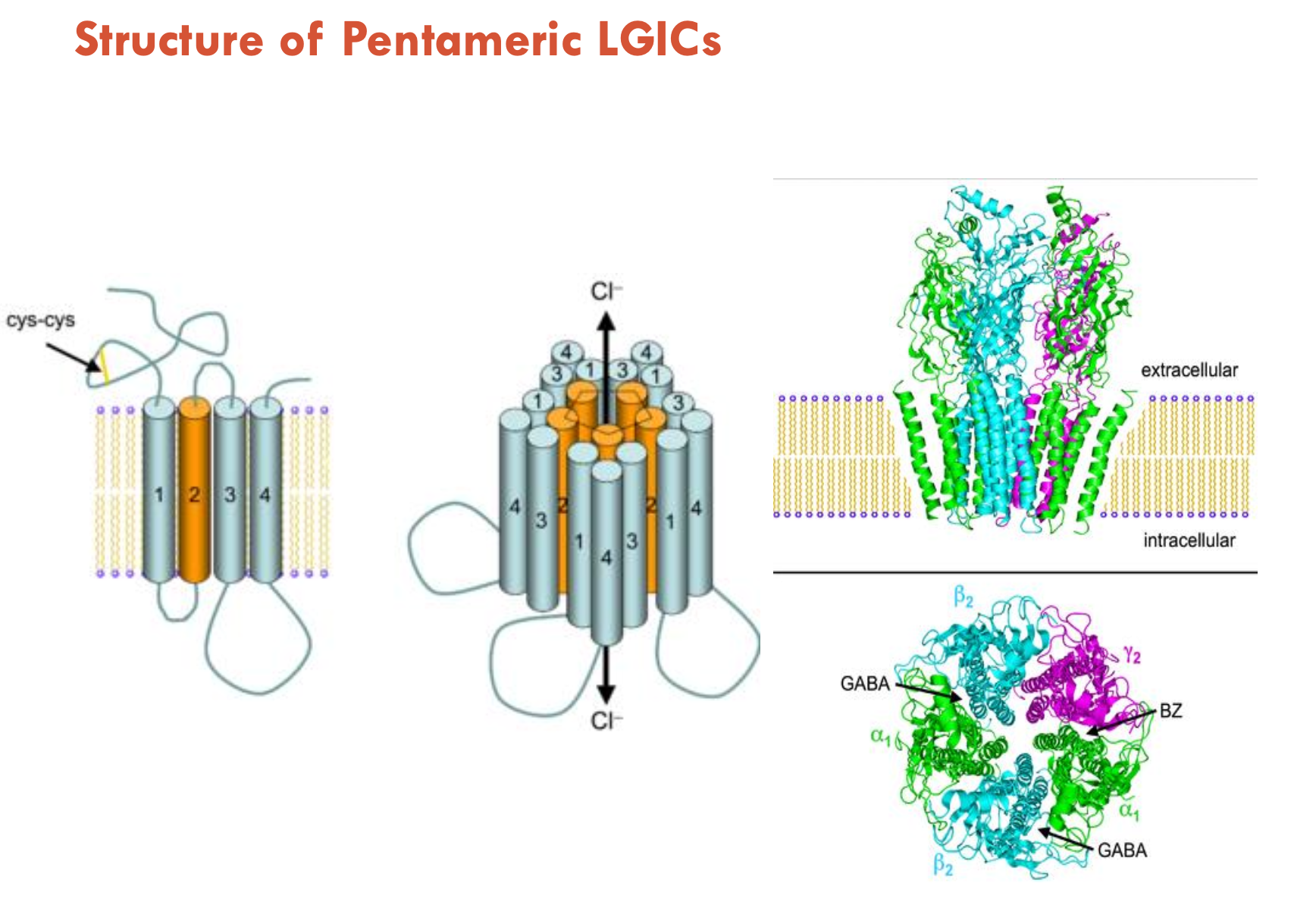
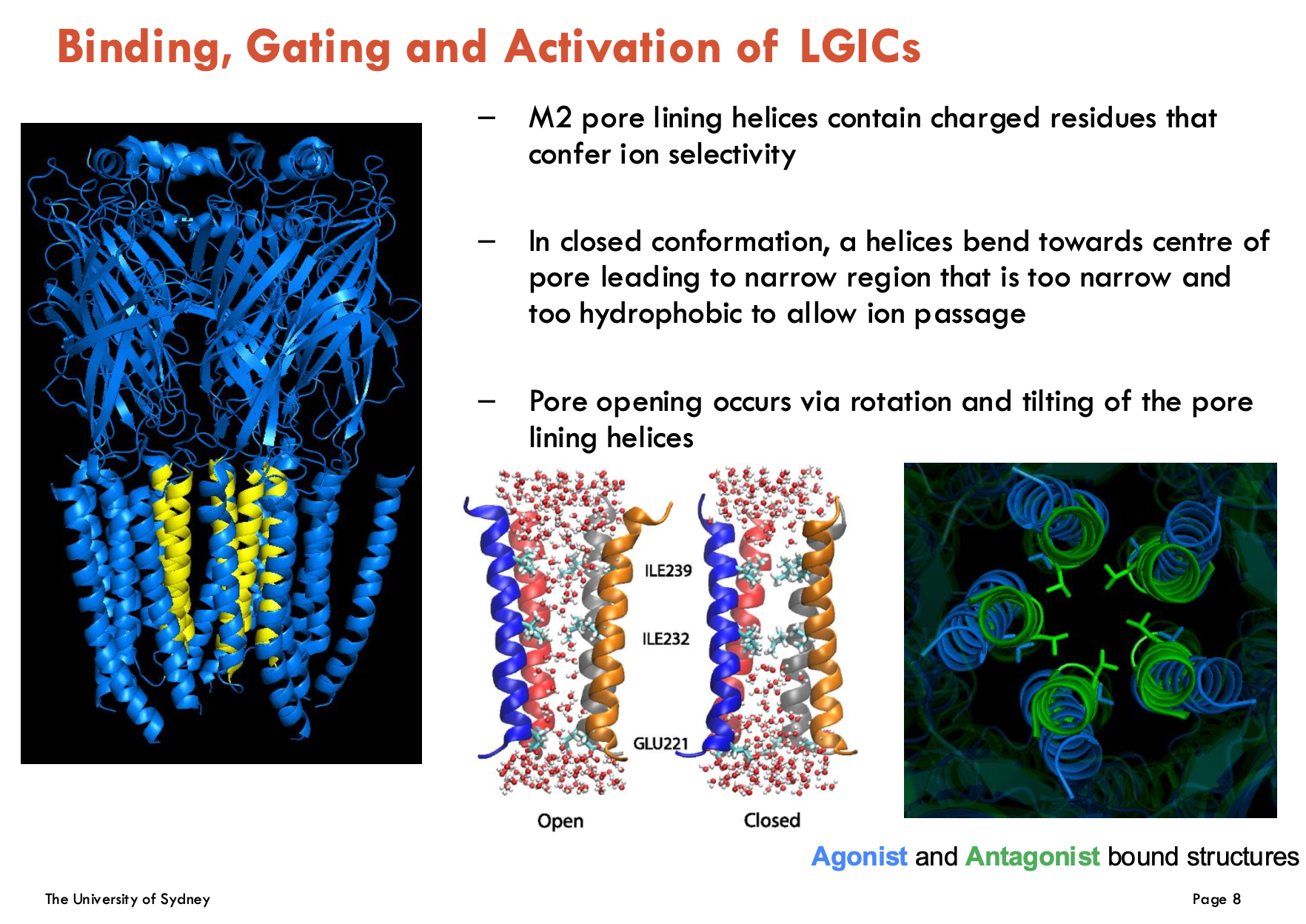
Describe the structure of pentameric LGICs
TM proteins consisting of 5 subunits
each subunit has an extracellular (blue in diagram) and transmembrane (yellow in diagram) domain
How is ion passage controlled in LGICs?
M2 pore lining helices contain charged residues that confer ion selectivity
In closed conformation, α helices bend towards centre of pore → leads to narrow region that is too narrow and hydrophobic → does not allow ion passage
Pore opening occurs via rotation and tilting of the pore lining helices
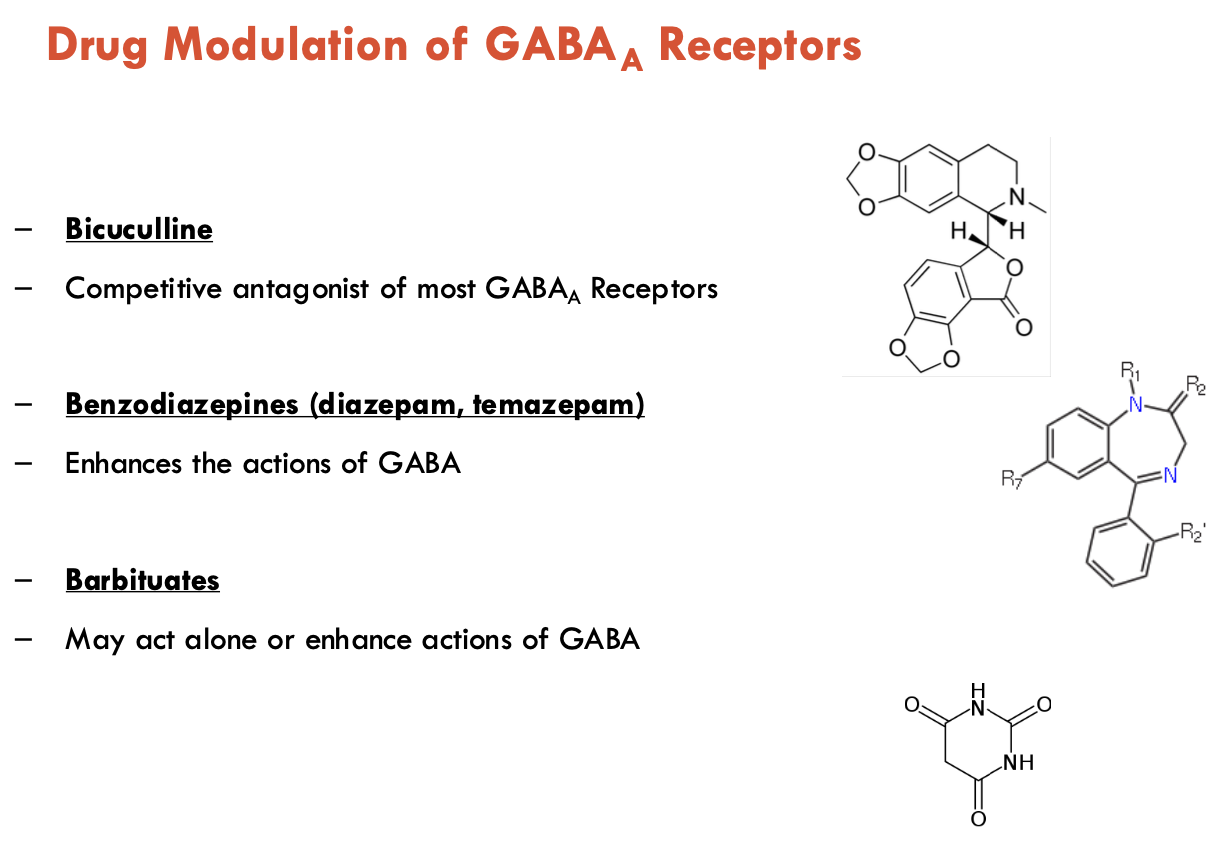
Identify 6 drug modulations of GABAA receptors
Bicuculline: competitive antagonist → blocks GABA binding
Benzodiazepines (diazepam, temazepam): Positive allosteric modulators (PAMs) → ↑ GABAA efficacy
Barbituates: PAMs, but also direct activator at high doses
General Anaesthetics (e.g. propofol): PAMs, but also direct activator at high doses
Ethanol: Prolongs channel opening → enhances GABAA currents
Neurosteroids (e.g. allopregnanolone): Promote channel opening (especially α/δ-containing receptors)
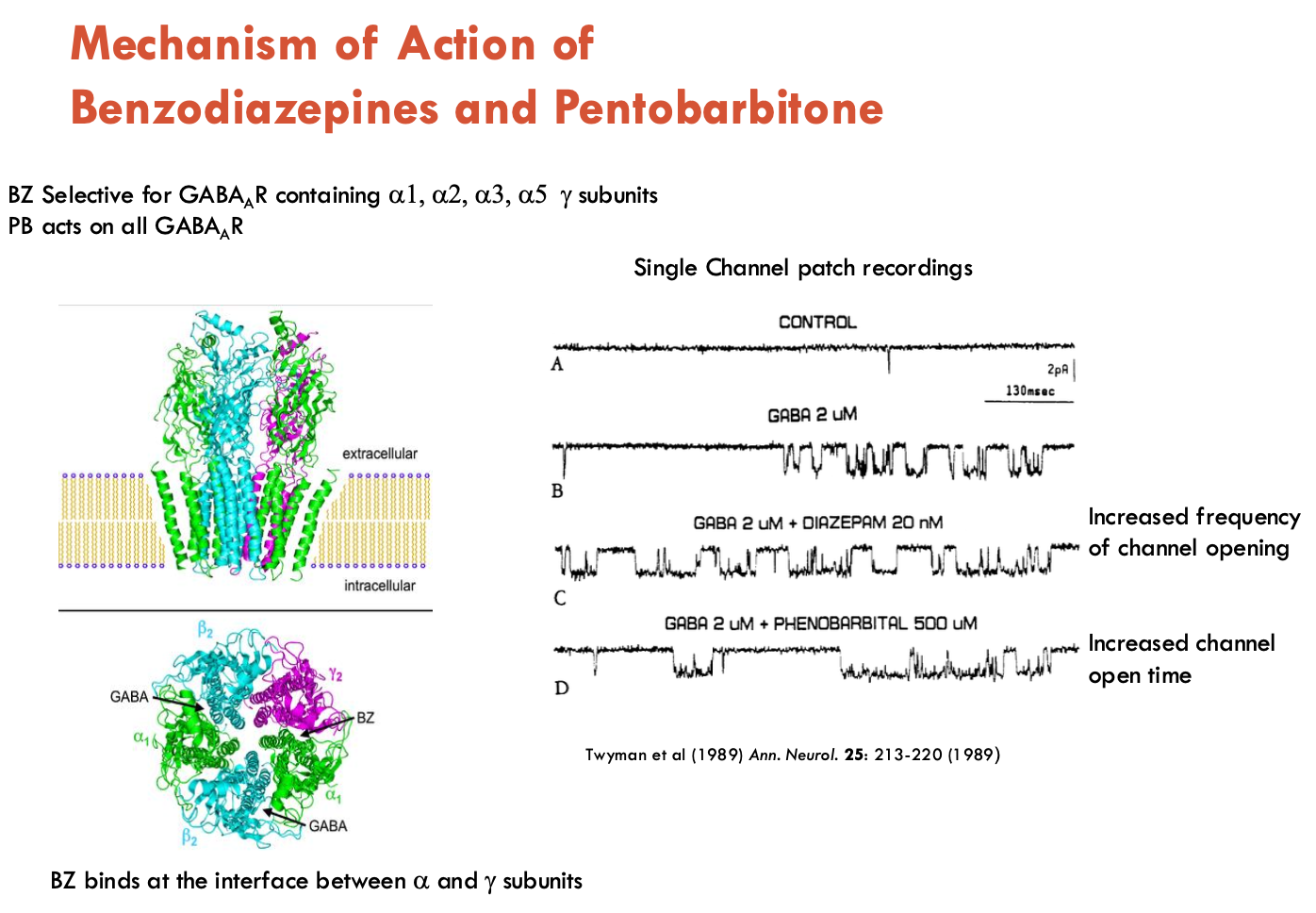
What is the MOA of benzodiazepines and pentobarnitone?
Benzodiazepines (e.g. diazepam)
Target: GABAA receptors with α₁/α₂/α₃/α₅ + γ subunits.
Binding Site: Interface of α and γ subunits (allosteric)
Action:
↑ channel opening frequency → ↑ GABA’s inhibitory effects, BUT no direct activation
Relatively safe
Pentobarnitone
Target: All GABAA subtypes (no subunit selectivity)
Action:
↑ duration of GABA-induced channel openings
At high doses: Directly activates receptors (GABA-independent) → contributes to sedative and anticonvulsant effects
Risks:
Respiratory depression → lethal overdose
Tolerance/dependence
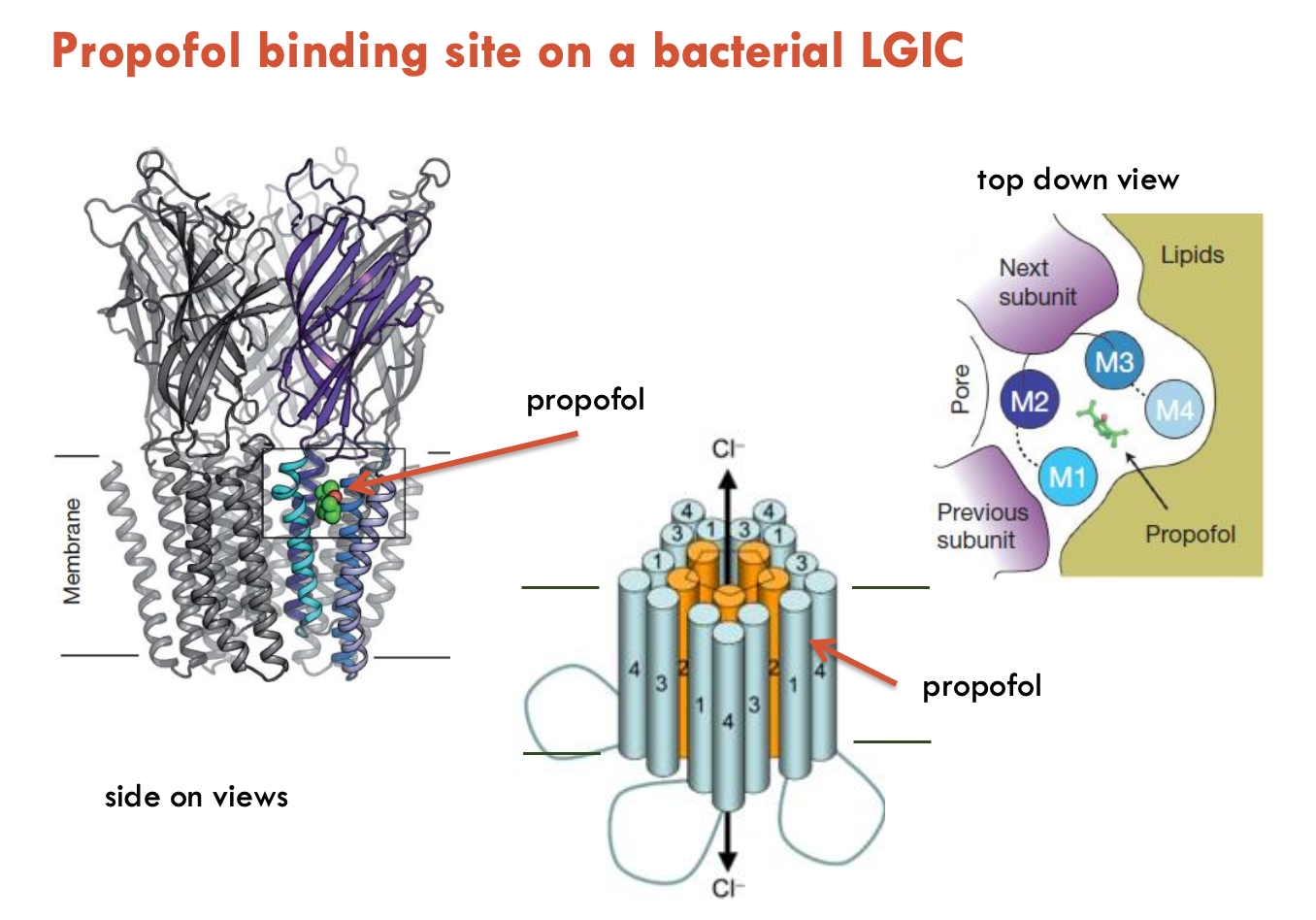
Describe the MOA of propofol on LGIC
Propofol is a general anaesthetic that binds to a distinct allosteric site (not BZ or GABA) → increases duration of Cl- channel opening → enhances GABA effects
short-acting drug
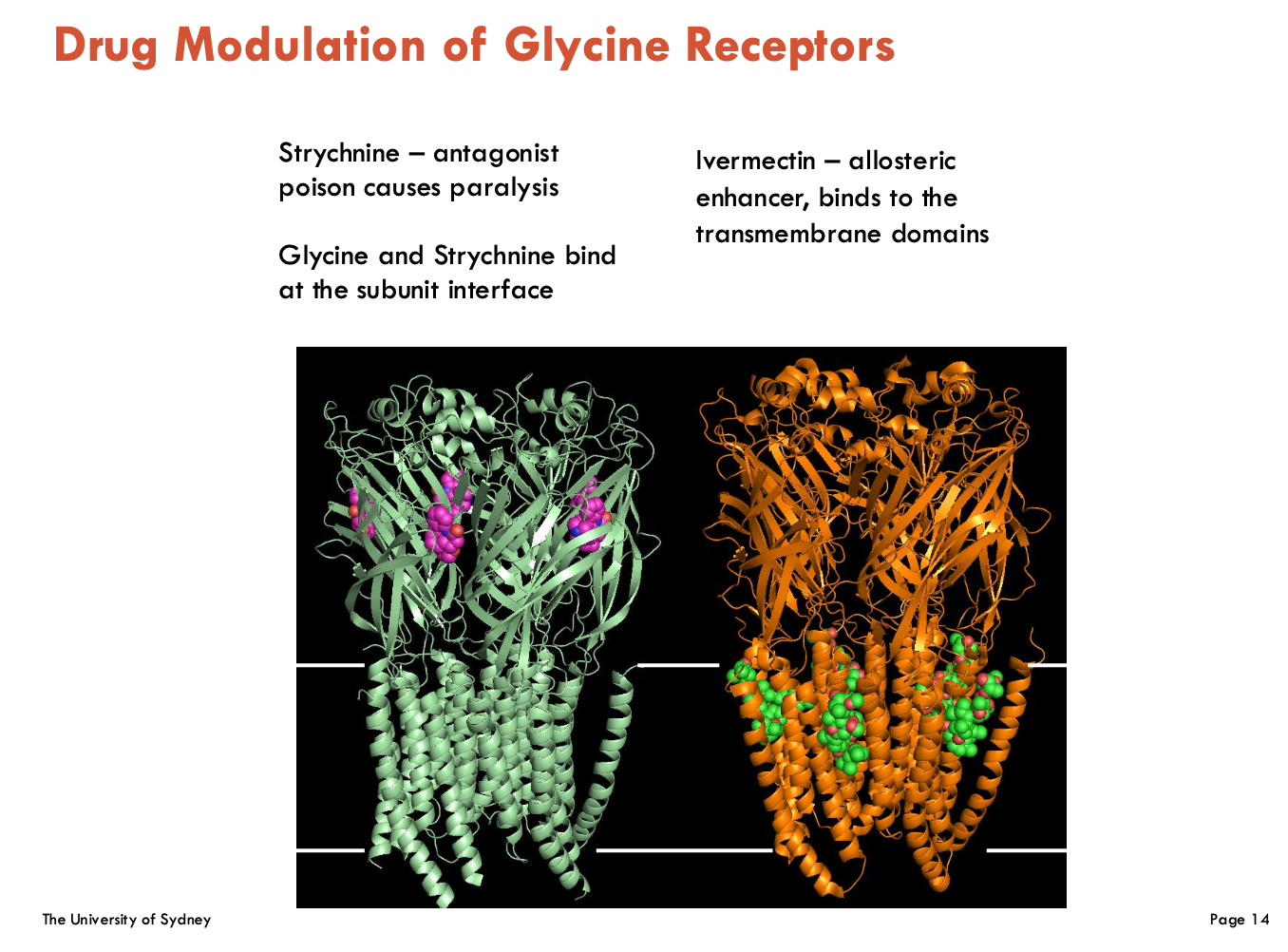
Describe drug modulation of glycine receptors (2)
Strychnine:
Competitive antagonist → binds at subunit interface, blocking glycine → stops inhibitory neurotransmission → causes uncontrolled muscle contraction (paralysis)
Ivermectin:
Allosteric agonist → binds TM domains, irreversibly locks GlyR open (permanent)
What are the subtypes of ionotropic glutamate receptors (2)?
NMDA-R: requires glutamate and glycine for activation
AMPA-R: requires only glutamate for activation
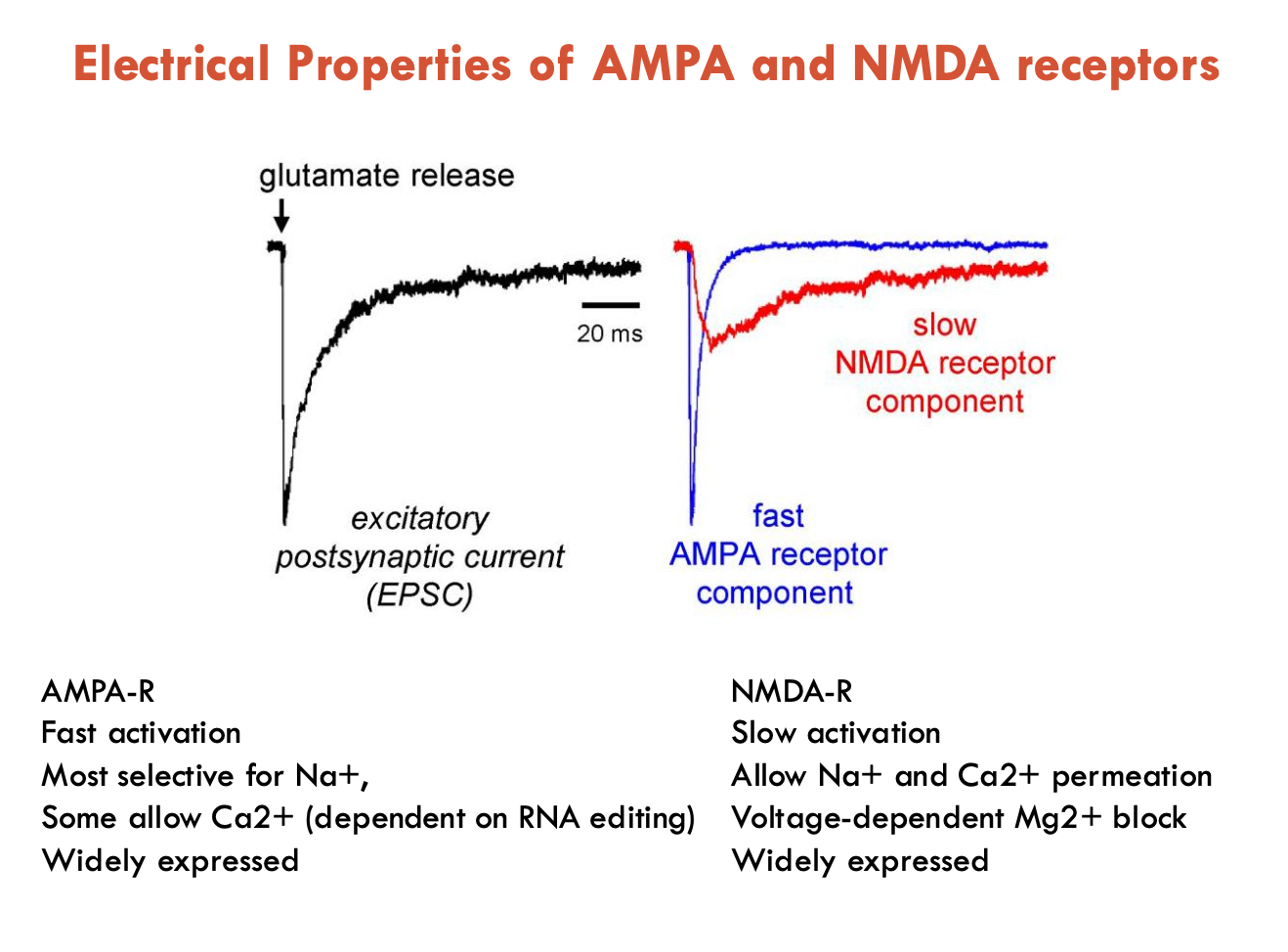
Distinguish between the electrical properties of AMPA and NMDA receptors
AMPA-R
fast activation and desensitisation (inactivation)
most selective for Na+
some allow Ca2+ (dependent on RNA editing)
widely expressed
NMDA-R
slow activation and desensitisation
allow Na+ and Ca2+ permeation
voltage-dependent Mg2+ block
widely expressed
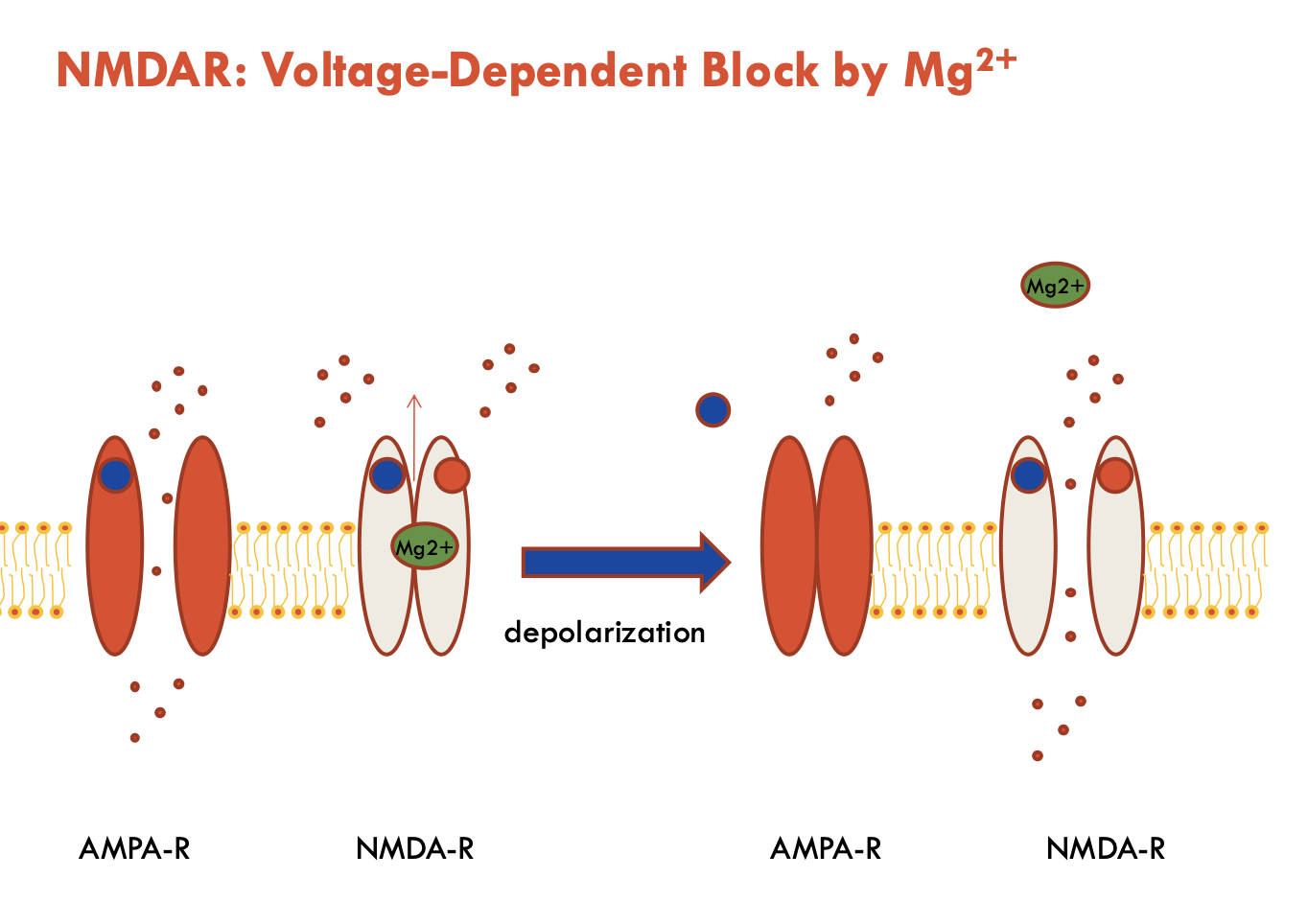
What is the voltage-dependent block mechanism of NMDAR by Mg2+?
At resting membrane potential:
Mg²⁺ blocks the NMDAR channel pore
Even when glutamate + glycine are bound, no ion flow occurs
Upon Depolarisation (via AMPA-R activation):
Mg²⁺ is expelled from the pore → opens NMDAR → allows ion flow
Therefore, NMDAR requires both:
Presynaptic glutamate release (ligand binding)
Postsynaptic depolarisation (Mg²⁺ removal)
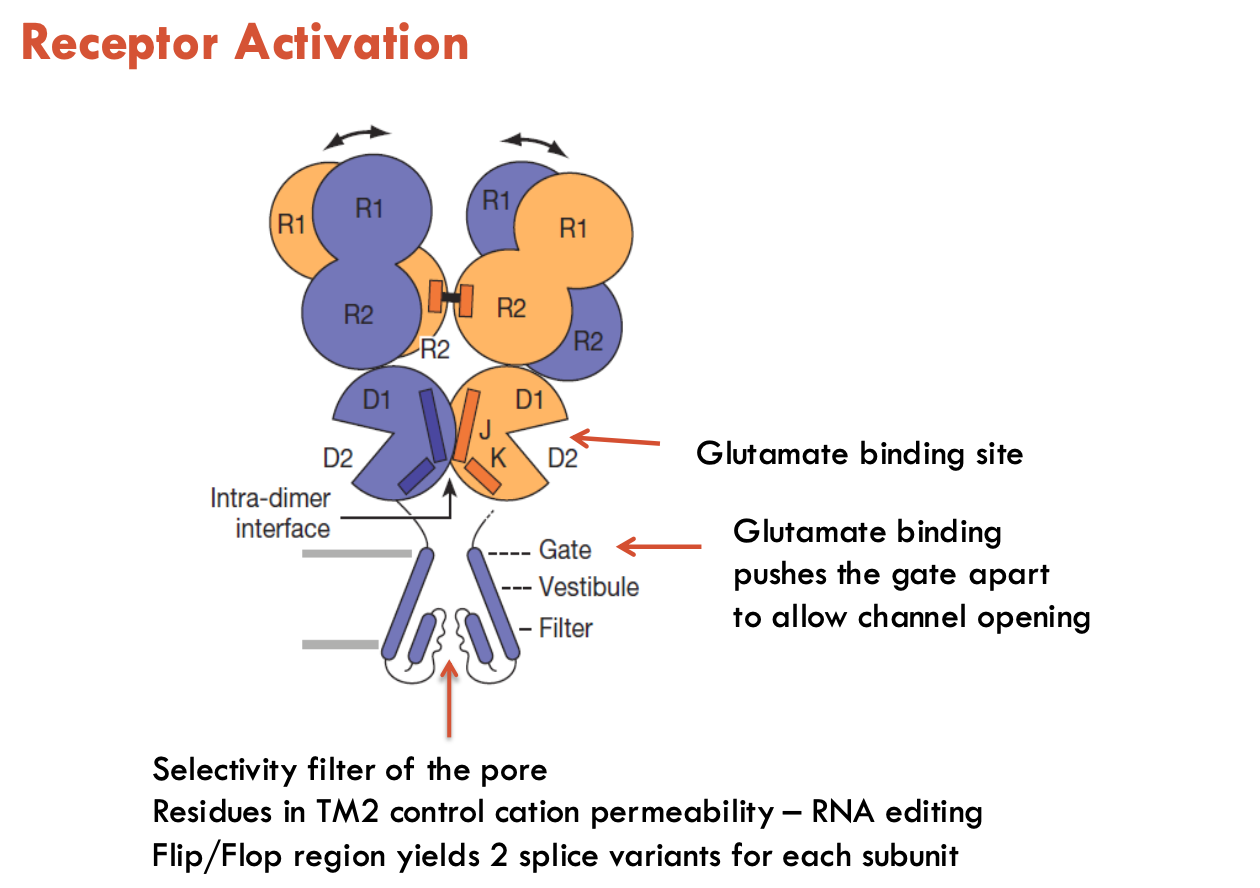
Describe the structure of excitatory glutamate receptors
4 subunits; each has 4 TM domains
has really large extracellular domains
How does the structure of excitatory glutamate receptors aid in its function?
Ligand-binding domain (LBD) (aka “venus flytrap” domain)
when ligand binds, LBD engulfs the ligand
triggers domain rotation → pulls open ion channel gate
Selectivity filter region
Pore residues in TM2 adjust to control cation permeability (RNA editing alters this)
Flip/flop region
Alternative RNA splicing yields 2 splice variants: flip and flop
Flip: slower desensitisation (stays open longer → sustained current)
Flop: shuts off more quickly
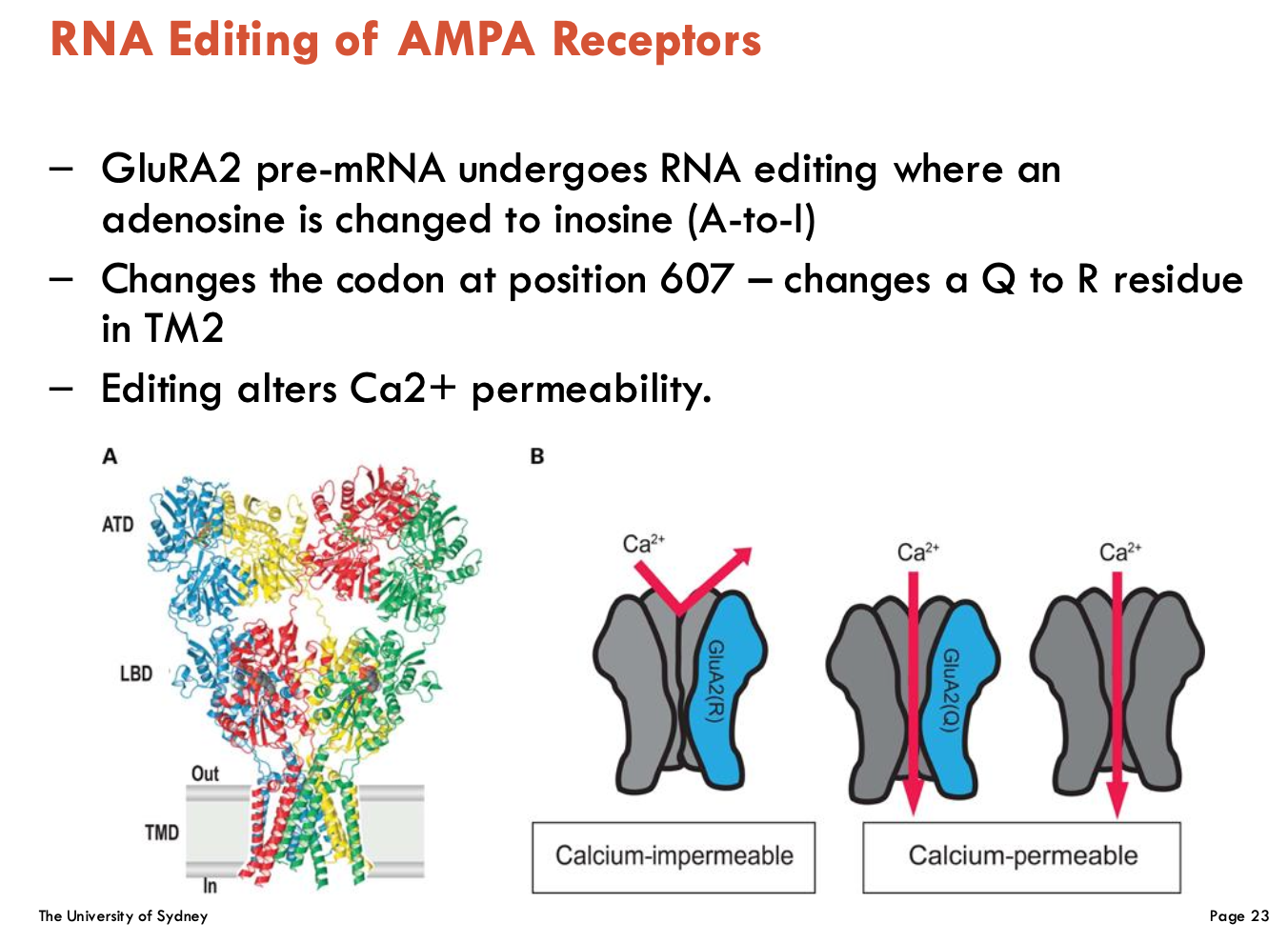
How can AMPA-R be affected by RNA editing?
A-to-I editing in GluA2 pre-mRNA (position 607, Q/R site)
Converts a glutamine (Q) codon to arginine (R)
editing alters Ca2+ permeability → blocks Ca2+ influx to reduce excitotoxicity
Which VGICs cause depolarisation and repolarisation?
Depolarisation: voltage-gated Na+ channels
Repolarisation: voltage-gated K+ channels

Describe the family of VGICs
143 members of the family
7 major groups
mostly K+ channels

What conditions can arise from VGICs in disease states?
Channelopathies - mutations associated with pathological conditions
Conditions: episodic ataxia, paralysis, myotonia, long QT syndrome, seizures

Describe the structures of VGICs
Na+ channels:
4 sets of 6 TM domains (similar but not identical)
ion channel is at the interface of the 4 sets joining
each set has an S4 TM domain: contains positively charged residues, unusually located at the periphery to serve as a voltage-sensor
Pore Loop: between S5 and S6 (selectivity filter)
Ca2+ channels:
similar to Na+ channels
K+ channels:
consists of 6 TM domains, then 4 identical copies are made to form the channel
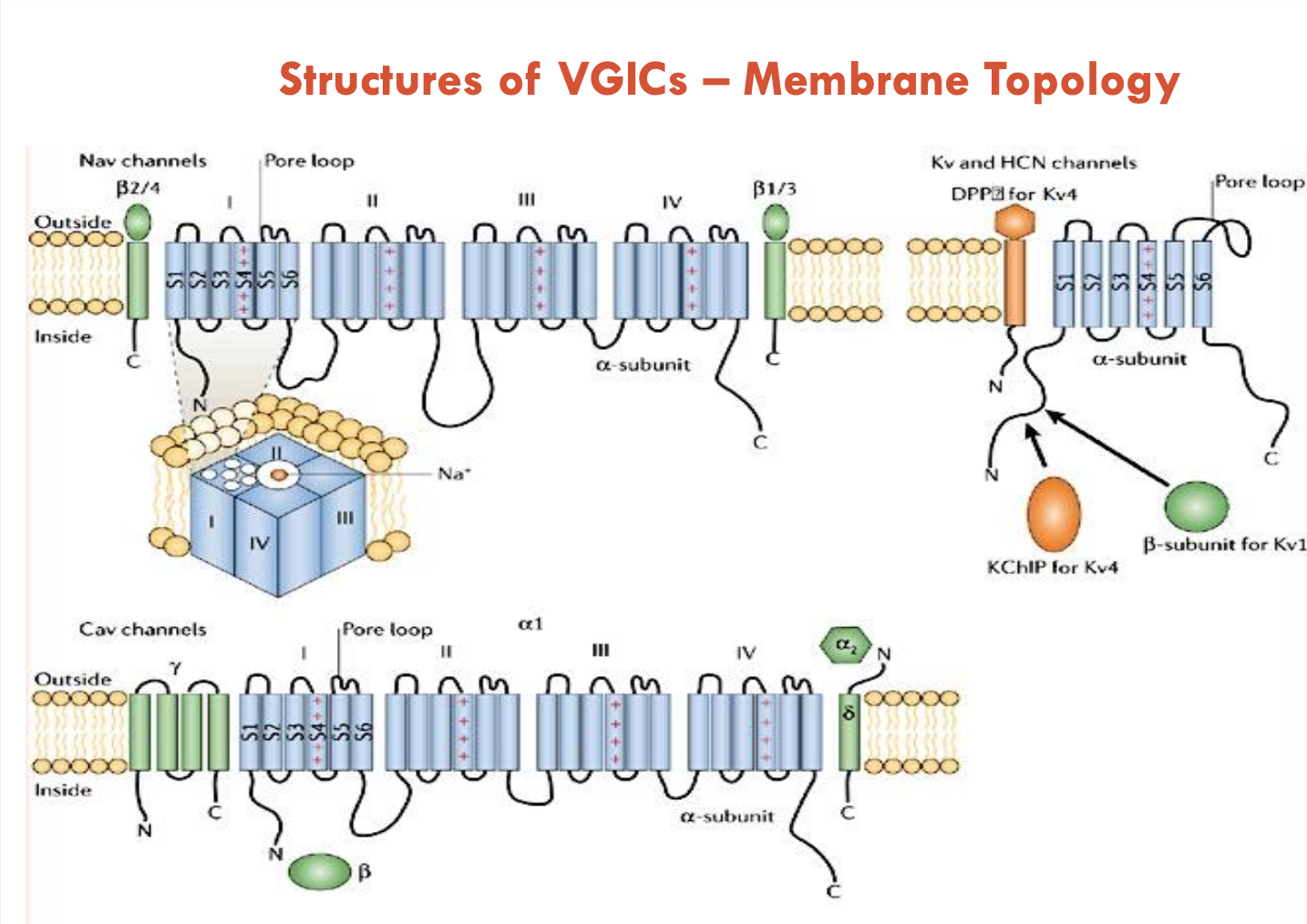
Describe the accessory subunits of VGICs
The α subunits of the Na+, K+, and Ca2+ channels show considerable structural similarity
α subunits can function on their own
accessory of β subunits are more diverse and modulate the function of the α subunit; β subunit
can regulate expression levels, location, and trafficking
can alter voltage dependence of activation or inactivation
can bind drugs that modulate function
phosphorylation of β subunit can regulate VGIC functions
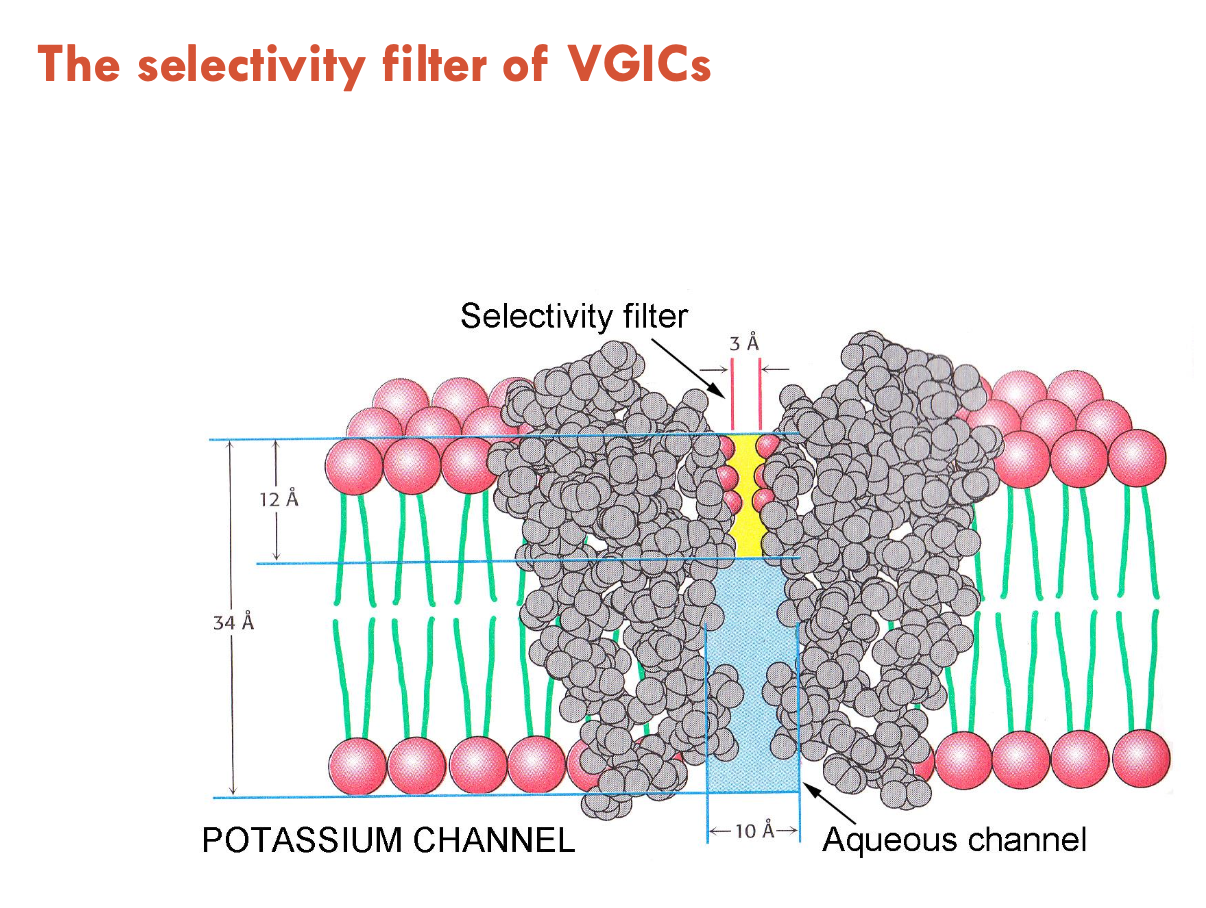
Describe ion selectivity of VGICs
VGICs contain an aqueous pore that controls selectivity for Na+/K+/Ca2+ ions
all VGICs have a similar pore structure
K+ channels are 100 - 1,000 fold selective over Na+
Na+ channels are 10 fold selective for Na+ over K+
Ca2+ channels are 1,000 fold selective for Ca2+ over other cations
How do VGICs open?
VGICs open in response to changes in membrane potential
VGICs contain a voltage sensor, which moves in response to changes in membrane potential
regulatory channels in some related channels can regulate opening of channels (e.g. Ca2+ activated K+ channels, cAMP regulated K+ channels)

How do VGICs close (2)?
Deactivation: Membrane repolarisation (back to resting state) reverts channel to closed state
Inactivation: voltage-dependent Na+ channels closes shortly after opening, even if the membrane is still depolarised
Ball-and-chain mechanism: an intracellular domain swings in to block the pore and stop ion flow
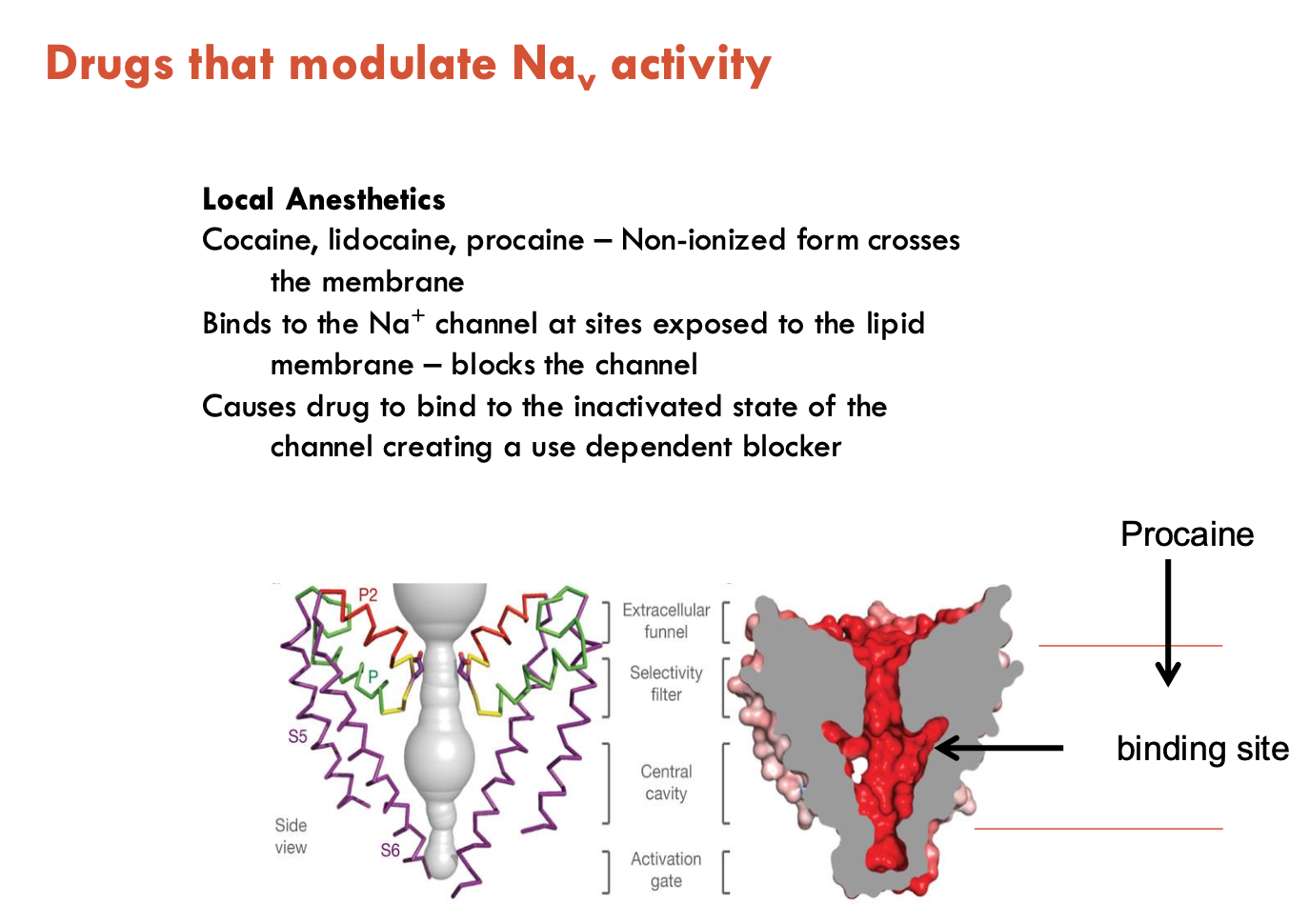
How do drugs modulate Na+ activity?
Local anesthetics (e.g. cocaine, lidocaine, procaine)
Cross the membrane in their non-ionised form
Ionised form binds to intracellular side
Preferentially bind to the inactivated state of the channel
Act as use-dependent blockers (more binding with repeated activation)
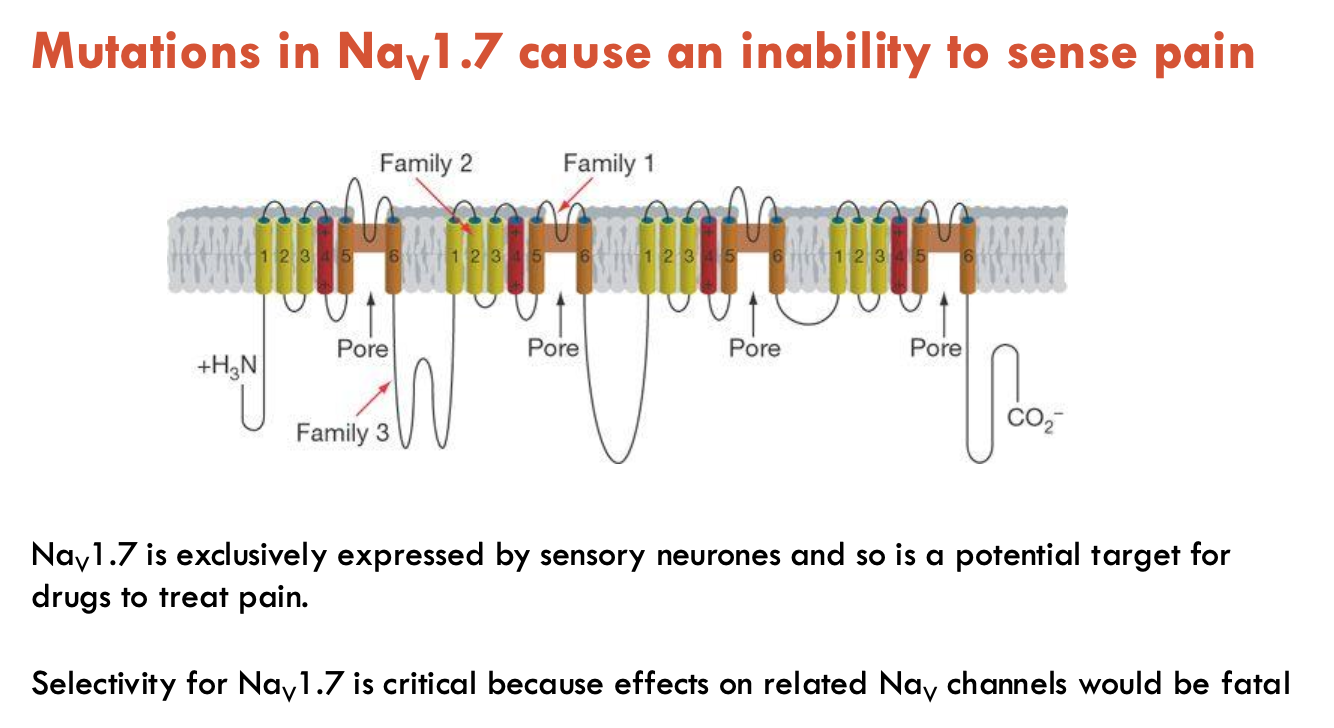
What do mutations in Nav1.7 do?
Causes an inability to sense pain
Nav1.7 is exclusively expressed by sensory neurones
potential target for drugs to treat pain
selectivity for Nav1.7 is critical because effects on related to Nav channels would be fatal
How do Kv channels act as a stabilising force (3)?
set cells’ membrane potential
repolarising the cell after an AP
control the cell’s firing rate and shape of AP
What are some main types of K+ channels (4)?
Note: There are 6 TM K+ channels and 2 TM K+ channels
Delayed rectifiers (6TMDs): slow activation → repolarises APs
A-type channels (6TMDs): transient current after hyperpolarisation → reduces firing rate (neurons)
Ca2+-activated K+ channels (6TMDs): opens with Ca²⁺ → prolongs hyperpolarisation
Inward rectifiers (2TMDs): Conducts K⁺ inward at hyperpolarised potentials → stabilises resting potential

What are the mechanisms of Ca2+-activated K+ channels (3)?
KCa open upon binding intracellular Ca2+ (typically after depolarisation-triggered Ca2+ influx)
Remains open for a long period (~few seconds)
Causes ‘long after hyperpolarisation’ (hyperpolarisation after an AP) → can slow AP firing rate
What are the functions of CaV channels (6)?
Extracellular Ca2+ is ~1-5 mM
Intracellular is 0.1 - 0.2 µM → but may rise to 100 µM after channel opening
Ca2+ entry can trigger many intracellular processes:
muscle contraction
NT release
activation of second messenger systems
alteration in gene expression
apoptosis (cell death)
depolarisation - Ca2+ spikes
What are the subtypes of CaV channels and their functions (4)?
L-Type (Cav1.1-1.4)
Cav1.1: skeletal muscle
Cav1.2: cardiac muscle, smooth muscle, brain (cell body and proximal dendrites)
In cardiac muscle, inhibitors can be used to increase blood flow in the heart
P/Q, N, and R channels (Cav2.1-2.3)
trigger NT release at synapses
N-type blockers are being developed to treat chronic pain
T channels (Cav3.1-3.3)
Low threshold, pacemaker activity (thalamus, heart)
Causes repetitive neuron firing, hormone secreion
What are TRP channels?
Transient receptor potential (TRP) channels
respond to varied sensory stimuli: temperature, touch, pain, osmolarity, pheromones, taste, and other stimuli
plays major roles in pain perception: heat, cold, sensitive to capsaicin
What activates TRPV1 (6)?
a variety of ligands:
vanilloids (e.g. capsaicin - found in chilli)
cannabinoids
ginsenosides
heat
various animal-derived toxins (e.g. VaTx1, VaTx2, and VaTx3 - found in tarantula venom)
endovanilloids (e.g. leukotriene B4, 12-S-HPETE, anandamide)
What are the physiological roles of GPCRs?
GPCRs are the largest and most diverse group of receptors in eukaryotes
transmit messages (light energy, NTs, peptides, lipids, sugars, proteins)
inform cells about life-sustaining light or nutrients in their environment or convey info sent by other cells
~50% drugs in the market act on GPCRs
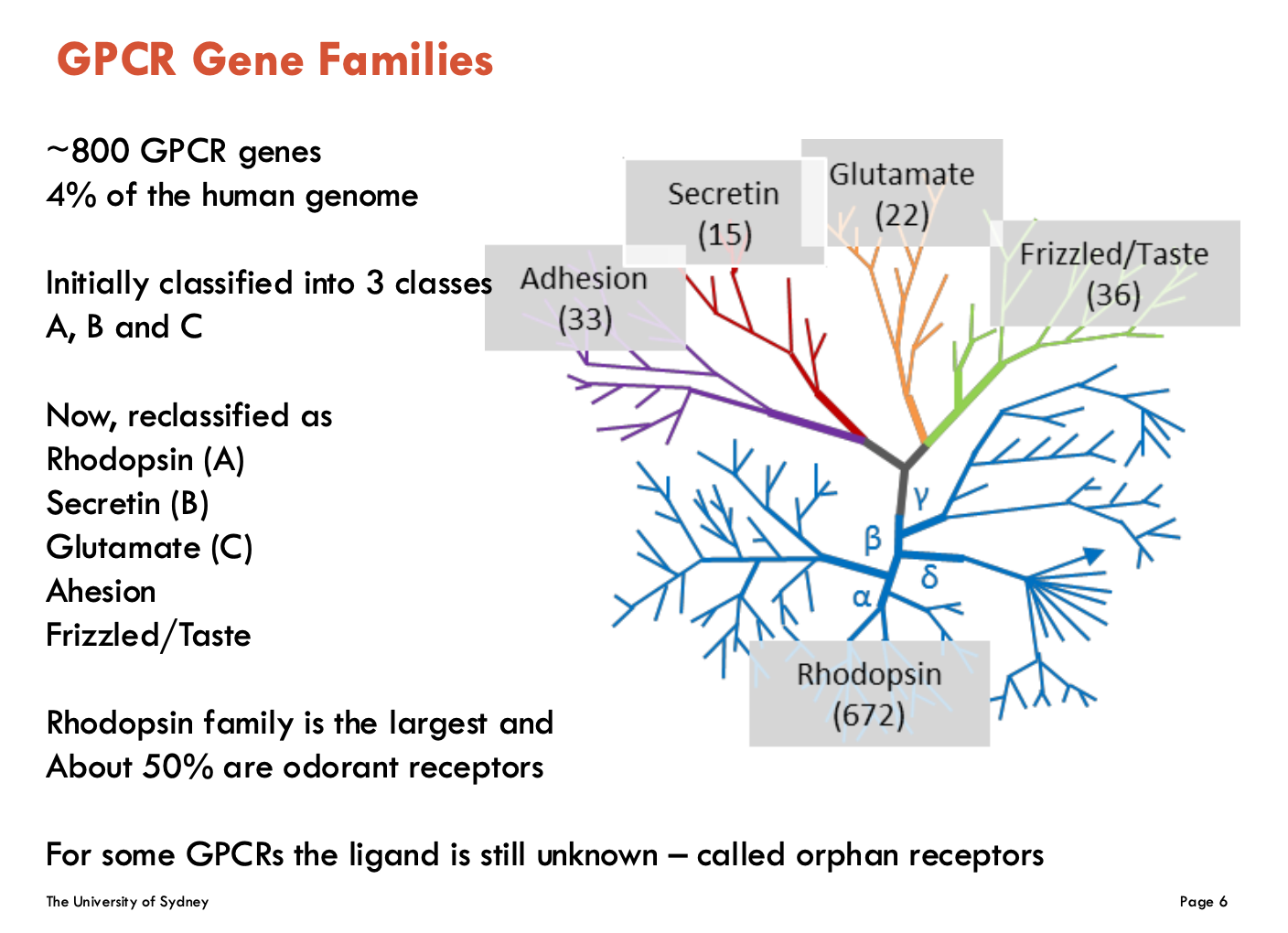
What are the GPCR classes (5)?
Classified as:
Rhodopsin (A)
largest family and about 50% are odorant receptors
Secretin (B)
Glutamate (C)
Adhesion
Frizzled/Taste
Some GPCRs have unknown ligands - “orphan receptors”
What are the structural differences between GPCR classes?
Class A
7 TM core with short extracellular and intracellular loops
includes β2 Adrenergic Receptor, μ and δ opioid Receptors, 5HT1A Receptor
Class B1 and B2:
large-ish extracellular domains, where ligand binds
includes GLP1 receptor
Class C:
is a dimer (two GPCRs that come together to signal)
large extracellular domain - contains “venus flytrap domain”
includes metabotropic glutamate receptor
Class F
frizzled/taste receptors
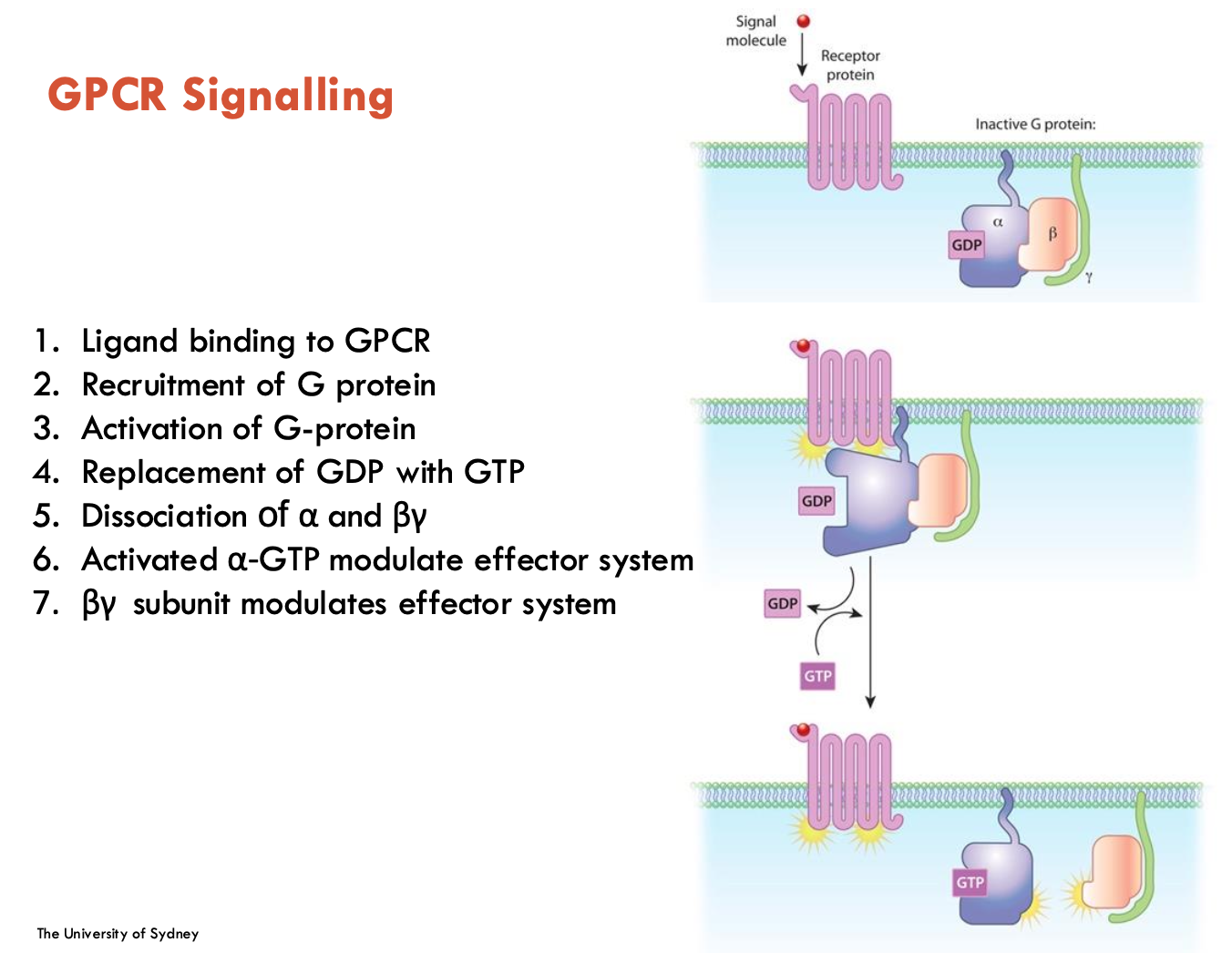
List the 7 steps of GPCR signalling
ligand binding to GPCR
Recruitment of G protein
Activation of G-protein
Replacement of GDP with GTP
Dissociation of α and βγ
Activated α-GTP modulate effector system
βγ subunit modulates effector system
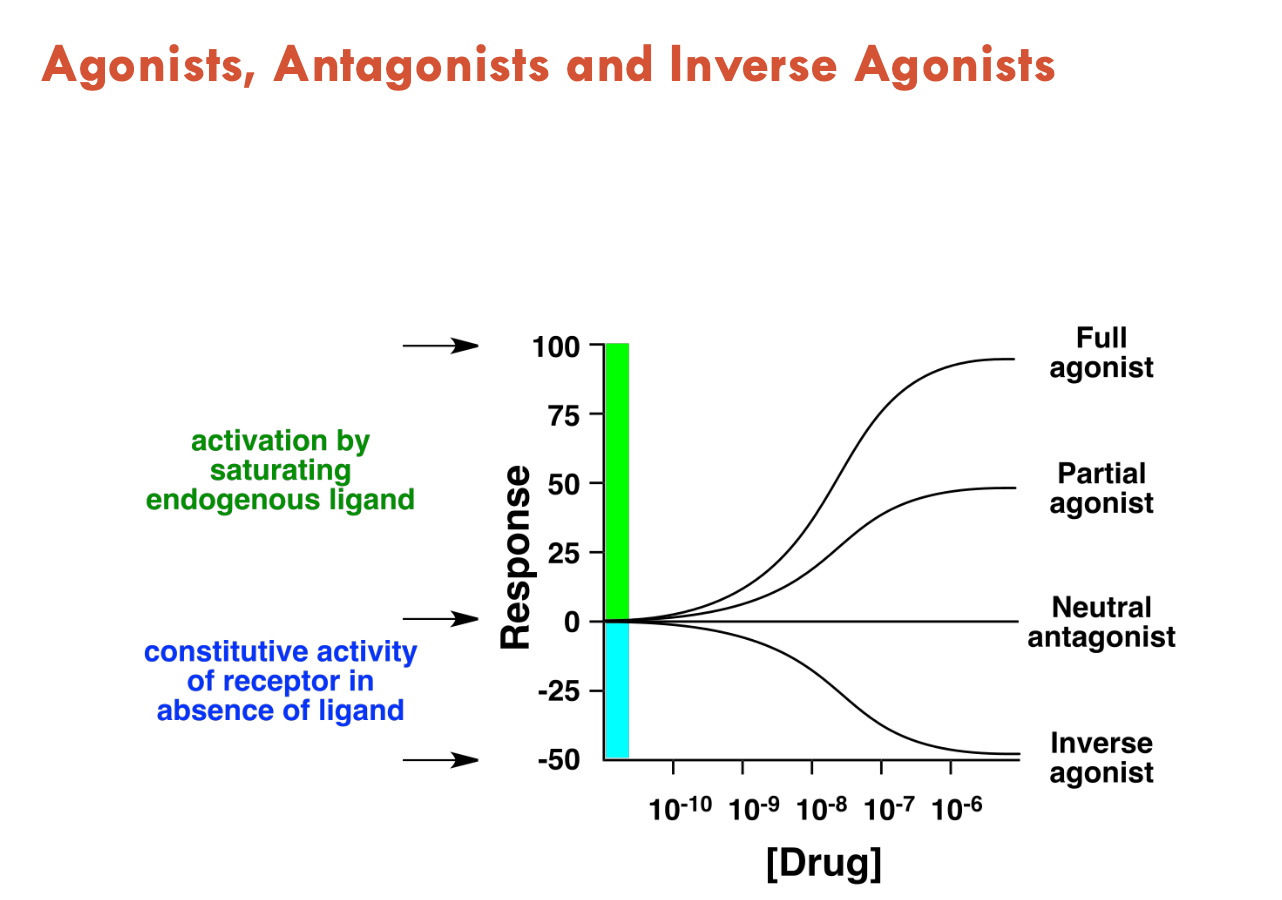
What can activate GPCRs (3)?
Agonists (activate GPCR, mimics effect of natural ligand)
Antagonists (do not activate, just blocks agonists from binding - neutral effect)
Inverse agonists (reduce constitutive receptor activity)
Features of β2 adrenergic receptor (3)
Adrenergic receptors are a prototypical GPCR model
βARs play roles in pulmonary and CV physiology → are the target of many drugs
GPCRs have to undergo conformational changes to achieve function
even in absence of an agonist, they have basal activity
How was β2AR structure solved (2)?
Initial Attempt (3.7 Å):
Used Fab5 monoclonal antibody to stabilise extracellular loops
Inverse agonist locked receptor in inactive state
Screened 1,000s of crystallisation conditions
Findings / Challenges:
7 TMDs + 8th helix visible
IL3 loop was too flexible, destabilising crystals
EC loops unresolved
Ligand density poorly resolved
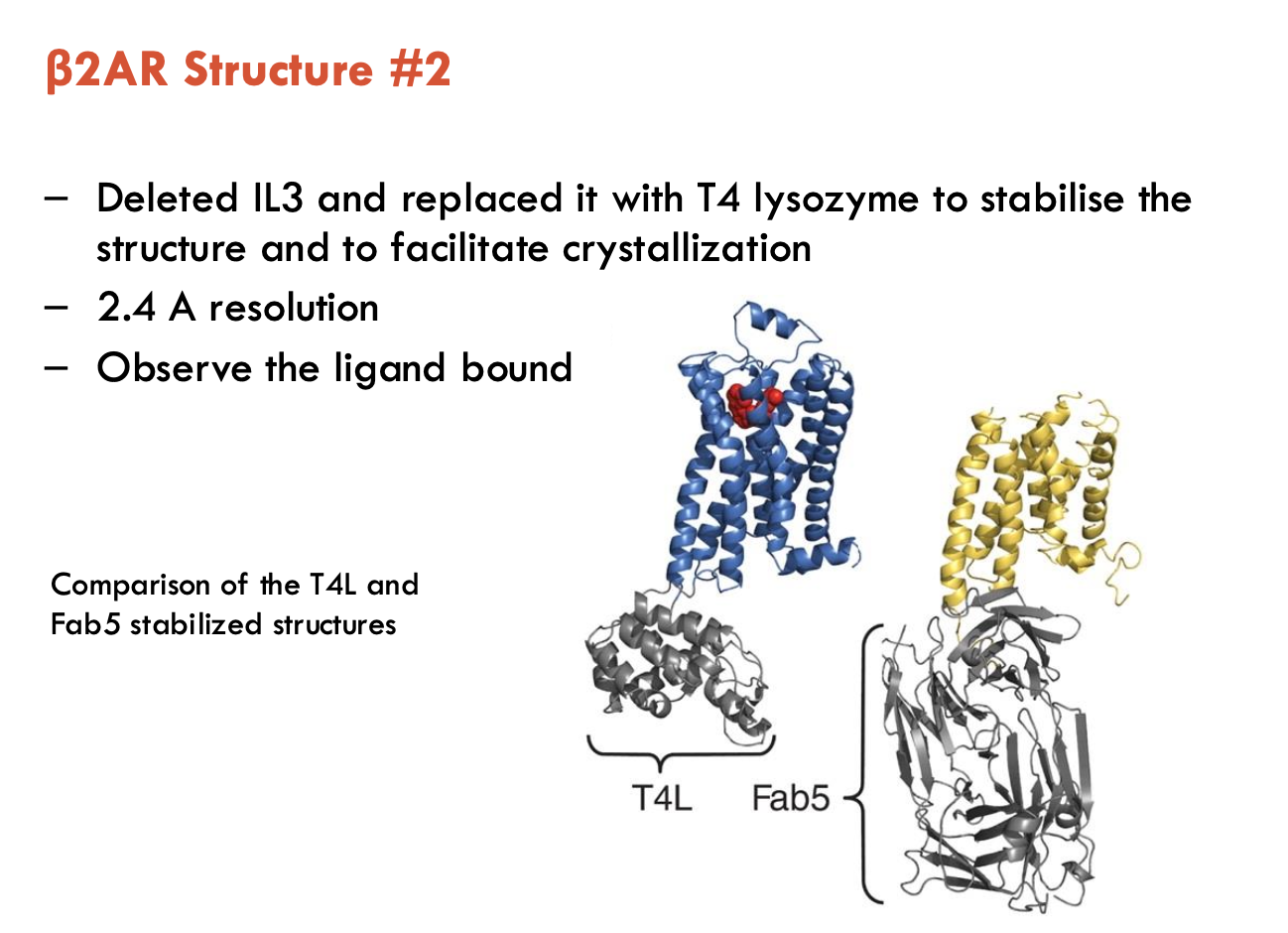
Breakthrough (2.4 Å):
Replaced flexible IL3 loop with T4 lysozyme (rigid scaffold)
Achieved higher resolution (2.4 Å)
Bound ligand clearly visible
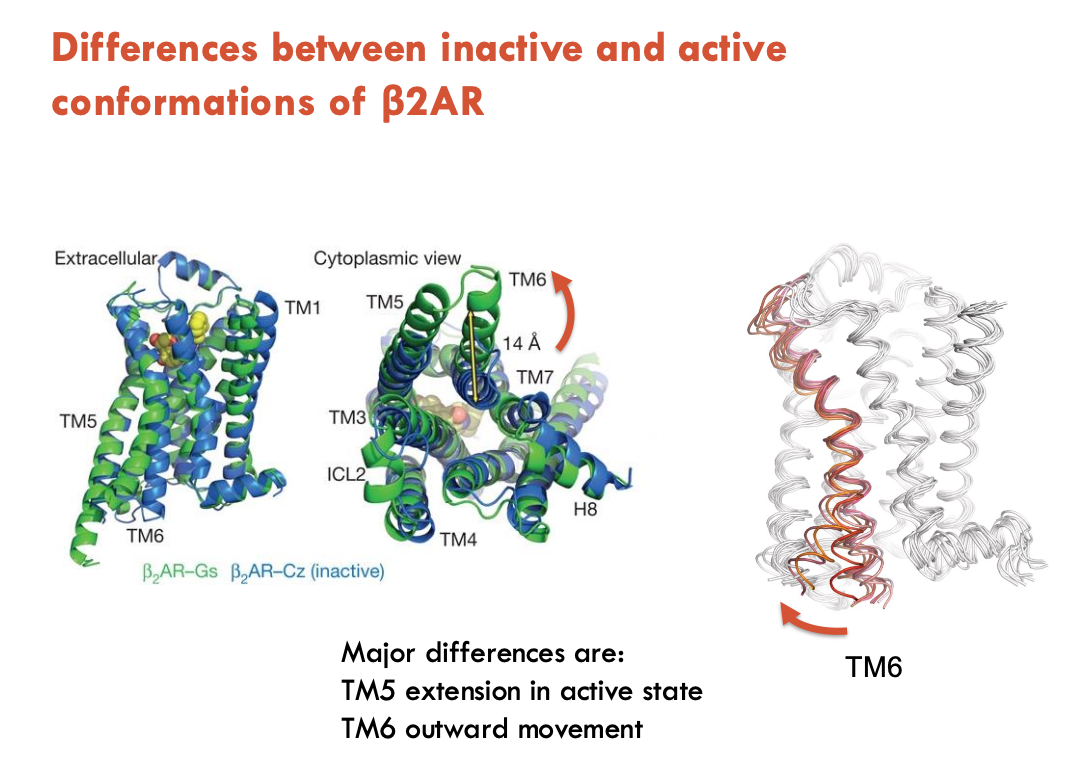
How were the inactive and active conformations of β2AR compared?
A nanobody (Nb80) was selected for its affinity for the agonist-bound structure of β2AR
facilitated crystal formation
Findings of the active state:
TM5 extension in active state
TM6 outward movement
How can a stable β2AR-Gs complex be formed (4)
β2AR couples efficiently in lipid membranes but not so well in detergents used to solubilise and purify these proteins
replace N-terminus with T4 lysozyme (improves stability)
crosslink β2AR to Gs → forms β2AR-Gs complex used to immunise llamas
llamas produce Nb35 (nanobody = single-chain antibody)
Nb35 binds to the β2AR-Gs complex and prevents dissociation

Features of a GPCR-G protein complex using β2AR as an example (4)
Active β2AR (ICL2, TM5, TM6) interact extensively with Gsα (α5 helix, α4 helix, β3 strand)
No contact between β₂AR and Gβγ subunits
ICL2 loop of β₂AR may determine Gα subtype binding - but more work required
β₂AR can form dimers in cells, but not observed in crystal structure
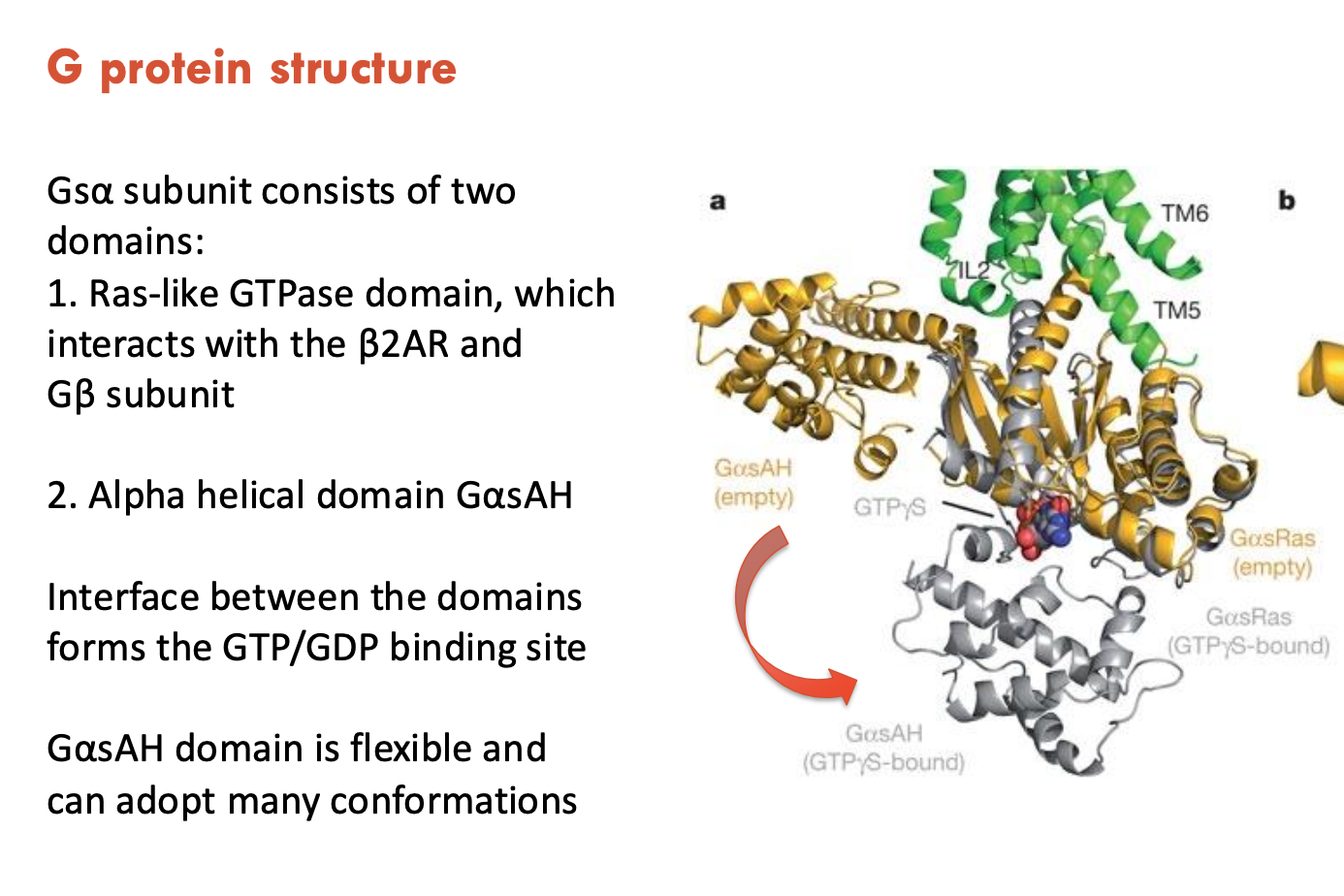
Describe the structure of Gsα subunit
Gsα subunit consists of 2 domains:
Ras-like GTPase domain, which interacts with the β2AR and Gβ subunit
α helical domain GαsAH, which is flexible and can adopt many conformations
The interface between domains forms the GTP/GDP binding site
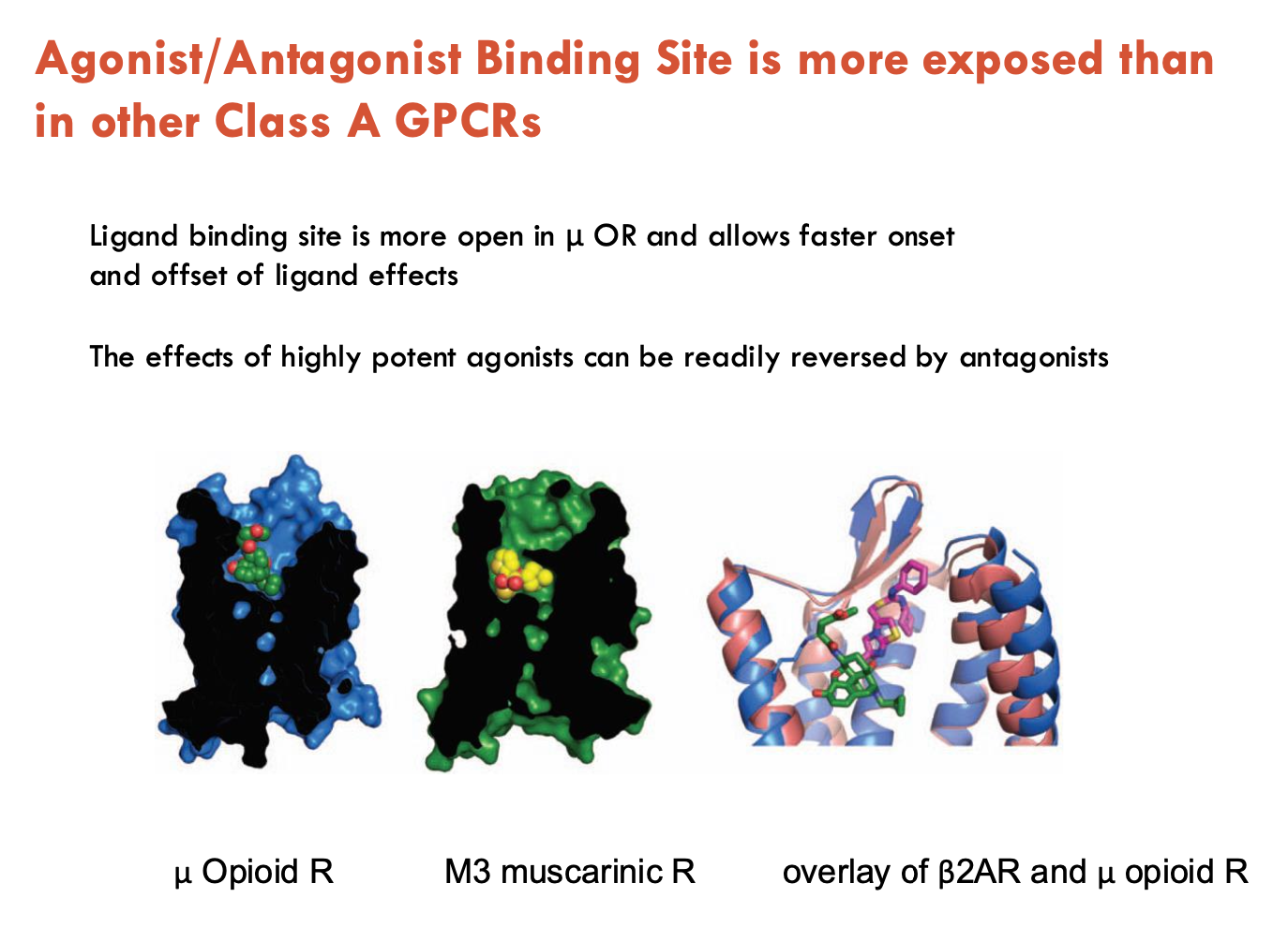
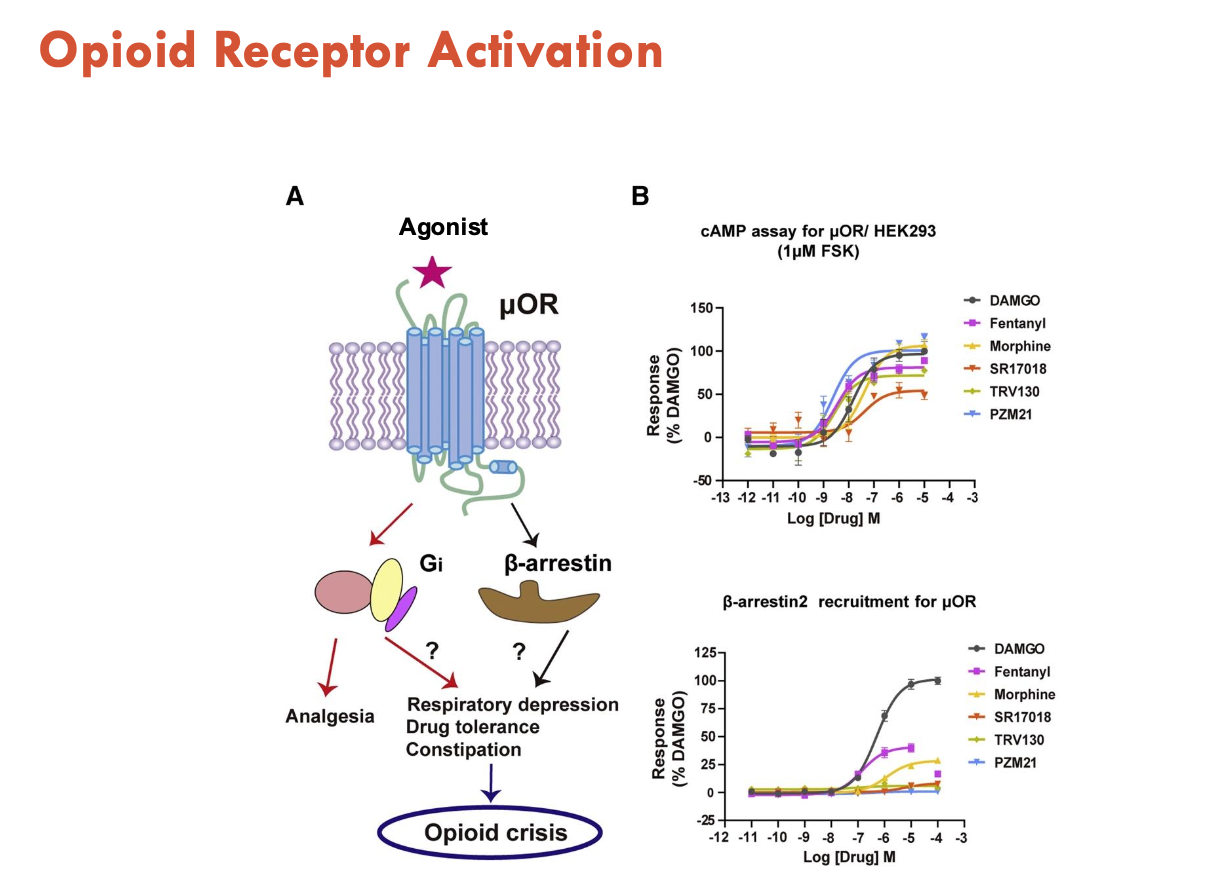
How is the μ opioid receptor (μOR) different to other Class A GPCRs?
Ligand binding is more open in μOR → allows faster onset and offset of ligand effects
effects of highly potent agonists (e.g. morphine, codeine, heroin) can be readily reversed by antagonists
What effects do μOR agonists have?
Activate two pathways:
Gi → analgesia
β-arrestin → respiratory depression, drug tolerance, constipation → opioid crisis
therefore, activation of this pathway is trying to be inhibited
changes in receptor activation may allow design of opioids with less side effects
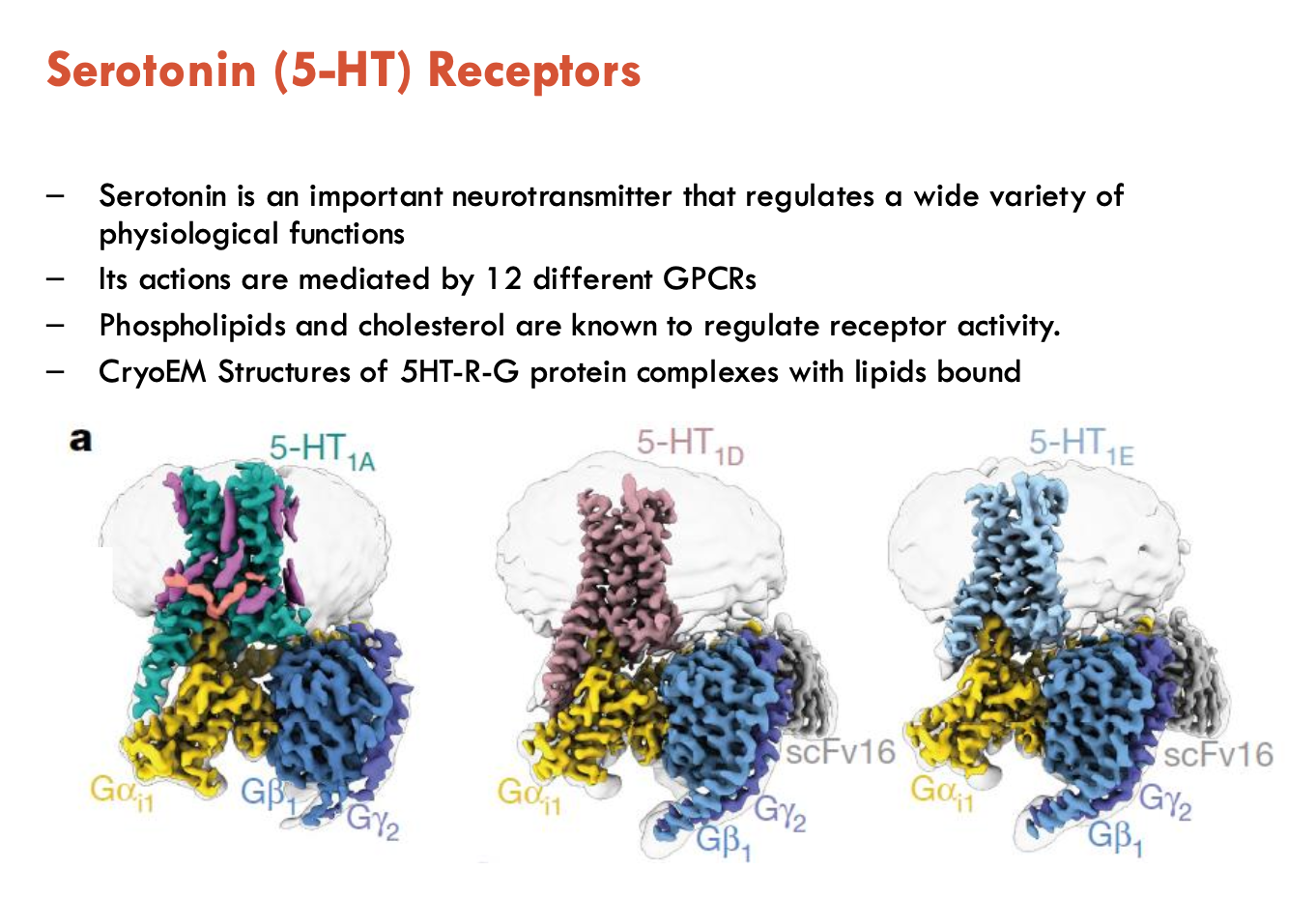
Features of serotonin (5-HT) receptors (4)
Serotonin receptors that are GPCRs are Type 1, 2, and 4
serotonin actions are mediated by 12 different GPCRs
phospholipids and cholesterol regulate receptor activity
CryoEM structures of 5HT—G protein complexes with lipids bound
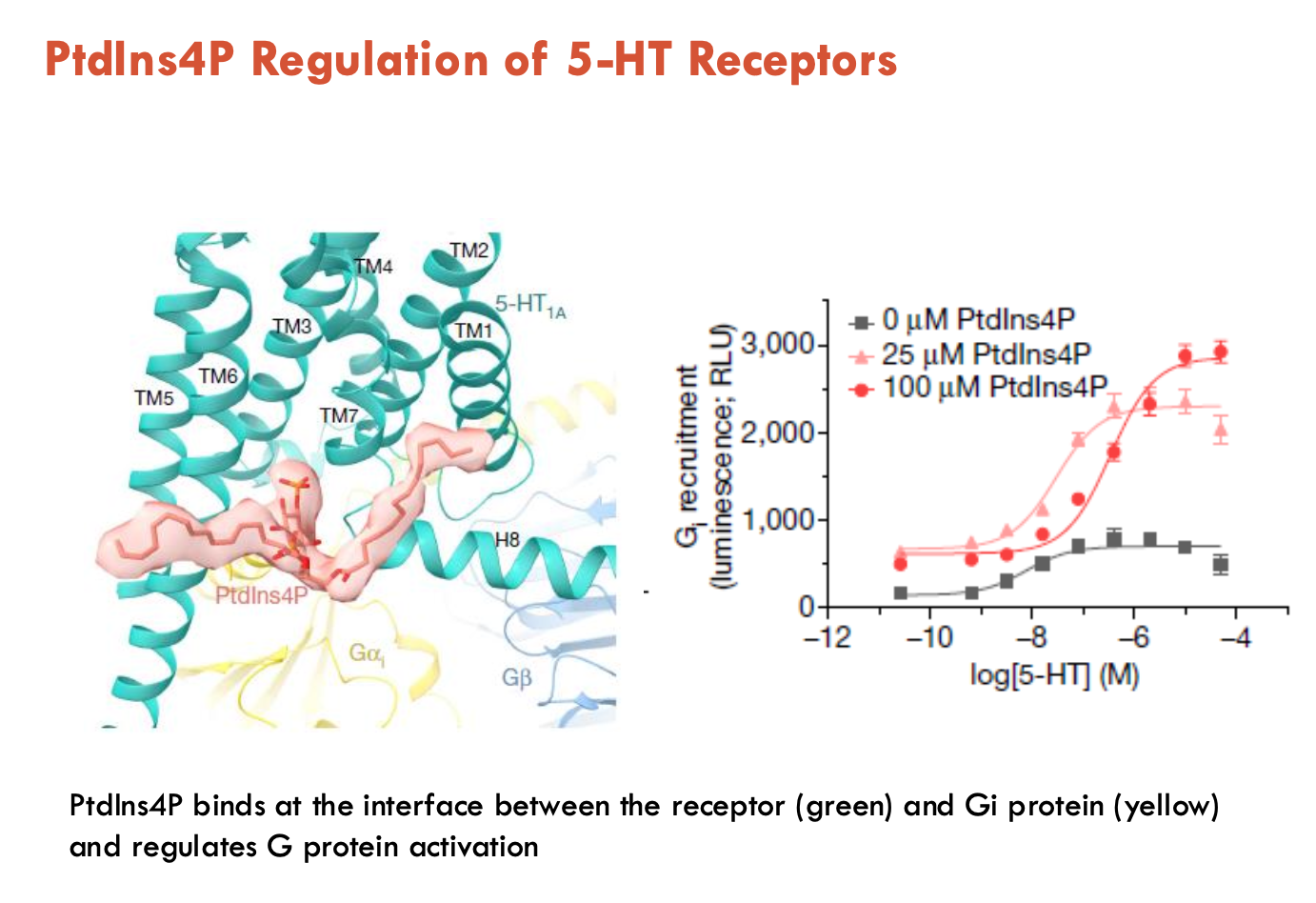
How are 5-HT receptors regulated by lipids?
Phosphotitydl-inositol-4-phosphate (PtdIns4P) binds at the interface between the receptor and Gi protein → regulates G protein activation
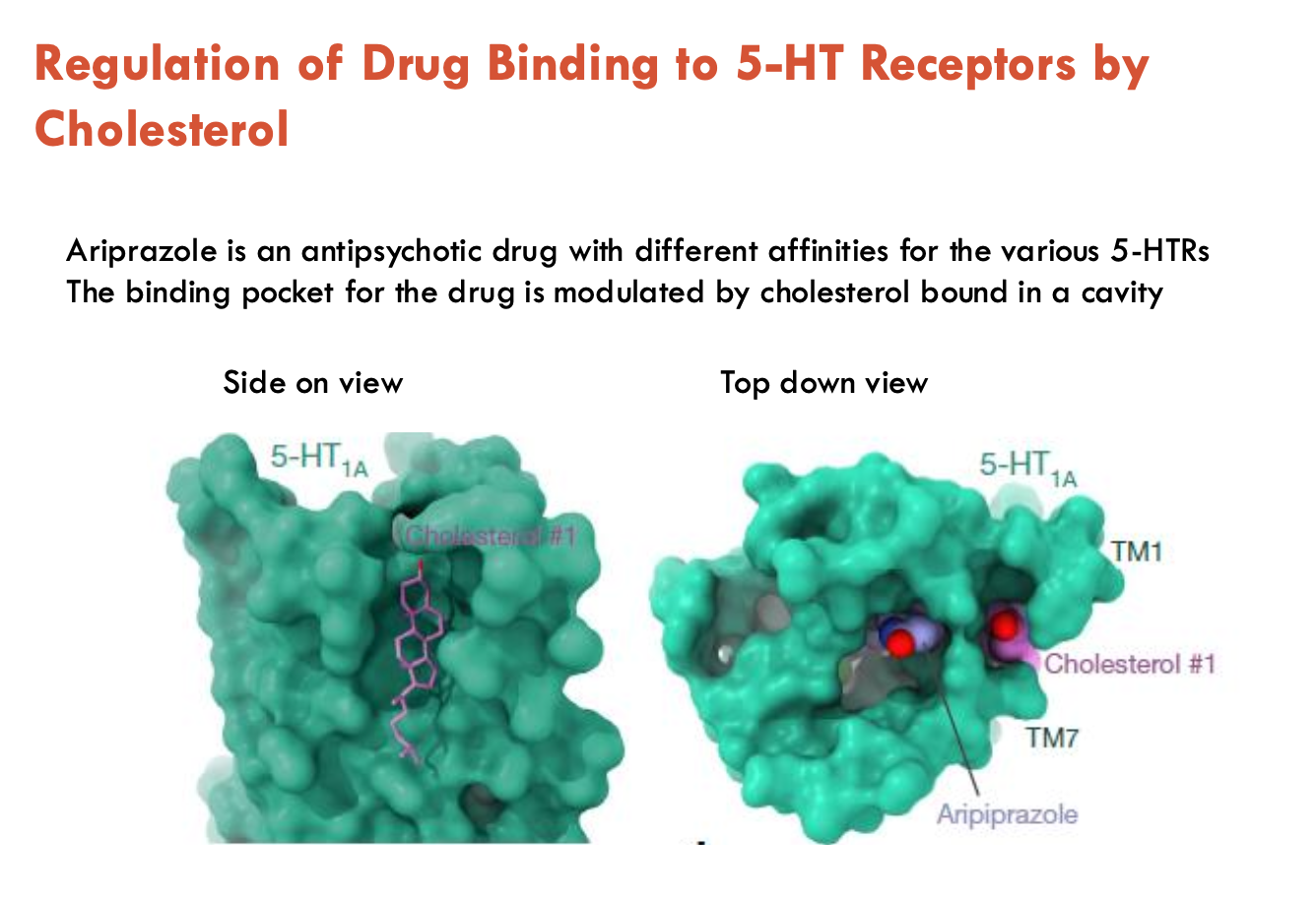
How does cholesterol influence 5-HT receptor function (e.g. ariprazole)?
Ariprazole is an antipsychotic drug with different affinities for the various 5-HTRs
binding pocket for the drug is modulated by cholesterol bound in a cavity
Features of GLP1 receptor (2)
GLP is a hormone released primarily in GIT in response to food intake and controls secretion of insulin from pancreatic islets in a glucose-dependent manner
GLP1-R is a class B GPCR
GLP1-R binds with peptide agonist