BPK 305 Midterm 3
1/113
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
114 Terms
Myocardial Ischemia
-Lack of blood flow to heart
-Local vs. global --> global = all of heart is affected (probably not pumping)
-Location --> vessels affected (e.g. left anterior descending, or circumflex arteries) defines areas at risk
-Transmural (3-D) location --> vessels go into heart, feed tissue throughout the heart
-Location and extent of block largely determines extent of cardiac damage
-Vessels go from epicardium --> endocardium; subendocardium is particularly sensitive to (partial) ischemia (affected by any upstream blockage)
-Transmural ischemia requires low blood supply across entire wall (e.g. thrombus)
Myocardial ischemia - effects
-Supply/demand mismatch
-Anoxia/hypoxia (anoxia = total lack of O2, hypoxia = not enough O2)
-Decreased free fatty acids
-Decreased glucose
-Build up of waste products (CO2, lactate, K+, H+)
Myocardial Ischemia Consequences
-Metabolic Effects
-Bursts of free radicals
-Increased [K+]_o
-Electrical remodelling
-Reduced force of contraction
-Impaired calcium handling
-Post-ischemic contractile dysfunction
Myocardial Ischemia Consequences - Metabolic effects
-Less β-oxidation due to less O2 availability --> less ATP production
-Less glucose oxidation --> more lactic acid --> decreased pH (acidosis)
-Extracellular lactate and H+ accumulation inhibits lactate/H+ cotransporter --> increased intracellular H+ (acidosis) (pumped out into extracellular fluid, but no blood to clear it)
-In cytoplasm: increased lactate, H+ (acidosis); decreased O2
Myocardial Ischemia Consequences - Free radicals
-A lot of damage occurs when flow is re-established (e.g. upon reperfusion of tissues)
-Molecular oxygen can damage tissues --> reactive oxygen and nitrogen species (ROS/RNS) generated by NADPH oxidases and mitochondria cause injury (e.g. O2-, H2O2)
-Peroxidation of lipid membranes (cell, SR, mitochondrial)
-Direct effects on cell proteins e.g. ion channels and exchangers
-Oxidative cellular damage and impaired ion homeostasis
Myocardial Ischemia Consequences - Increased [K+]_o
-Decreased Na-K pump activity --> low intracellular ATP and low pH; K+ accumulates outside cell
-Increased conductance of ATP-dependent K+ channels (normally closed by ATP) --> increases extracellular K+
-Greater repolarizing current during action potential --> shorter AP duration, less Ca2+ influx, less contractility --> less demand for O2 and ATP = cardio protective
-Higher [K+]_o depolarizes RMP (changes E_K to -60 mV) --> closer to threshold; dangerously over-excitable state
Myocardial Ischemia Consequences - Electrical remodelling
-Altered ion channel activity --> decreased Na+ channel activity, decreased voltage-gated K+ channel activity, decreased Ca2+ channel activity
-If conduction pathways are affected (e.g. bundle branches), may be proarrhythmogenic (promote arrhythmia)
Myocardial Ischemia Consequences - Reduced force of contraction
-Acidosis causes increased [Ca2+]_in but outweighed by decreased myofilament Ca2+ sensitivity (TnC site II), decreased cross-bridge cycling (decreased ATPase activity)
-Troponin can't bind to Ca2+, myosin ATPase aren't as active --> decreased contractility
-Cardiac force generation and thus cardiac output are decreased
Myocardial Ischemia Consequences - Impaired calcium handling
-Multiple effects of acidosis
-Decreased calcium current
-Less SR Ca2+ release (RyR is Ca2+ gated; during acidosis, less Ca2+ can bind to RyR)
-Less NCX Ca2+ extrusion
-Na/H exchange --> more Na+ inside cell, Ca2+ entry via NCX
-Ca2+ bound to cytoplasmic buffers is released (the cause of large changes)
-SERCA inhibition --> less SR uptake of Ca2+
-Net effect = cytoplasmic Ca2+ elevation/overload
-Ca2+ is elevated even during diastole --> contraction during diastole --> tonic contracture --> myocardial damage
-Elevated Ca2+ activates protease enzymes (e.g. Calpain = causes protein breakdown)
-Mitochondrial Ca2+ overload can trigger apoptosis
Myocardial Ischemia Consequences - Post-ischemic contractile dysfunction
-Reversible --> stunning (decreased contractility for a few days) and hibernation (state of hypocontractility lasting weeks/months; involves revascularization)
-Irreversible = total ischemia > 30 mins --> necrosis (disorganized cell death; scar tissue --> collagen, doesn't function nicely, doesn't conduct electricity) and apoptosis (programmed cell death)
Osmolarity vs. Tonicity
-Osmolarity = total number of particles/solute (includes dissociation)
-Tonicity = only solutes that do not readily diffuse across membrane
Stages of myocardial injury (ischemia)
Osmotic Overload
-Large increase of small molecules in cytoplasm (ATP --> ADP + Pi, phosphocreatine --> creatine + Pi, glycogen --> lactate (via glucose))
-Osmolarity can increase from 300 mOsm/L to 400 mOsm/L (water tries to enter cell)
-Upon reperfusion with 300 mOsm/L, water enters causing cells to swell/rupture; intracellular organelles also swell/rupture (mitochondrial rupture = cell death?)
Immune Response
-Neutrophils are attracted to ischaemic areas, permeate tunica intima
-Release reactive oxygen species (ROS), causes damage to heart itself
-Complement system can be activated by ischemia --> produces membrane attack complex (MAC); kills cells by penetrating their membrane (normally bacteria but damages heart instead)
pH Paradox
-Acidosis --> increased lactic acid, imbalance in ATP breakdown and production
-Decreases contractility (-ve inotrope), ion channel function, myofilament Ca2+ sensitivity (TnC site II), ATPase activity (less crossbridge cycling); affects Ca2+ handling proteins
-Must relieve acidosis to restore contractility/ion handling but rapid relief of acidosis causes additional damage to myocytes
Myocardial Ischemia - pH Paradox
-Ischemic event causes high levels of H+
-Inhibits N/K-ATPase --> accumulation of Na+/H+ in extracellular space
-Reperfusion increases drive of Na/H exchanger (more Na+ in)
-High Na+ levels causes NCX to pump Na+ out (brings in Ca2+)
-Ca2+ overload activates proteasese, causes mitochondrial Ca2+ overload
-Causes cell death
Rapid reperfusion causes cell death
-NHE blockers (e.g. amiloride) induce acidosis at rest; (sort of) protect against reperfusion injury
Ventricular depolarization/repolarization
-Endocardium depolarizes slightly sooner because conduction system is on the inside
-Epicardium depolarizes slightly earlier
-Occurs during QRS complex (early depolarization) or T-wave (early repolarization)
-ECG only sees differences in membrane potential (e.g. systole) --> diastole = has even potentials
Subendocardial ischemia - injury current
Diastole
-Injured subendocardium has elevated RMP
-Persistent flow of charge into neighbouring regions
-LV endocardial injury = elevated T-Q segment --> V_endo > V_epi during diastole
Systole
-If ischemic injury prevents normal depolarization, then V_epi > V_endo during AP plateau
-Causes S-T depression
Transmural infarction
-Some endocardial depolarization persists due to increased preconditioning in endocardium
-Bump in epicardial AP disappears; contributes to S-T elevation
-Deep Q (due to dead tissue) --> pathological Q = >25% of R
-Endocardium is used to a little bit of ischemia (during systole, blood flow stops) --> recovers better
Determining Injury Current
-ECG has no "zero" --> use end of depolarization (QRS) as reference point or J point
-Injury current is determined by diastolic voltage - J
-If J is higher, injury current is negative; if J is lower, injury current is positive
-Tail of injury vector originates at the injury
ECG changes with myocardial infarction
After recovery
-Heightened T-waves, followed by T-wave inversion (altered directionality of repolarization)
-ST segment elevation due to injury current
-Deep (dirty, pathological) Q waves --> reflects dead (left) myocardium
-Q = sepatal and RV depolarization (if part/all of LV is dead/electrically invisible)
Serum changes with myocardial infarction
-Some people experience T-wave inversion (e.g. athletes)
-Serum changes within 48-72 hours are often used in diagnosis
-LDH (lactate dehydrogenase)
-CK (creatine kinase)
-TnI (troponin I) --> calpain degrades TnI; TnI levels rise during infarction --> can distinguish between unstable angina and infarc
Myocardial infarction - Pre/postconditioning
Preconditioning
-Reduces damage of subsequent more severe infarction; reduces infarction size
-Mechanism = brief and mild ischemia
-Increases K_ATP activity (plasma membrane and mitochondria); increases vasodilator metabolites (adenosine, CO2, hypoxia --> also increases VEGF, HIF to stimulate blood vessel proliferation)
-Releases noradrenaline, bradykinin, opioids --> activate G-proteins, protein kinases (PKC, MAP, P13-K)
-Multiple angina episodes may offer some protection
Postconditioning
-Restarts the flow in brief bursts rather than all at once
-Shorter bursts of reperfusion produce less damage --> smaller infarct size, less chance of arrhythmia
-Can be applied clinically (unlike preconditioning --> event must be predicted for preconditioning)
Organization of respiratory tracts
Upper --> Nasal cavity, pharynx, tongue, vocal cords, larynx, esophagus
-3 sinuses (frontal, sphenoid, maxillary)
Lower --> Trachea, right/left lungs, right/left bronchus, diaphragm
Division of the lungs
-Lobes are defined by fissures (3 right lobes and 2 left)
-Bronchopulmonary segments = basic anatomical units; defined by regions supplied by segmental bronchus and can be surgically isolated
-Lobar bronchi supply each lobe
-Conducting airways = ~150 mL; nose/mouth to non-respiring bronchioles
-Terminal bronchioles = smallest bronchioles without alveoli; no cartilage
-Respiratory unit (total = ~2500 mL) = basic physiological unit; respiratory bronchioles, alveolar ducts, and alveoli
Functions of the respiratory system
-Gas exchange --> O2 uptake and CO2 release
-Conditioning of inspired air --> warming and moisturizing (if not warmed, more likely to get air bubbles in blood); filtering particles >10μm
-Secretes mucus --> clears debris from airways
-Filters small emboli from blood
-Secretes surfactant and ACE (angiotensin converting enzyme)
-Acid/base balance of blood --> CO2/HCO3- buffering
-Vocalization (at larynx)
-Olfaction --> nerve endings in roof of nose extend from olfactory epithelium to olfactory bulb
-Heat loss
Components of the respiratory system
-Air pump --> upper airways/conducting airways, lungs, chest wall
-Surface for gas exchange --> alveoli
-Mechanism to carry O2/CO2 --> hemoglobin
-Tissue diffusion --> capillary endothelium
-Central mechanisms of control --> chemoreceptors, drive to breath
-Local/peripheral mechanisms of control --> chemoreceptors, Hering-Breuer reflex
-Circulatory system --> heart, vasculature
Physiological Dead Space
Anatomical dead space (conducting airways) + alveolar dead space (alveoli that are ventilated but not perfused)
Respiratory epithelium - Large conducting airways
Goblet cells --> secrete mucin, sialic acid --> makes mucus
Submucosal glands --> secrete water, ions, mucus
-Secrete bacteriocidal lysozymes (lactoferrin, antileukoprotease) --> immune function
Sol layer
- Periciliary layer of fluid
-Produced by columnar epithelial
-Allows free movement of cilia --> moves mucus towards mouth
Mucus layer
-Traps airborne particles
-Discontinuous blanket
Airway epithelium
-Thin and becomes more in alveoli
-Lose cartilage, submucosal glands, goblet cells (around generation 12)
Cilia
-200-250 per columnar epithelial cell
-Contain ATPase thought to mediate beating motion
-Sweep mucus out of airways
Microvilli
-Brush cells
-Increase surface area for secretion
Airway dynamics
-Air moves by convection in conducting airways; by diffusion in alveolar airways
-Convection = moving from high to low pressure areas --> driving force = difference between atmospheric (P_B) and alveolar pressure (P_A)
-Velocity × cross sectional area (CSA) = constant
-Small decrease in CSA between trachea and bronchi (small increase in velocity)
-Alveolar airways facilitate diffusion --> huge surface area (~80 m^2 or a tennis court); slow velocity of air
Terminal respiratory unit
-Airway stemming from a single terminal bronchiole
-Terminal bronchiole = no alveoli
-Respiratory bronchioles have alveoli
Alveolar pneumocytes
Type I --> flat and elongated
-95% of alveolus surface area
-Thin cytoplasm; primary site of gas exchange
-Fused to endothelium
Type II --> small and cuboidal
-2% of alveolus surface area
-Role in regeneration of Type I cells
Type III --> brush cells
-Found throughout the lungs; closely associated with nerves
-Possible role as chemoreceptors
Pores of Kohn/Canals of Lambert
-Inter-alveolar pores/canals
-Allow gas diffusion between alveoli and bronchioles
-Prevent alveolar collapse due to surface tension (especially if an alveoli is congested)
Pulmonary blood supply
-Lungs receive the entire CO from the RV
-Receives 2 blood supplies: pulmonary artery and bronchial arteries
-Pulmonary arteries/arterioles are thinner and have larger diameter than systemic circulation --> increases compliance
-Can contain a large volume of blood, reduce pulse pressures; distensibility protects against edema
Pulmonary artery
-Carries deoxygenated blood
-Capillaries have ~5μm radius (~80 m^2 area)
-Enhanced gas exchange
-Single file RBC passage; slow flow due to high cross-sectional area (~750 ms passage time for a RBC)
-Very close to alveolus (short diffusion distance); almost like a sheet of blood surrounding alveolus
Bronchial arteries
-Supplies larger bronchi with oxygenated blood from left side of heart
-Oxygenated blood supply to bronchioles
-1/3 drains to bronchial veins (and to right atrium)
-2/3 drains to pulmonary veins (to left atrium) --> acts as a shunt (from right to left side of heart without gas exchange)
Pulmonary blood pressure/volume
-Pulmonary vasculature = low pressure system (mean pressure in pulmonary artery = 15 mm Hg, 8 in left atrium --> driving force = 7 mm Hg)
-Pulmonary blood ~10% (500 mL) of total blood volume
-Can decrease 50% or increase 200% (ranges between 250 and 1000 mL)
-E.g. resting capillary blood volume ~75 mL; maximal exercise capillary blood volume ~200 mL
Pressure and Flow during Inspiration
-Boyle's Law: P ∝ 1/V (when temperature/mass are constance)
-Changes in volume of lungs during inspiration and expiration generate the pressure changes required for ventilation
-Lung volumes increase (alveoli expand) --> causes decrease in pressure (based on Boyle's law) --> creates pressure difference --> draws air in
Muscles of breathing
Inspiration
-Diaphragm (flattens, increases thoracic volume) and external intercostals (lift rib cage --> like bucket handle when looking from above)
-Scalenes and sternocleidomastoid used during exercise (accessory muscles) --> scalenes lift first 2 ribs, SCM lifts sternum
-Upper respiratory tract muscles decrease airway resistance
Exhalation
-Passive at rest
-Internal intercostals (opposite of external intercostals), rectus abdominus (compresses thoracic cavity), and external obliques are accessory muscles
Innervation of muscles of breathing
-Inspiration is innervated from C1 - T12; expiration is from T1 - T12
-Spinal cord injury above T12 influences respiratory function (e.g. during exercise); above C5 renders inspiration dependent on accessory muscles; above C3 requires artificial ventilation
-C1/C2 = trapezius, sternocleidomastoid
-C3-C5 = diaphragm
-C4-C7 = scalenes
-T1-T12 = Lateral external intercostals, internal intercostals
-T6-T12 = rectus abdominus, external and internal oblique, transversus abdominus
Quiet vs. Forced breathing
Quiet
-Inspiration = diaphragm, external intercostals
-Expiration = passive (no muscles used)
Forced
-Inspiration = Scalenes, sternocleidomastoid, neck and back muscles (e.g. trapezius), upper respiratory tract muscles (relax to maximize air flow)
-Expiration = abdominal muscles, internal intercostals, neck and back muscles
Intrapleural space
-Parietal pleura = stuck to chest wall; visceral pleura = stuck to lungs
-Pleural cavity is filled with pleural fluid --> allows lungs to slide over chest wall; sticks lungs to chest wall
-Negative pressure in the (intra)pleural space --> counters elastic recoil of the alveoli and keeps them open
-Greater pressure when stretched (e.g. when lung volume increases, becomes more negative)
-Pneumothorax = puncture of pleural space; lung collapses, alveoli collapse = atelectasis
Lung compliance
-Lungs are highly distensible
-Compliance refers to ease with which they can expand under pressure --> ΔV/ΔP
-Determined by elastin and collagen fibres in lung parenchyma
-Lung inflation elongates fibres --> more elastic force --> elastic recoil; lungs revert to initial size
-Elastic recoil is the reason for collapse during pneumothorax
-Decreased compliance = more resistance to distension (e.g. pulmonary fibrosis)
-Measured under static conditions --> glottis open, no air flow, P_A = 0)
-Hysteresis (not the same in both directions) --> more pressure is needed to open an airway than to collapse
-Inflation has to overcome elastic recoil and surface tension --> collapsed alveoli have high surface tension and less surfactant
-Therefore, inspiration is active; expiration is passive
-In adult lungs, volume changes ~200 mL for each cm H2O change in intrapleural pressure
Compliance and pathology
Emphysema = increased compliance --> difficult to expire
-Smoke --> immune system response releases elastase --> breaks down elastin --> decreased elastic recoil --> have to actively exhale during quiet breathing
Fibrosis = decreased compliance --> difficult to inspire
-Particulate matter triggers immune response --> macrophages secrete growth factors causing proliferation of fibroblasts --> increases collagen deposition
Surface Tension
-LaPlace's Law --> P = 2T/r --> Inward pressure trying to collapse a bubble is equal to twice its surface tension divided by radius
-Takes twice the inspiratory pressure to keep a small alveolus open compared to one twice the size (larger r = smaller P)
-At low lung volumes (mostly small alveoli), harder to inflate (takes more inspiratory pressure)
-The smaller an alveoli gets, the more it collapses --> forces air into a larger alveoli
-Prevention of alveolar collapse is due to interdependence (mechanical tethering) and surfactant (reduces surface tension)
Pulmonary Surfactant
-SURFace ACTive AgeNT --> reduces surface tension
Composition
-90% lipid --> dipalmitoylphosphatidylcholine (DPPC) and phosphatidylcholine (hydrophobic)
-10% protein --> albumin, IgA, apoproteins (hydrophilic)
-Some apoproteins also have immune function
-Secreted by Type II cells as lamellar bodies; forms tubular myelin rich in surfactant apoproteins
Pulmonary Surfactant Function
-Reduces surface tension
-Minimizes tendency for small alveoli to collapse --> maximizes surface area for gas exchange
-Increases compliance and decreases elastic recoil --> easier to inflate lungs
-Keeps alveoli moist
-Minimizes fluid accumulation in alveoli (preventing pulmonary edema)
-Maintains alveolar size (helps equalize size of alveoli) --> prevents expansion at different rates
-In rapidly expanding alveoli, surfactant becomes more dispersed --> less able to decrease surface tension --> counteracts the rapid expansion (slower expansion; "slower" alveoli can catch up)
-Reduces hysteris --> therefore hysteresis must be due to properties of surface tension
Infant respiratory distress syndrome
-Most common in births <28 weeks; due to reduced/absent surfactant
Symptoms
-Shortness of breath, grunting sounds
-Lungs are stiff and hard to inflate (alveoli collapse during exhalation)
-Unusual breathing movement (due to soft ribcage --> flailing chest)
Treatment
-High oxygen and humidity
-Artificial ventilation
-Synthetic/animal-derived surfactant
Respiratory Cycle - Pressure/Volume/Flow
Pressure
-P_IP (intrapleural pressure) --> controlled by brain; driven by thoracic volume
-P_TP (transpulmonary pressure) --> a "static" parameter; reflects elastic recoil of lungs, and is determined by V_L --> lags behind P_IP slightly
-Alveolar pressure changes --> P_A (alveolar) is variable; P_B = 0
-ΔP = P_A = P_IP + P_TP
-If no volume changes, P_IP and P_TP must be equal and opposite
-If P_IP is more negative than P_TP is positive, lungs inflate; if less negative, then lungs deflate
Volume
-Initial changes are more linear (during inhalation and exhalation) before plateauing
Flow
-Dynamic; can only be measured when air is flowing
-Flow (Vdot) = ΔV/ΔT = ΔP/ΔR --> ΔR = airway resistance
V = P_A/R
Pressure/Volume of breathing
-Work of breathing = ΔP×ΔV
-0ABCD = total mechanical work of inspiration (sum of 0ACD = work to elastically stretch lungs and ABCE = work to overcome non-elastic resistance)
-0AECD = inspiratory work to overcome elastic resistance --> potential energy available for passive expiration
-ABCE = inspiratory work to overcome non-elastic resistance (airway resistance)
-AECF = energy required to overcome resistance to airflow during expiration --> if AECF is less than 0AECD, then expiration can be passive
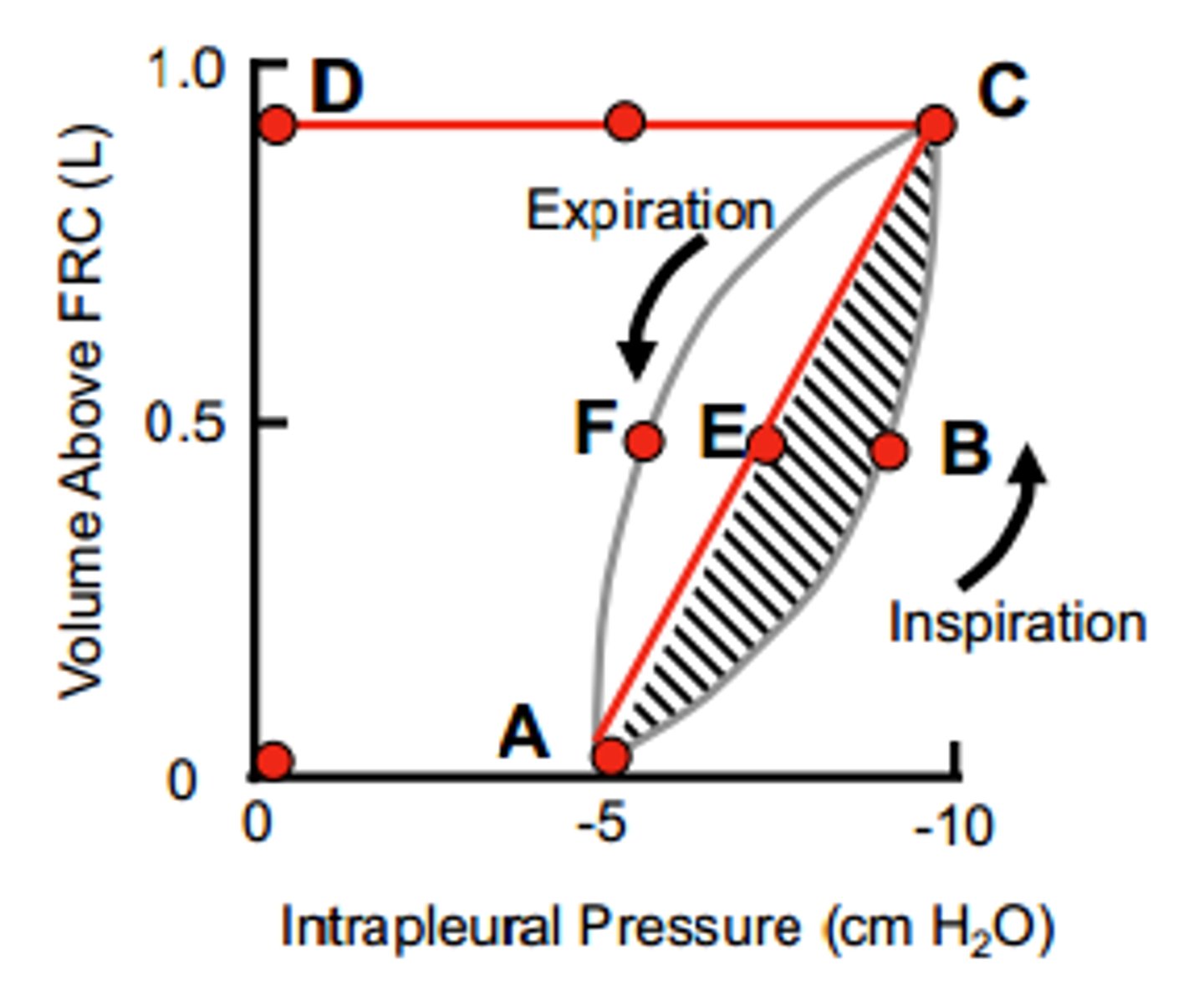
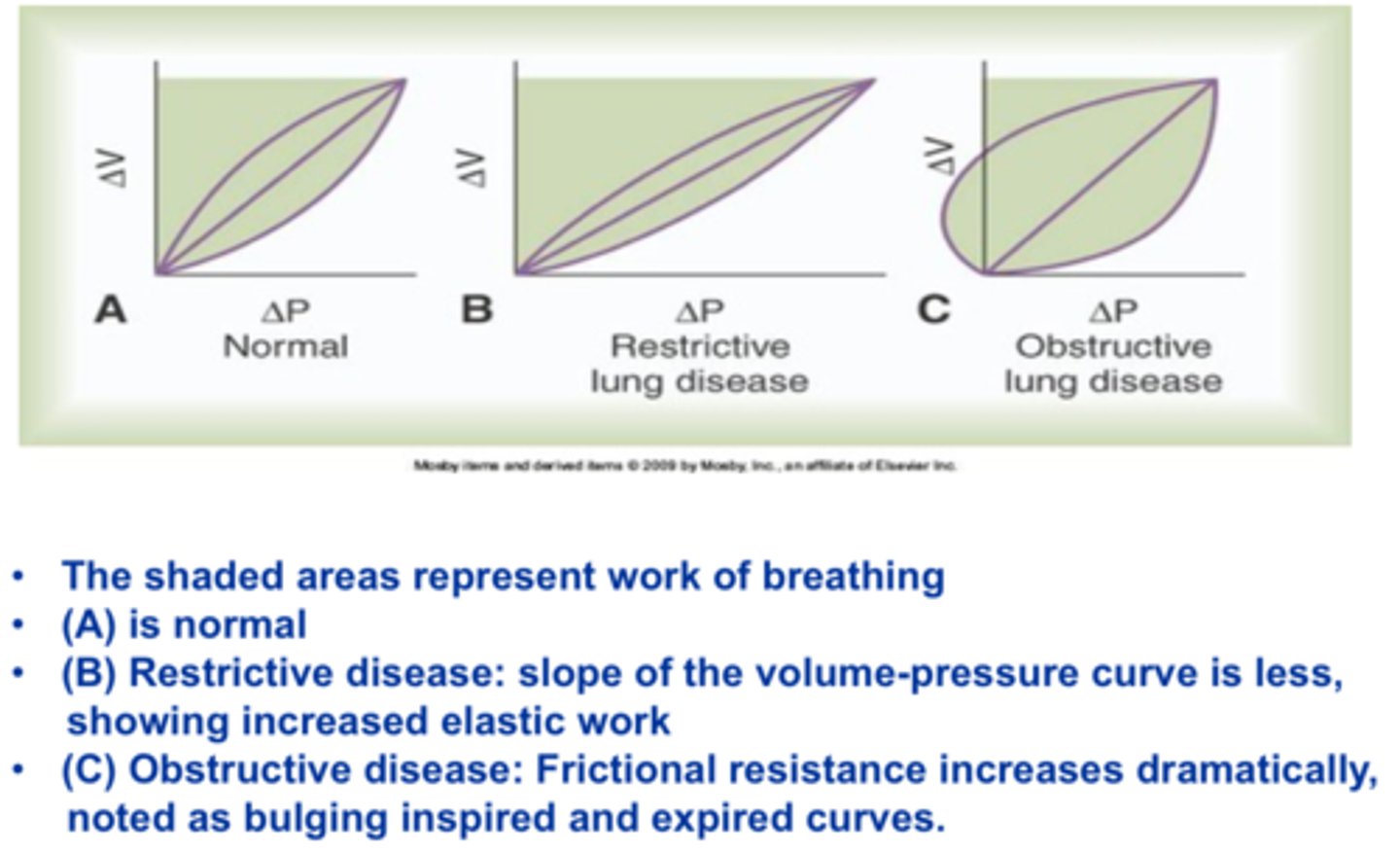
Work of Breathing - Pathology
Restrictive lung disease (e.g. pulmonary fibrosis)
-Decreased compliance
-Increased work of breathing (shallower slope)
-Little change to overcome airways resistance, but have to overcome more elastic resistance
Obstructive lung disease (e.g. asthma, bronchitis)
-Increased resistance
-Increased inspiratory and expiratory work
-Not much change in compliance, but much more resistance to airflow
-Positive intrapleural pressure --> have to have active (forced) exhalation
-Left side>right side --> compressive forces during exhalation; smaller airways (resistance)
Poiseuille's Law
-Airway resistance is proportional to viscosity of gas, length of tube and inversely proportional to r^4 --> R_Aw = 8ηl/πr^4 for laminar flow; primary site of resistance = large bronchi
-Reynold's number (Re) = 2rvd/η --> depends on radius, velocity, density, viscosity
-2000 < Re < 3000 = transitional (generations 1 - 6); Re > 3000 = turbulent flow (trachea during cough)
Factors influencing airway resistance
-Lung volume --> decreased volume = increased resistance (and vice versa --> larger volume = decreased resistance due to mechanical tethering opening nearby alveoli)
-Mucus --> decreases airway calibre (r) (especially in smaller airways)
-Edema --> decreases airway calibre
-Density of the air --> increases when diving --> increases turbulence; at altitude, less density, turbulence
-Smooth muscle contraction/relaxation --> noradrenaline from sympathetic nerves, adrenaline stimulate AC pathway, bronchodilation; ACh from vagus nerve inhibits AC, bronchoconstricts; Histamine activates IP3, causes bronchoconstriction (allergy)
-Local effects of CO2 --> increased upper airway CO2 (hypercapnia) = increased ventilation, decreased resistance; decreased upper airway CO2 (hypocapnia) = increased resistance (especially in asthmatics --> increased CO2 sensitivity?)
-Cold/Hypoxia --> airway congestion, increased secretions, decreased mucociliary clearance, pulmonary vasoconstriction, decreased chemosensitivity, decreased ventilation, bronchoconstriction
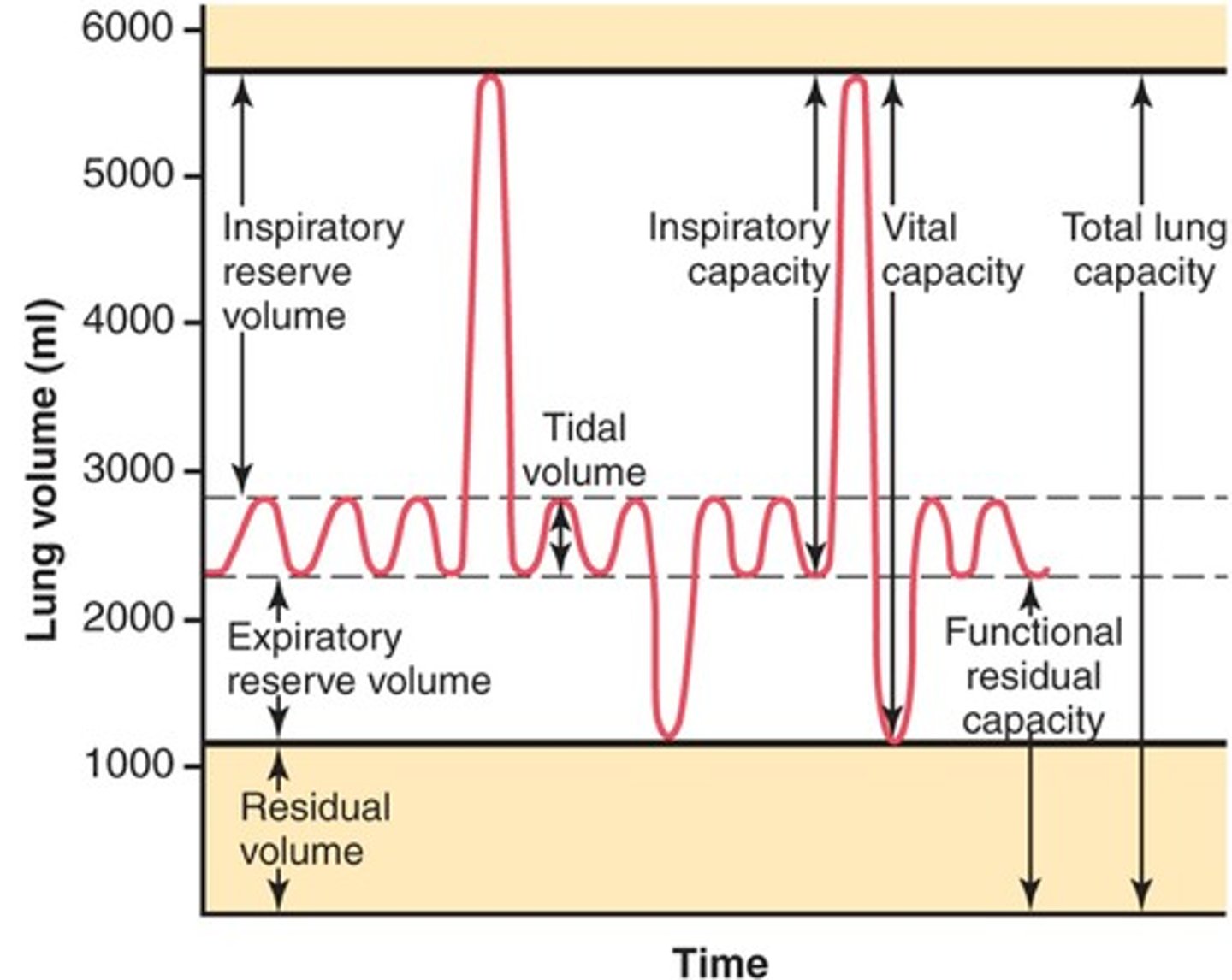
Lung Volumes and Capacities
-TV = Tidal volume = easy respiratory effort (in/out) --> ~500 mL
-IRV = inspiratory reserve volume = extra amount of air that can be inspired in maximal effort
-ERV = expiratory reserve volume = extra amount of air that can be expired in maximal effort
-RV = residual volume = amount of air that cannot leave lungs (keeps them inflated) --> 1.5 L
-TLC = total lung capacity = sum of all volumes/capacities
-VC = vital capacity = total amount of air that can be expired after maximal inspiration
-FRC = functional residual capacity = ERV + RV (volume of gas remaining after normal expiration)
-FEV1 = forced expired volume in 1 second
-V_E = minute ventilation = TV×frequency of breathing (500 mL/breath × 15 breaths/min = ~7.5L/min)
Flow-volume loop
-FVC = Forced vital capacity = width between RV and TLC
-FEV1/FVC <0.75-0.8 suggests obstructive pulmonary disease
-FEF_75/50/25 = forced expiratory flow at 75/50/25% of FVC; indicator of small airway function
-PEFR = peak expiratory flow rate --> gives information of patient's effort/consistency (want PEFR within ~10% between trials) --> increases with effort
-At low lung volumes, expiratory flow rate is the same regardless of effort ("effort independent"/"flow limited") --> only first 20% of expiration is effort dependent
-Due to increased airway resistance at low lung volumes and positive P_IP compressing airways
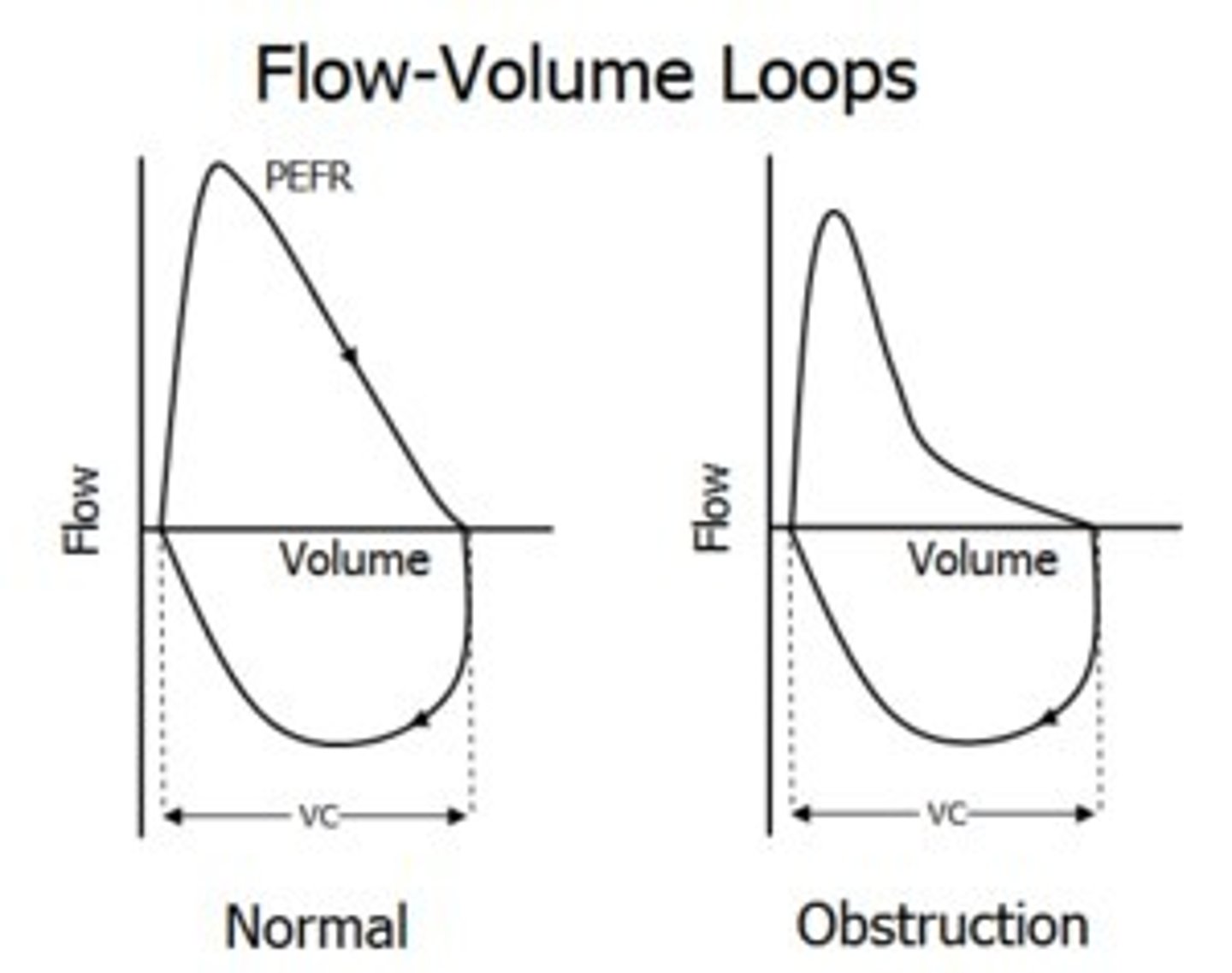
Flow-volume loops - pathology
Restrictive (e.g. pulmonary fibrosis) --> less compliant, stiffer --> cannot expand as much (total volume is lower, but same flow rates)
Obstructive (e.g. asthma, COPD) --> struggle to blow out through much narrower airways --> takes longer to blow out volume (same volume but much lower peak flow)
Helium Dilution
-Used to measure FRC
-Helium used because it has low water solubility, and low diffusion into blood
-Know initial concentration and volume (of container, V1, C1)
-After opening stopper, can measure C2 --> volume = (V1 + V2, V2 = lungs) --> solve for V2
Body Plethysmography
-Measures total thoracic lung volumes and capacities
-Utilizes Boyle's Law (P1V1 = P2V2)
-Advantage = includes volume trapped due to collapsed airways (unlike He dilution)
Measuring anatomical deadspace
-Use N2 washout to measure anatomical deadspace (does not participate in gas exchange)
-Normal inspiration/expiration --> normal quiet inhalation of 100% O2 (fill entire deadspace with O2; some mixes with alveolar air)
-Expire through a N2 meter --> initially, no N2 breathed out (pure O2 from dead space) --> rise in N2 exhalation (still some O2) --> only N2 exhaled from alveoli (plateau)
-V_Deadspace ~ 150 mL
-V_A = alveolar ventilation rate = Frequency × (TV - dead space) = 12 × (500 mL - 150 mL) = 4.2 L
Measuring physiological deadspace
-Compare arterial vs. expired CO2
-Based on perfused alveolar P_CO2 = arterial P_CO2; end expired P_CO2 = P_CO2 from perfused alveoli diluted by air from non-perfused alveoli
-If alveolar dead space, alveolar P_CO2 and arterial P_CO2 will not match
-Measure arterial P_CO2, expired P_CO2 --> measure tidal volume --> Apply Bohr Dead Space Equation
-Total Dead space ~ 250 mL
Equal Pressure Point
Before exhalation
-No flow, P_A = P_B = 0
-Intrapleural pressure (P_IP) = -30 cm H2O
-Transairway pressure (P_TA) = P_TP = -P_IP
-P_IP holds airways open against elastic recoil (P_L)
Early exhalation
-Expiratory muscles contract
-P_pl = +60 cm H2O, P_A = +90 cm H2O
-Airway pressure (P_AW) falls approaching mouth --> due to airway resistance, decreased cross-sectional area (acceleration of gas, thus decreasing pressure)
Late exhalation
-Equal pressure point = where P_AW = P_pl
-Moves towards alveoli with lower lung volumes and higher airway resistance
-Higher pleural pressure cannot increase flow due to matching of driving force by airway compression
-Usually in cartilagenous airways
-In obstructive lung disease, EPP is in non-cartilagenous airways due to higher airway resistance --> premature airway closure
Peak expiratory flow rate
-Represents the greatest flow rate during a maximal exhalation
-Commonly used to monitor asthma --> can detect abnormal function well before symptoms
-Normal values depend on age, gender, height, ethnicity, weight
-For average 175 cm, 70 kg man, PEFR = ~600 L/min
-In asthmatics, can be as low as ~250 L/min
Perfusion
-Blood flow through lungs is proportional to CO
-Affected by HR (sympathetic, parasympathetic, hormones) and SV (EDV, compliance, contractility, VR --> P_systemic filling, plasma volume)
-CO = MAP/TPR --> MAP affected by sympathetic; TPR = Poiseuille's Law --> mostly radius (compliance, VSM activity, etc.)
-Key determinant = distribution of blood throughout lung tissue
Zones of pulmonary perfusion
-Zones are determined by relationship between P_A, P_a and P_V (NOT ANATOMICAL --> can shift, e.g. with exercise)
-Do not account for phase of the cardiac or respiratory cycle
-Zone 1, 2 = apex; Zone 3 = middle; Zone 4 = bottom (base)
-Middle of lungs (zone 3) is about the same pressure as pulmonary arteries/veins
-Base has hydrostatic pressure --> slightly higher pressure than pulmonary vessels
-Apex has lack of hydrostatic pressure --> slightly lower pressure than pulmonary vessels
-Net results are due to decrease in alveolar vessel resistance and increase in extra-alveolar vessel resistance
Pulmonary Perfusion - Zone 1
-Apex
-P_A (alveolar) > P_a (arterial) > P_v
-Does not occur in normal physiology
-Can occur in certain situations (e.g. hemorrhage --> low P_a; positive pressure ventilation --> P_A is high, squishes capillary)
Pulmonary Perfusion - Zone 2
-Apex
-P_a > P_A > P_v
-Some capillary compression or closing; not a lot of blood flow at top of zone 2 or during diastole
-Bottom of zone 2/systole --> higher hydrostatic pressure in arteriole, capillary, vein --> less capillary closure (recruitment)
-Can have beat to beat pulsatile flow
Pulmonary Perfusion - Zone 3
-Middle of lungs
-P_a > P_v > P_A
-Capillaries should always be open (alveolar pressure is lowest)
-At bottom of zone 3, further increase in hydrostatic pressure in arteriole, capillary, vein --> distension
-Moving towards base, (P_a - P_v) is constant, but resistance decreases to perfusion increases
Pulmonary Perfusion - Zone 4
-Base of lungs
-P_a > P_v > P_A
-Vessels in thoracic cavity --> external pressure exposed to is intrapleural pressure
-Intrapleural pressure is less negative at bottom of lungs (gravity acts against elastic recoil) --> extra-alveolar vessels tend to collapse because P_IP is lower
Pulmonary vascular resistance
-Decrease in vascular resistance due to recruitment and distension
-Rapid increase in blood flow with decreasing resistance --> very large increases in flow with small changes in pressure (don't want large pressure changes) --> want to accommodate large changes in cardiac output with minimal changes in pressure
-Higher pressure at capillaries = more filtration = higher risk of edema
Recruitment
-Some vessels are open, but do not conduct blood --> closed vessels begin to open with smaller pressure rise (and in zone 2)
Distension
-Increases in pressure distend the vessels --> occurs with larger pressure rise or lower in lung (e.g. zone 3)
-Total resistance depends on alveolar and extra-alveolar vessels --> lowest at FRC, higher at RV and TLC
Alveolar vessels
-Expand during systole (and inspiration) --> higher pressure inside (P_capillary) and lower pressure outside (P_A)
-Compress during diastole (and expiration) --> lower pressure inside and higher pressure outside
-More compressed as alveoli expand/lung volume increases (more compression = more resistance)
Extra-alveolar vessels
-At increasing lung volumes, more negative intrapleural pressure --> pulls extra-alveolar vessels open (less resistance)
Local effects of perfusion on pulmonary blood vessels
Vasodilators
-High P_A(O2), low P_A(CO2), high pH, increased parasympathetic stimulation, histamine H2 agonists, β-adrenergic agonists (fight or flight)
Vasoconstrictors
-Low P_A(O2), high P_A(CO2), low pH, increased sympathetic stimulation, histamine H1 agonists, α-adrenergic agonists (daily stress)
Hypoxia
-low alveolar PO2 (<70 mm Hg); vasoconstricts adjacent pulmonary vessels --> increases resistance, decreases flow (opposite of systemic vessels)
-Shuts down flow to under-ventilated alveoli
-May involve chemoreceptors; involves mitochondrial O2 sensor (low O2 = less H2O2 from mitochondria, Kv1.5 inhibition, depolarize VSM, Ca2+ entry, contraction)
Hypercapnia (high CO2)
-Vasoconstricts pulmonary vessels (opposite of systemic)
-Less potent than hypoxia
-May act by decreasing pH (local effect on pulmonary VSM)
Autonomic Nervous System
-Sympathetic = less compliance, stiffens walls --> more resistance
-Parasympathetic = mild dilation
-ANS effects on pulmonary vessels are mild (more effect on airway resistance)
Metabolic factors
-Fairly unresponsive, but NO=vasodilator, β-adrenergic agonists = vasodilators, α-adrenergic agonists = vasoconstrictors
Pulmonary and interstitial fluid exchange
-Capillary hydrostatic pressure = +7 mm Hg
-Interstitial hydrostatic pressure = -8 mm Hg
-Interstitial fluid oncotic pressure = -14 mm Hg
-Plasma oncotic pressure = -28 mm Hg
Net EFFLUX = 29 mm Hg, INFLUX = -28 mm Hg
-Net = +1 mm Hg (filtration) --> increased capillary hydrostatic pressure has large effects on filtration --> lungs are designed to minimize changes in capillary hydrostatic pressure
Alveolar ventilation
Vdot_A = (TV-V_D)×breathing frequency (~5.25 L/min for 15 breaths/min --> ~70% of total ventilation)
-Can also measure alveolar ventilation from alveolar CO2 content (P_ACO2)
-Body produces ~200 mL/min of CO2 from oxidative metabolism --> CO2 produced in tissues diffuses into lungs at same rate as production
-Alveolar ventilation removes this CO2 from lungs
-Vdot_CO2 = 200 mL/min; P_ACO2 = 40 mm Hg; k= 0.863
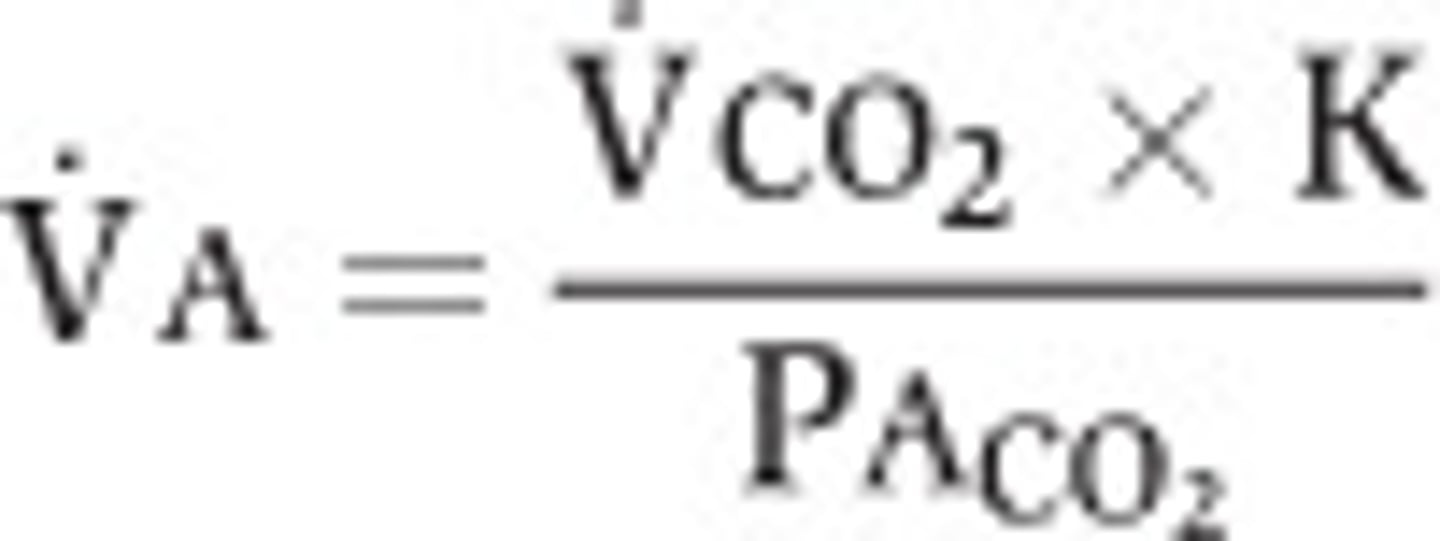
Alveolar ventilation and PCO2
-If alveolar ventilation is sufficient, P_CO2 is normal (~40 mm Hg)
-If insufficient, don't blow off enough CO2 --> P_CO2 rises (>45 mm Hg) = hypercapnia (due to hypoventilation)
-If in excess, blow off too much CO2 --> P_CO2 falls (<35 mm Hg) = hypocapnia (due to hyperventilating)
-Doubling alveolar ventilation halves P_ACO2
-At steady state, arterial P_CO2 = alveolar P_CO2
-Hyperventilation --> respiratory alkalosis --> cerebrovascular vasoconstriction --> dizziness
-Increasing alveolar ventilation also increases alveolar P_O2 --> asymptotically approaches P_iO2 (149 mm Hg)
Alveolar Gas Equation
-Amount of O2 depends on amount of CO2 --> P_AO2 = P_iO2 - P_ACO2 --> what is left of the inspired O2 after metabolism replaces some alveolar O2 with CO2
-Ratio of CO2 produced from O2 is not always 1 --> P_AO2 = P_iO2 - (P_ACO2/R) --> R = respiratory quotient; ratio of CO2 excreted to O2 taken up, depends on metabolic substrate (carbohydrate = 1, fatty acid = 0.7; usually assume R = 0.8)
-Normally P_AO2 ~ 100 mm Hg
Uneven distribution of ventilation
-At FRC, P_IP is lower at base, so alveoli are underinflated and more compliant --> a given ΔP_IP during inspiration causes greater ΔV near base (compared to apex) of lungs
-Alveolar ventilation increases from apex to base of lungs due to gravity and posture
Uneven ventilation distribution - pathology
Reduced compliance
-Stiff lung has lower volume at FRC and half the normal tidal volume (for reduction of compliance by half)
-Increased non-uniformity of ventilation
Increased airway resistance
-Obstructed lung has same volume at FRC, but slower filling/emptying
-Non-uniformity of ventilation increases with increased breathing rate
Reduced lung volume (e.g. reduced by a mass)
-Filled lung has half of normal volume at FRC and half of normal tidal volume
-Constant non-uniformity of ventilation
V-Q ratio
-V/Q ratio is 1 just above heart (~mid-lung height)
-Q falls off faster than than Vdot_A from base to apex
-Regional V/Q ratio affects alveolar gas composition
-V/Q = ∞ in inspired air since no contact with blood (0 perfusion)
-V/Q = 0 in mixed venous blood since ventilation = 0 (e.g. pulmonary artery)
-P_AO2 is higher at apex than base of lungs
V-Q Mismatch
Lack of perfusion (e.g. pulmonary emboli)
-Alveolar PO2 rises, PCO2 falls (V/Q --> ∞)
-Compensation to normalize mismatch --> perfusion of other lung tissue increases (increases V/Q non-uniformity)
-Low airway P_CO2 causes bronchiolarconstriction --> decreased ventilation --> higher V in neighbouring airways
-Decreased perfusion causes type II alveolar cells to secrete less surfactant --> less ventilation (smaller tidal volumes on blocked side)
Lack of ventilation (e.g. blocked airway)
-Alveolar P_O2 falls, P_CO2 rises (V/Q = 0)
-Acts like a shunt --> decreases systemic P_CO2 (only in severe blockage)
-Compensation to normalize mismatch --> increased ventilation of other lung (V/Q increases)
-Hypoxia (and hypercapnia) cause vasoconstriction of vessels, decrease perfusion (to blocked lung)
-Shifts perfusion to healthy lung; restores V/Q close to normal levels
Diffusion in the Lungs
V(dot)_net = D_L × (P1 - P2)
V(dot)_net = movement of the gas
D_L = diffusion capacity of the lungs
P1 - P2 = driving force --> (P_alveolus - P_capillary)
-Diffusion capacity depends on solubility (Henry's Law), MW/density (Graham's Law), tissue thickness, diffusion distance, gas concentration (Dalton's Law, Henry's Law)
-Concentration gradient depends on gas concentration (Dalton's Law, Henry's Law)
Determinants of diffusion capacity
1) Graham's Law --> Diffusion ∝ 1/√(molecular weight) --> bigger gas diffuses slower
2) Henry's Law --> [O2]_dissolved = solubility × PO2 --> solubility is proportional to 1/Temperature (decreases in deeper circulation; if warm is not warmed up, will come out as emboli)
3) Dalton's Law --> P_B = P_O2 + P_CO2 + P_N2 --> individual partial pressures change as barometric pressure changes
4) Tissue thickness
5) Diffusion distance (includes liquid barriers)
6) k --> how the gas interacts with the barrier
Fick's Law
V(dot)_net = [k((A×s)/a√(MW))]×(P_A - P_C)
-k = conversion factor, A = surface area for diffusion, s = solubility coefficient, a = tissue thickness
In Lungs
-Large surface area of alveoli and capillaries in close proximity minimizes diffusion distance
-Thin epithelial/endothelial layers to minimize tissue thickness
-Substantial partial pressure differences
-Gases with advantageous diffusion properties
Diffusion steps
Alveolar air --> water layer --> interstitial space --> blood plasma --> red blood cell cytoplasm
-Diffusion capacity ∝ 1/resistance --> 1/D_Membrane = 1/D1 + 1/D2 + ... + 1/D10
-Therefore, 1/D_lung = 1/D_M + 1/(θ×V_C)
θ = rate of O2 uptake by Hb, V_C = capillary blood volume
Measurement of pulmonary diffusion capacity
-Maximum inspiration (He and CO) --> hold breath for 10 seconds --> expiration --> greater reduction in CO = greater diffusion capacity
-Normally ~ 24 mL/min/mm Hg (for CO)
-Diffusion capacity is reduced in disease (e.g. pulmonary fibrosis --> 15 mL/min/mm Hg) --> due to inflammation and scar formation (barrier to diffusion)
Diffusion/Perfusion limits
-Alveolar-capillary diffusion rate is analogous to workers at rail station
-Pulmonary blood flow is analogous to train speed
-# of cars/train is analogous to [Hb]
-Not enough workers = not all Hb loaded
-Too many workers = train already full
-Increased train speed = not enough time to load
-Decreased train speed = too much loading
Diffusion/Perfusion limits of gas exchange
-Carbon monoxide is diffusion limited (does not equilibrate)
-Nitrous oxide is perfusion limited
-O2 and CO2 are perfusion limited (equilibrate with alveoli ~1/3 of the way)
-Fast O2 equilibration is due to 75% Hb saturation in mixed venous return; large P_AO2 and P_aO2 gradient; high D_L (compared to CO) because O2 binds to Hb faster (higher θ)
-Total transit time of blood ~0.75 seconds; to get more O2 in blood, increase blood flow (more O2 in blood per unit time)
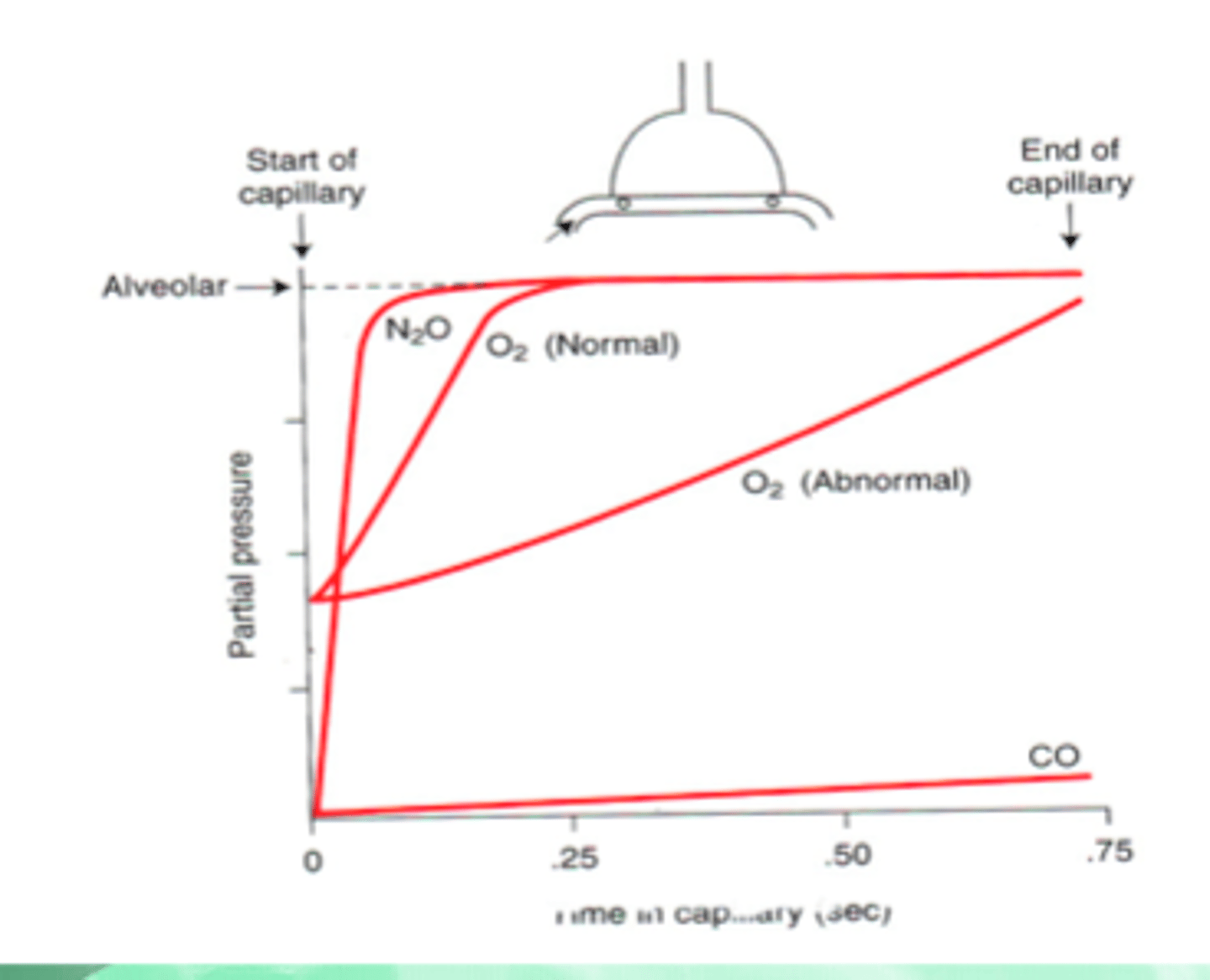
Diffusion/Perfusion limits of gas exchange - external factors
Exercise
-Higher CO = lower pulmonary capillary transit time (down to about 250 ms)
-Higher CO = more recruitment/distension --> more surface area = higher D_L
-O2 is still perfusion limited (equilibrates near end) except in elite athletes
Altitude
-Lower P_BO2 and P_vO2 (venous) --> lower O2 gradient
-Lower P_vO2 = Hb loading at steeper part of O2 dissociation curve --> more O2 uptake before equilibrium (smaller change in PO2 = more uptake)
-Net result = same shape, but lower partial pressures in capillary
Altitude + Exercise
-O2 diffusion can become diffusion limited (same shape as normal exercise, but lower partial pressures)
Oxygen transport
-2% dissolved in plasma and RBC cytoplasm --> can only supply about 15 mL/min (demand ~250 mL/min)
-98% bound to hemoglobin (increases O2 carrying-capacity by 65 fold)
Hemoglobin
-92% of hemoglobin is adult hemoglobin (HbA)
-Binds 4 O2 molecules
-2α and 2β subunits
-Each RBC has 280 million Hb molecules
-When 1 oxygen binds, Hb flattens, causes other affinities to increase (cooperative binding)
Sickle Hemoglobin (HbS)
-Genetic variant common in subsaharan Africa
-Mutation in β globin gene of Hb; causes polymerization of Hb --> distorts into a sickle shape; can occlude small vessels (sickle cell crisis)
-Prone to haemolysis (chronic hemolytic anemia), but resistant to parasites (e.g. malaria)
Fetal Hemoglobin (HbF)
-Has 2α and 2γ subunits
-Has increased O2 affinity
-Not inhibited by 2,3-DPG
-Newborns have both HbA and HbF; HbF levels decrease to adult normal (1-2%) by age 1
-Too much Hb = jaundice (kidneys produce too much bilirubin)
Oxygen content and delivery
-Hemoglobin bound O2 = 204mL O2/L
-Dissolved O2 = 2.9 mL O2/L
-Total arterial O2 content = 206.9 mL O2/L
-O2 delivery = cardiac output × arterial O2 content = 1034.5 L O2/min
-O2 consumption = cardiac output × arterial/venous O2 concentration difference (4 mL/dL) = 200 mL O2/min
-Arterial oxygen extraction = (CaO2 - CvO2)/(CaO2) = (20 - 16)/(20) = 20% --> 20% of available O2 is used
Oxyhemoglobin dissociation curve
-P_AO2 = 103 mm Hg
-P_aO2 = 90-95 mm Hg (~98% saturation of Hb)
-P_cO2 = 40 mm Hg
-P50 = pressure at which 50% of Hb binding sites are filled = 27 mm Hg
-Sigmoid shape is due to cooperative binding
-Low PO2 = low O2 affinity; Hb in T-state (tensed)
-Moderate PO2 = Hb shifts towards R-state (relaxed); higher O2 affinity
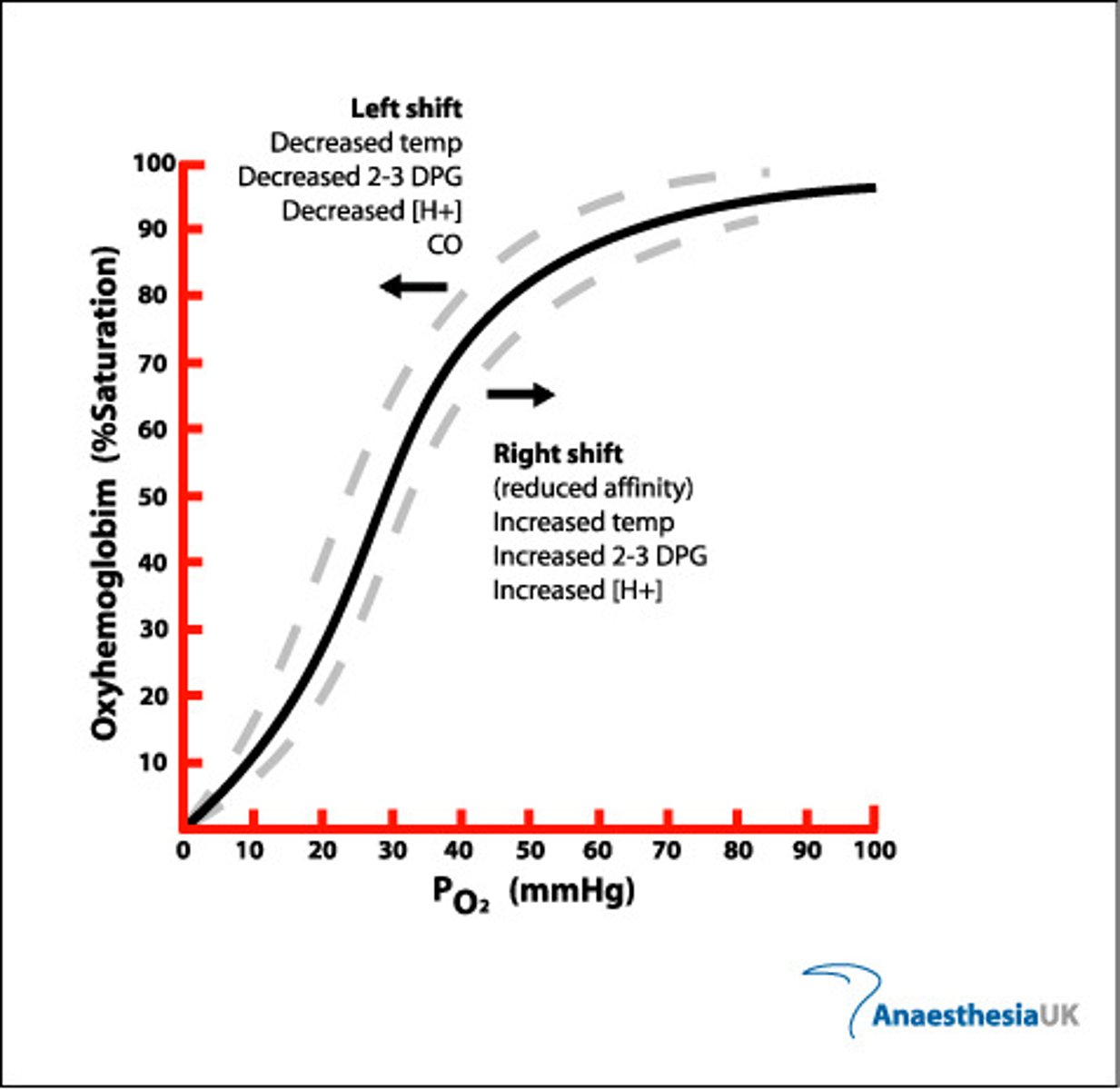
Factors that affect the oxyhemoglobin dissociation curve
-Physiological affinity changes have minimal effect on Hb saturation at normal P_aO2 (80-100 mm Hg), but left shift can reduce offloading at tissues
pH and CO2
-Decreasing pH (acidosis) or increasing CO2 --> right shift; reduced Hb saturation at a given PO2
-Decreased affinity aids O2 release at metabolically active tissues
Temperature
-Increasing temperature --> right shift; increases O2 release at tissues during exercise, reduces O2 release at extremities in cold
2,3-DPG (diphosphoglycerate)
-2,3-DPG is a metabolic intermediary
-Accumulates in RBC; due to anaerobic metabolism of RBC (no mitochondria)
-Reduces Hb affinity for O2
-Increasing 2,3-DPG causes a right shift
-2,3-DPG increases with exercise, altitude, anemia, pregnancy and alkalosis
CO2 transport - arterial
-89-90% transported as bicarbonate ions (HCO3-)
CO2 + H2O --> H2CO3 (carbonic acid) --> HCO3- + H+ (catalyzed by carbonic anhydrase)
-H+ is buffered by plasma proteins of Hb; venous blood is normally slightly more acidic (pH = 7.35) compared to arterial (pH = 7.4)
-HCO3- leaves cell in exchange for Cl- (chloride shift); H2O also enters to maintain electrostatic and osmotic balance
-5% of CO2 is transported dissolved in plasma/cell cytoplasm
-5-6% of CO2 is transported as carbamino compounds
CO2 transport - venous
68% = HCO3-
22% = Carbamino compounds
10% = dissolved
-(deoxy)Hb transports CO2 better than (oxy)Hb --> Haldane effect
CO2 dissociation curve
-Deoxygenated Hb has greater affinity for CO2, more readily forms carbamino Hb
-As hemoglobin releases O2 at tissues, more likely to pick up CO2
-At the lungs, high PCO2 causes CO2 to unload more easily
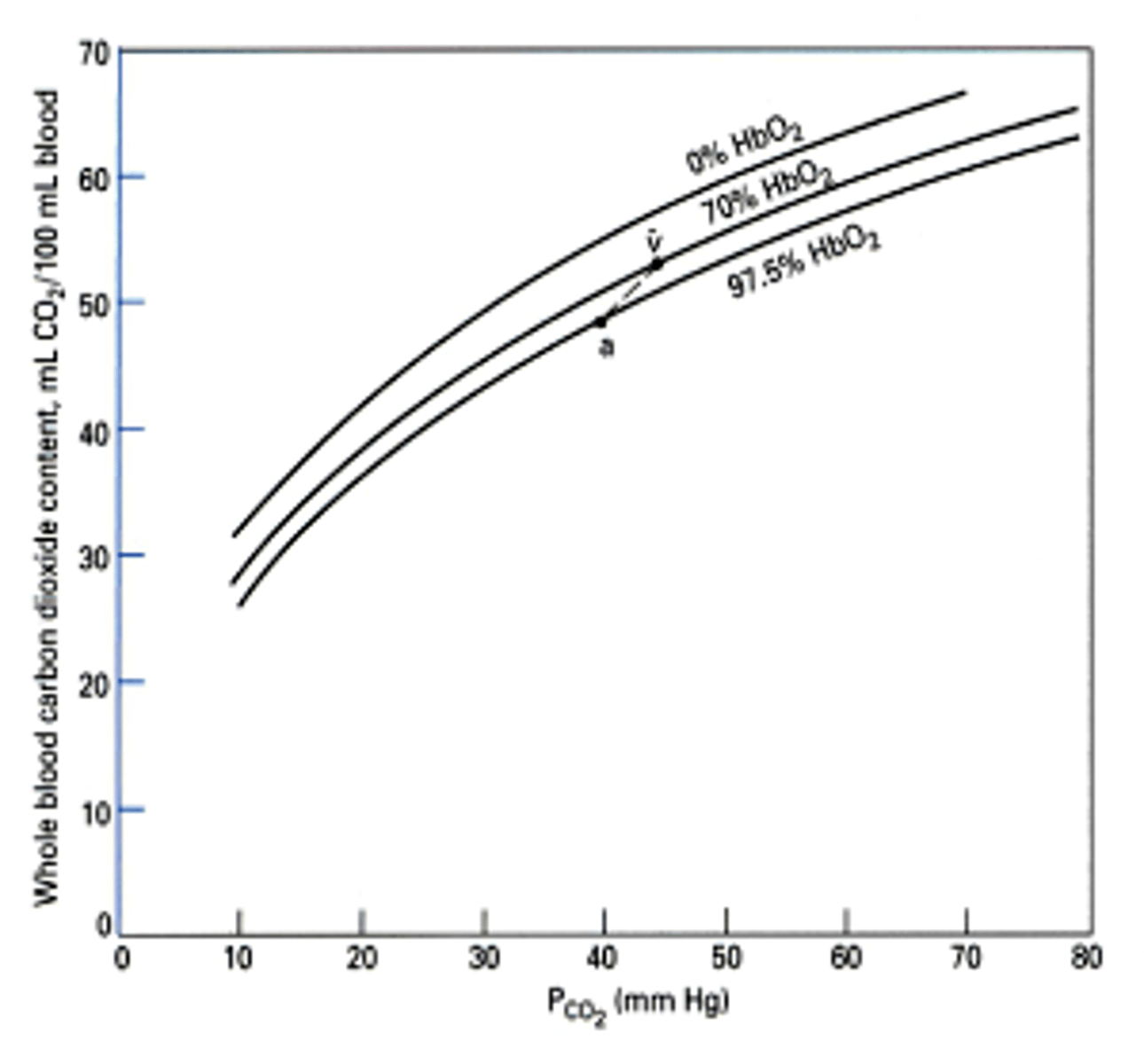
Increasing CO2 carrying capacity
-Increase PCO2 --> (Henry's Law/solubility, increased HCO3-, increased carbamino hemoglobin)
-Increase plasma protein concentrations --> increases pH buffering, drives HCO3- production
-Increase Plasma pH --> increases HCO3-
-Increase hemoglobin concentration --> increased carbamino hemoglobin, increased pH buffering, increased HCO3-
-Decrease PO2 --> increases carbamino hemoglobin
pH Regulation
-Normal plasma [H+] = 40 nM; pH = 7.4 --> sensitive to very small changes; almost all enzyme/protein activity is regulated by H+
-Precisely regulated --> pH < 7.35 = acidosis; pH > 7.45 = alkalosis
-Buffered by blood proteins and hemoglobin; buffers reversibly bind H+ and stabilize pH
-Buffer power β = # moles of strong base/1 pH unit change
-pH is lower in venous blood than arterial --> due to more CO2
Henderson-Hasselbalch Equation
pH = 6.1 + log [HCO3-]/(.03×P_aCO2)
-When pH is disturbed, due to change in [HCO3-] or P_aCO2
-Changes in [HCO3-] = metabolic disturbances; changes in P_aCO2 = respiratory disturbances
-HCO3- controlled by kidneys (takes hours-days); PaCO2 controlled by lungs --> independent control; powerful buffer
Compensation of acid-base disturbance
-Chemical (intra/extracellular) buffering --> fast but limited capacity (<1 second)
-Changes in ventilation --> slower but less limited (takes minutes to occur)
-Change in kidney secretion --> slowest but greatest capacity (hours - days)
-Metabolic disturbance (change in [HCO3-]) = respiratory compensation (change in PaCO2); respiratory disturbance (change in PaCO2) = metabolic compensation (renal change in [HCO3-])
-Low [HCO3-] or high PaCO2 = acidosis
-High [HCO3-] or low PaCO2 = alkalosis
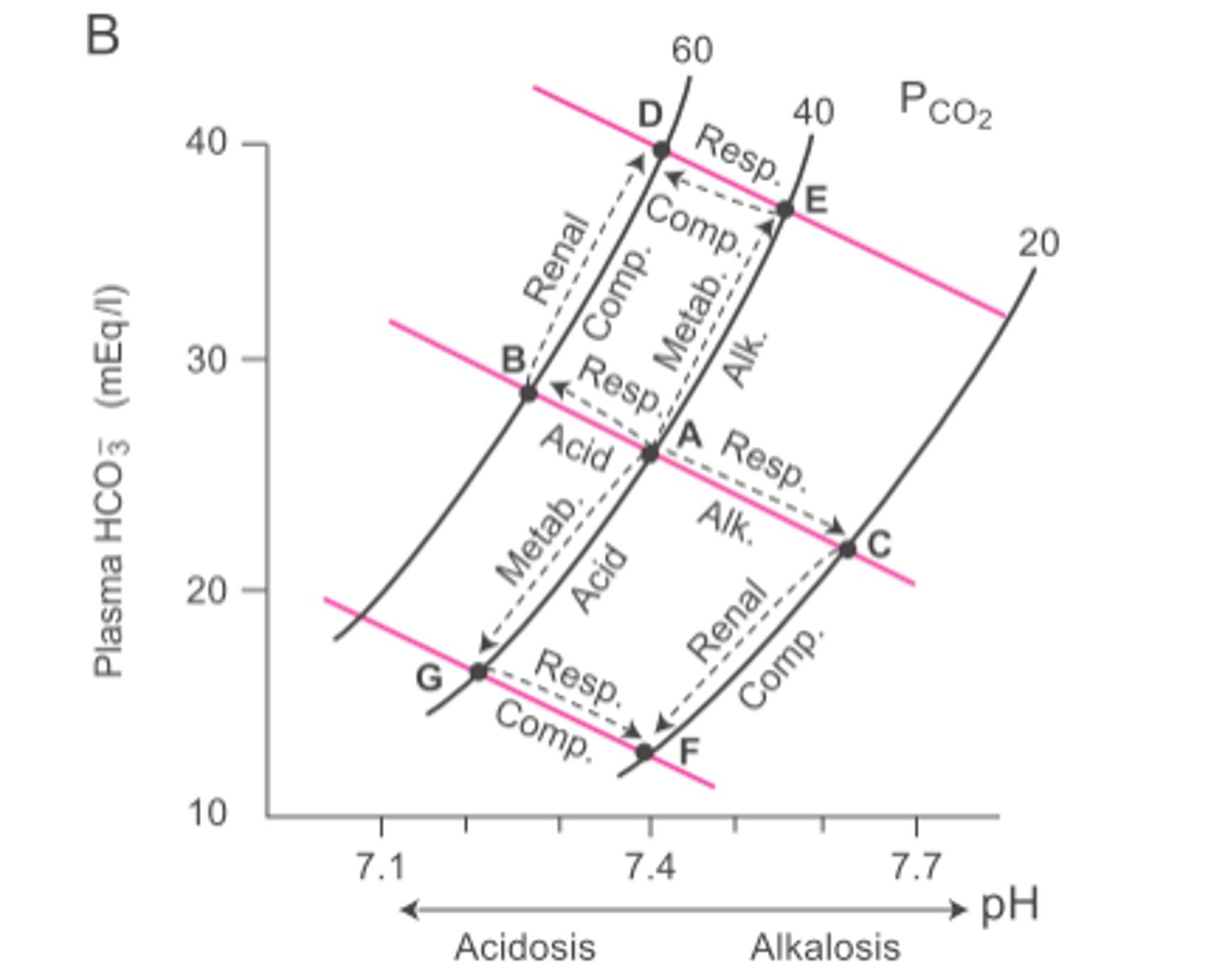
Davenport diagram
-Helps estimate HCO3- and non-HCO3- buffer equilibrium --> HCO3- buffers most of the H+ changes, B^(n) buffers (non-HCO3- buffers) most of the remainder
-Displays pH against [HCO3-] for a given PaCO2 (isopleth) --> different PCO2 = different isopleth; higher PCO2 = higher [HCO3-] for a given pH
-Multiple buffers that are not HCO3-; covers a wide range --> total gives slope (buffering power) of ~25 mM/pH unit
-Diluted blood = reduced buffering power (flatter slope)
![<p>-Helps estimate HCO3- and non-HCO3- buffer equilibrium --> HCO3- buffers most of the H+ changes, B^(n) buffers (non-HCO3- buffers) most of the remainder</p><p>-Displays pH against [HCO3-] for a given PaCO2 (isopleth) --> different PCO2 = different isopleth; higher PCO2 = higher [HCO3-] for a given pH</p><p>-Multiple buffers that are not HCO3-; covers a wide range --> total gives slope (buffering power) of ~25 mM/pH unit</p><p>-Diluted blood = reduced buffering power (flatter slope)</p>](https://knowt-user-attachments.s3.amazonaws.com/f71602b1-ec23-498c-a91f-c9d72a603688.jpg)
Respiratory acidosis
-Develops when lung gas exchange is impaired (e.g. barbituates, pneumonia, emphysema)
-PaCO2 increases, decreased pH, increased [HCO3-]
-Metabolic compensation (renal) = increased renal H+ secretion --> drives renal reabsorption of HCO3-
-PaCO2 is still elevated, but acid disturbance is mostly compensated
Net result: high PaCO2, low pH, high HCO3-
Respiratory alkalosis
-Develops due to hyperventilation (e.g. altitude, anxiety)
-PaCO2 decreases, pH increases, decreases HCO3-
-Metabolic compensation (renal) = decreased renal H+ secretion --> decreases plasma [HCO3-]
-PaCO2 is still low, but base disturbance is mostly compensated
Net result: low PaCO2, high pH, low HCO3-
Metabolic acidosis
-Develops from ketoacidosis, diarrhea (loss of HCO3-)
-Decreases [HCO3-], decreases pH
-Respiratory compensation = increased [H+] stimulates peripheral (directly) and central (indirectly) chemoreceptors --> results in hyperventilation (decreases PaCO2, brings pH back to normal)
-PaCO2 is low, but acid disturbance is mostly compensated
Net result: low PaCO2, low pH, low HCO3-
Metabolic alkalosis
-Develops from vomiting (loss of H+ in gastric juice)
-Increases pH, increases [HCO3-]
-Respiratory compensation = decreased [H+] reduces chemoreceptor stimulation --> results in hypoventilation (causes plasma CO2 retention, pH to fall to normal)
-PaCO2 is high, but base disturbance is mostly compensated
Net result: high PaCO2, high pH, high HCO3-
Davenport diagram/Acid-Base Nomogram
Acid-Base Nomogram --> shaded areas show normal compensatory limits for each condition; clinically useful
-Only requires measurement of blood pH and PaCO2 for diagnosis
Davenport Diagram
-Shows compensatory mechanisms for primary respiratory and metabolic changes in plasma pH
-Moving to new isopleth = respiratory problem --> shift along isopleth (metabolic compensation) to return to normal pH
-Shift along isopleth = metabolic problem --> move to a new isopleth (respiratory compensation) to return to normal pH