Cell Bio Exam 2
1/196
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
197 Terms

Both
1957 Ribonuclease Experiment
Denature by heat (breaks noncovalent bonds)
Denature by reducing agents (breaks disulfide bonds)
Protein spontaneously refolds when these elements are removed
Significance: folds in vitro, solely by primary amino acid seq interactions
Can you predict the structure of proteins?
With AI models like Alphafold2
Chaperones use ATP hydrolysis to drive protein folding of hydrophobic domains
does not drive, more passive
Chaperones
Bind hydrophobic regions
Protein misfolding, aggregation
Prevent premature interactions/binding
Co-translational binding, before interacting domains are synthesized
Bound before subcellular targeting is initiated
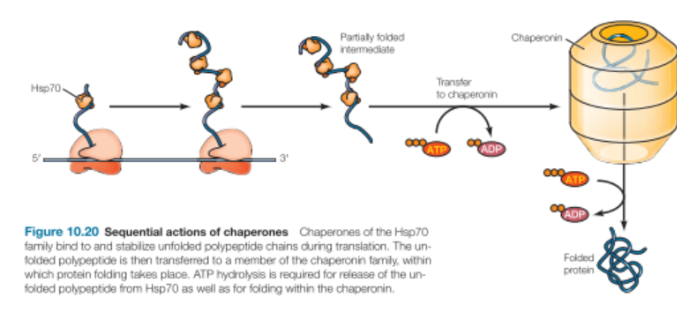
Hsp70
Bind hydrophobic parts of unfolded regions
Release and rebind with ATP hydrolysis
Cycles on and off until hydrophobic regions find each other in the correct fold
When hydrophobic domains are no longer available, Hsp70 no longer binds
Found in cytosol and in subcellular organelles
The unfolded polypeptide will then be transferred from Hsp70 to chaperonin to fold
Chaperones definition
Proteins that facilitate the folding of other proteins
Act as catalysts that facilitate assembly w/o being part of the assembled complex
How does chaperones function?
By binding to and stabilizing unfolded or partially folded polypeptide chains that are intermediates along the pathway leading to the final correctly folded state
This binding stabilizes the amino-terminal portion in an unfolded conformation until the rest of the polypeptide chain is synthesized
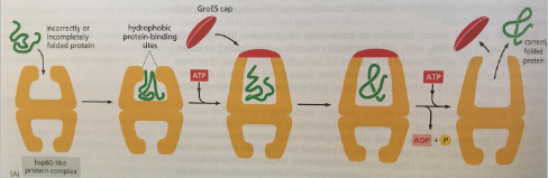
Chaperonins
Hsp60 Family
Chamber protein within which protein folding takes place
Consists of multiple protein subunits arranged in 2 stacked rings to form a double-chambered structure
Shields unfolded polypeptide chains from the cytosol within its chamber
ATP binding is required for
Association of cap
Removal of cap and release of protein
How does the chaperonin work?
E. Coli GroEL subunits rotate hydrophobic to hydrophilic surfaces, promoting protein folding
cytosol
the aq component of the cytoplasm of a cell, within which various organelles and particles are suspended
Ubiquitin
Small protein
Links to lysine in proteins
Attachment can mark proteins for degradation
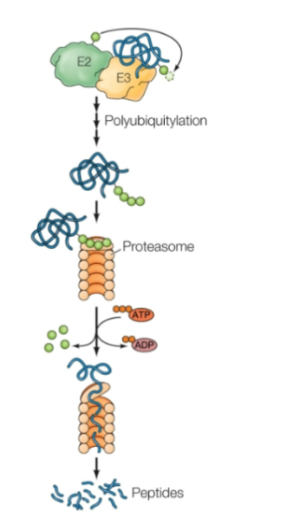
Ubiquitin-Proteasome Pathway
Major mediator of regulated protein degradation
Ubiquitin is activated by E1 with ATP
The ubiquitin is then transferred to E2
The ubiquitin is then transferred to the target protein by E2 complexed with a third protein, called ubiquitin ligase (E3).
Proteins targeted for degradation are marked by the addition of multiple ubiquitins to form a polyubiquitin chain, which is catalyzed by some E3s. These are recognized and degraded by the proteasome (a large, multisubunit protease complex)
How many E3s in mammalian cells?
About 600 to confer target specificity
Proteasome lid
Binds polyubiquitin chain
Removes ubiquitin subunits (ATP hydrolysis)
Unfolds and translocates protein into center chamber
Central barrel complex
hollow core lined with proteases that cleave peptide bonds to chop into peptide fragments
Recycling
peptides later digested by cytosolic peptidases into amino acids for cellular recycling
Angelman’s Syndrome:
Genetic disorder of nervous system
Developmental and cognitive disabilities, lack of speech, overly happy syndrome
Cause:
Ch 15 deletion, includes UBE3A
E3 ligase needed for targeted protein degradation in neuron development
Protein activity regulation
Binding small molecule regulators
Post-translational modifications
Protein-protein interactions
Complexes
Subcellular organization: condensates membrane-less organelles (ex: nucleoulus)
Protein Kinase A
Inactive: when regulatory subunits (R) are bound to the catalytic subunit (C)
Active: when 2nd messenger molecule, cAMP binds the regulator and it changes conformation/lets go of the now active catalytic subunits
cAMP dependent protein kinase
A tetramer consisting of 2 regulatory and 2 catalytic subunit
Cyclic AMP binds to the regulatory subunits, leading to their dissociation from the catalytic subunits. The free catalytic subunits are then enzymatically active and able to phosphorylate serine residues on their target proteins.
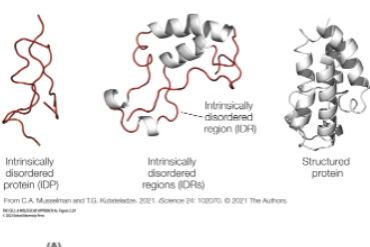
Intrinsically disordered regions
polar and charged domains, available for interactions
bind other proteins, RNA Concentrate and separate into discrete domains
Participate in membrane-less compartments
Phospholipids
Amphipathic molecules consisting of two hydrophobic fatty acid chains linked to a phosphate-containing hydrophilic head
Spontaneously form bilayers in aq solutions bc their fatty acid tails are poorly soluble in water so the hydrophobic tails will be buried in the interior of the membrane and the polar head grps will be exposed on both sides in contact w water
Amyloids
Fibrous aggregates of misfolded proteins
Misfolded proteins aggregate to form insoluble amyloid fibrils characterized by beta-sheet structure
Alzheimer’s disease
Characterized by amyloid plaques in the brains of patients. These form due to aggregation of amyloid beta protein
Neurodegenerative, common cause of dementia
Alzheimer’s may be caused from the toxicity of soluble forms of ABeta oligomers rather than accumulation of amyloid plaque
The amyloid beta protein plaques were a correlation w alz
The mutations in amyloid beta protein genes cause disease (causation)

Prions
Misfolded proteins that are capable of self-replication
Misfolded proteins that stimulate further misfolding
Inherited Alzheimer’s Disease may be caused by…
Mutations in amyloid precursor protein gene
Mutations in proteinase (catalyzes proteins into smaller polypeptides, so w/o this amyloid proteins are less regulated)
Aduhelm Trial
Cleared plaques
Did not effect symptoms (as in cognitive decline)
Very expensive
Donanemab
Clears plaque and slows cognitive decline
Verrrry expensive, serious side effects
So what can we learn from all the Alzheimer’s Disease research mishaps?
We should critically evaluate
We should be more open to other hypotheses outside of the amyloids
Can look at Tau protein
Stabilizes microtubules in neurons
Mutations can cause dementia
Mouses may not be a relevant model for human disease
Transmissible spongiform encephalopathy
AKA prion diseases
Kuru
To shake
1950s New Guinea unusual human disease, rare and fatal
Cannibalism practice of eating tissue of deceased elders
Long incubation period
Prion disease
Scrapie
Sheep
Progressive
Fatal
Brain deterioration
Persistent scratching
Related to chronic wasting disease
Creutzeldt-Jakob Disease
Human
Rare inherited and sporadic disease
Mad Cow Disease
Contaminated feed (probably containing scrapie sheep tissues)
4 million cows killed to prevent spread to other cattle and humans
1980s-1990s spread to humans
Stanley Prusiner: Basis for Prion Diseases
So at first they thought it was a virus, BUT it was resistant to nucleases
Sensitive to proteases → proteins only hypothesis
Once sequence identified → single proteins → prions (encoded in genome)
PrPC
Prion protein normal fold
Susceptible to proteases
Remains monomeric
Not disease related
α-helical form
PrPSc
Forms a misfolded amyloid structure
Can propagate by inducing the misfolding of PrPcs to the amyloid PrPSc
Can infect a cell and replicate by inducing autocatalytic amyloid formation of endogenous PrPc
More resistant to proteases (an enzyme which breaks down proteins and peptides)
Polymeric
Disease-related
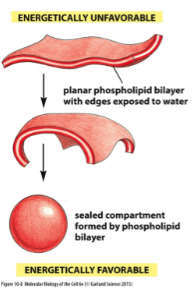
Phospholipid Bilayer
Forms a stable barrier bw 2 aq compartments and represent the basic structure of all biological membranes
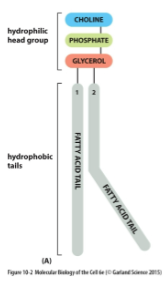
Phospholipid Structure
Hydrophilic head
Hydrophobic tail - fatty acid tail
Fatty acid chain length and degree of saturation varies
Typically, one chain is saturated, one is unsaturated
Net negative charge inner surface of bilayer due to PS in inner leaflet
How are phospholipid membranes built and distributed?
Synthesized on the Endoplasmic Reticulum - cytosolic leaflet where most phospholipids are made
Scramblases distribute lipids symmetrically bw leaflets
Cholesterol
Abundant in animal cell membranes
Rigid ring structure
Hydrophilic hydroxyl head grp
Hydrophobic tail
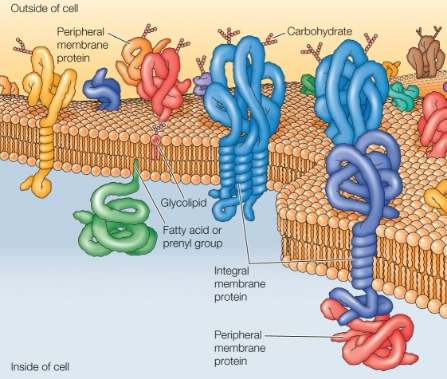
Fluid Mosaic Model
Proteins are inserted into the lipid bilayer
Membrane proteins carry out the specific functions
Integral membrane proteins: embedded directly within the lipid bilayer
Single pass
Multi-pass
Peripheral membrane proteins: membrane association is indirect, through protein-protein interactions
How do we dissociate integral membrane proteins?
Via detergents
Hydrophobic tails bind hydrophobic regions of integral membrane proteins, forming detergent-protein complexes that are soluble in aq solution
How do we dissociate peripheral membrane proteins?
Dissociate from membrane by extreme pH or high salt
Soluble in aq solutions
Alpha helixes and beta barrel folds
Integral membrane protein transmembrane domains
Rich in hydrophobic amino acids
Typically folded as alpha helix or beta barrels
Folds that shied polar peptide bond thru H bonding
Hydropathy Analysis
Can predict potential alpha-helical transmembrane domains from primary aa seq, rich in hydrophobic amino acids
Temperature and Membrane Fluidity
👆 thermal energy = 👆 fluidity
Fatty acid chains associate less tightly, more fluid
Saturation and membrane fluidity
Lipids containing unsaturated fatty acids increases kinks in the fatty acid chains due to the dbl bonds ▶ more fluid membrane
Fatty Acid Chain Length and Fluidity
Interactions bw shorter fatty acids chains are weaker than those w longer chains ▶ membranes w shorter fatty acid chains are less rigid and more fluid
Cholesterol and Membrane Fluidity
At low temp, increases fluidity (separates phospholipid fatty acid chains, loosens packing)
At high temp, decreases fluidity (rigid structure among fatty acids, restricts fluidity)
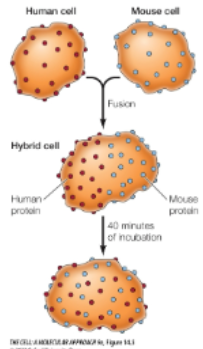
How do we know proteins are mobile in membranes?
1970s researchers
Used anti-human and anti-mouse antibodies labeled w diff fluorescent dyes
Formed human/mouse hybrids
Labelled human and mouse proteins intermingled over cell surface within 40 min of incubation
“This lateral movement of membrane proteins was first shown directly by Larry Frye and Michael Edidin in 1970. Frye and Edidin fused human and mouse cells in culture to produce human–mouse cell hybrids (Figure 15.5). They then analyzed the distribution of proteins in the membranes of these hybrid cells using antibodies that specifically recognize proteins of human and mouse origin. These antibodies were labeled with different fluorescent dyes, so the human and mouse proteins could be distinguished by fluorescence microscopy. Within 40 minutes after fusion, the mouse and human proteins became intermixed over the surface of hybrid cells, indicating that they moved freely through the plasma membrane”
FRAP
Fluorescence Recovery After Photobleaching
“. In this technique, a region of interest in a cell expressing a GFP-labeled protein is bleached by exposure to high-intensity light. Fluorescence recovers over time due to the movement of unbleached GFP-labeled molecules into the bleached region, allowing the rate at which the protein moves within the cell to be determined”
The Fate of a newly synthesized protein
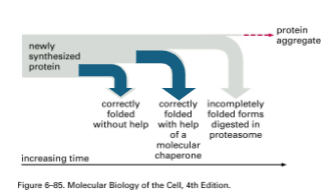
Heat Shock Proteins
Chaperones & chaperonins are actually heat-shock proteins
They accumulate in response to exposing cells to high-temperature
“Many HSPs function as molecular chaperones to protect thermally damaged proteins from aggregation, unfold aggregated proteins, and refold damaged proteins or target them for efficient degradation.”
Explain the 2 different possible consequences if protein folding fails in cells, and how each could contribute to disease
Misfolded protein aggregation → neurodegenerative diseases like Alzheimer’s
Loss of protein function → cystic fibrosis
“Cystic fibrosis is caused by mutations in CFTR (responsible for the transport of chloride ions across the plasma membranes of several types of cells, such as those lining the respiratory tract). “Defective chloride transport as a result of these mutations leads to obstruction of the respiratory tract by thick plugs of mucus, leading to recurrent infections and the death of most patients from lung disease.”
Mouse Model for Alzheimer’s Disease
They did a mouse model w mutant human APP (amyloid precursor protein) gene
Observe plaques
Neurodegenerative symptoms
Test and develop alz therapeutics
Vaccination against ABeta peptide
But does this really represent humans w alz?
Describe why transmembrane domains frequently have alpha-helical or beta barrel structures.
1. Alpha-Helical Transmembrane Domains
Most common in eukaryotic and prokaryotic membrane proteins
🔹 Why Alpha-Helices?
Hydrophobic Amino Acid Side Chains: The outer surface of the helix contains nonpolar residues that interact favorably with the lipid bilayer.
Internal Hydrogen Bonding: Backbone hydrogen bonds (between C=O and N-H groups) are satisfied within the helix, avoiding unfavorable interactions with the hydrophobic membrane.
Versatility: Multiple helices can assemble into channels, transporters, or receptors, allowing for diverse functions.
🔹 Examples:
G protein-coupled receptors (GPCRs) – Seven transmembrane helices for signaling.
Ion channels (e.g., K⁺ channels, aquaporins) – Helices form selective pores.

Nucleus
A key feature that distinguishes bw eukaryotic and prokaryotic cells
Functions of the Nucleus
Hosts/Protects the DNA genome
Specialization of nuclear vs cytoplasmic functions provides opportunity for transcription & translation regulation (ex: alternative splicing)
Mechanical stability
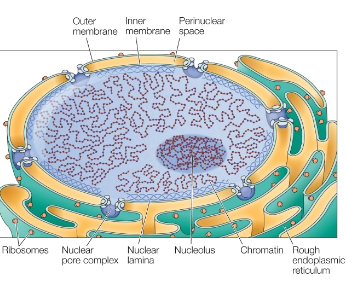
Nucleus Structure
Double membrane (nuclear envelope)
Nuclear pores (composed of proteins)
Nuclear lamina (nuclear skeleton) composed of lamins (intermediate filaments)
Subnuclear organization and membrane-less organelles (nucleolus)
What’s the evolutionary advantage of having a nucleus? Examples?
Protection of the genome
Separate synthesis/processing of RNA from cytoplasmic events; provides opportunities for gene expression regulations
Examples:
Protein nuclear import & export enables transcriptional regulation
Splicing & alternative splicing enables greater complexity from a single gene
Regulation of RNA transport enables translational regulation
Evolutionary origin of the nucleus
Membrane invagination → double membrane w pores that formed at membrane junctions
Interior of nucleus and cytosol originated from the same compartment
Topological Relationship of the Cell 🤪
Nuclear Compartment → topologically related to the cytosol
The cytoplasm and the nucleus are said to be topologically equivalent because the outer and inner nuclear membranes are continuous with one another, so that the flow of material between the nucleus and cytosol occurs without crossing a lipid bilayer.
The ER, Golgi, lysosome, & vesicular compartments → topologically related to cell exterior
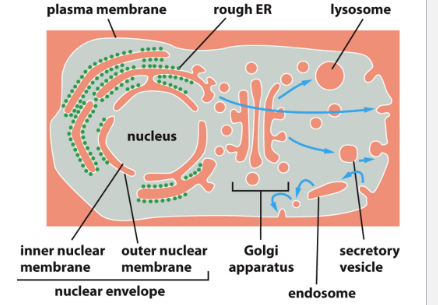
Nuclear Envelope
Outer Membrane
Continuous w rough ER membrane
Inner Membrane
Integral membrane proteins bind nuclear lamine proteins

Cell exterior
Nuclear Pore Complexes
Only channels for import and export
Selective and regulated
Huge molecular machine
Abundant (~3-4k of em)
Formed by nucleoporin proteins (NUPs)
Asymmetric: distinct cytoplasmic & nuclear features, important for directional trasnport
Central Channel
Lined by FG-rich proteins
NUPs are highly conserved in eukaryotes
IDR
Interact w transport machinery, restrict diffusion, promote selective transport
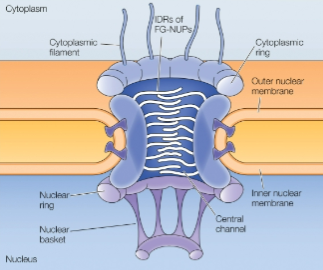
Molecular Traffic through Nuclear Pore Complexes
Two Transport Mechanisms:
Passive diffusion: small molecules pass freely thru the nuclear pore and equilibrate across envelope
Selective transport: most proteins and RNAs, recognized by specific signals, selectively transported across nuclear pore and accumulate in one compartment
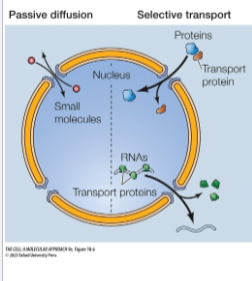
Nuclear Localization Signals
Selective nuclear transport relies on restriction of passive diffusion by Nuclear Pore Complex Interior
Specific import and export seqs on cargo protein:
Import: Nuclear Localization Seqs (NLS)
Export: Nuclear Export Seqs (NES)
Specific receptor proteins function as importers/exporters:
Bind and release NLS (or NES) in appropriate locations
Direct transport thru the Nuclear Pore Complex
Discovery of NLS
1980s
Butel and Lanford noted that SV40 T- Antigen viral protein that normally is transported to the nucleus stays in the cytoplasm if a particular sequence is mutated
Alan Smith’s grp identified a small essential region by deletion studies in SV40 T Antigen
They fused their presumptive NLS to a normally cytosolic protein
NLS was linked to gold particles and injected into the cytosol
Cells were fixed @ various times
Gold particles visualized in EM
NLS is sufficient for nuclear import!!!
Import occurs thru nuclear pores!!
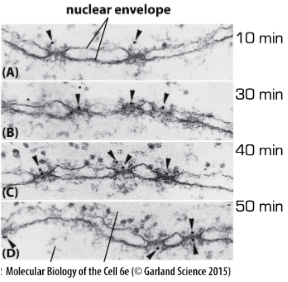
Characteristics of NLS
Can be
A continguous stretch of amino acids
Bipartite patch
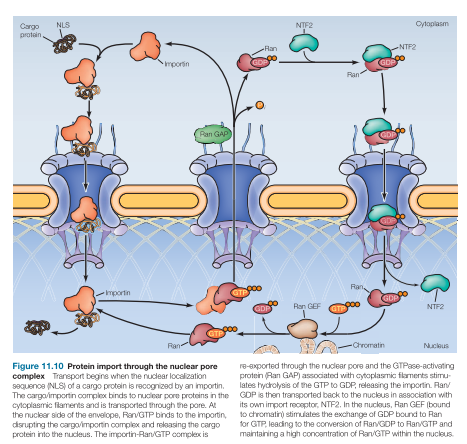
Importins
Nuclear import receptor
In the cytosol, importins bind: NLs of cargo then outer NPC cytoplasmic filaments
Transported thru pore/channel proteins
At the nuclear side of the envelope, importin releases cargo protein into the nucleus
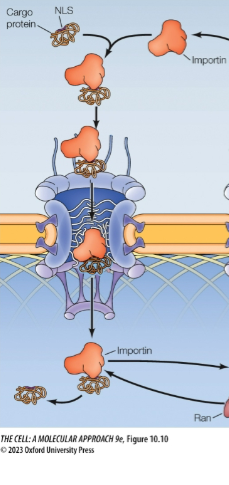
Problem: So importins have to bind cargo outside the nucleus but it also has to release cargo inside the nucleus. How’s it gonna get back out?
Solution: Ran-GTP in nucleus binds importin, makes importin release cargo (after conformational change), and transports cargo less importin back to the cytosol
Ran has 2 forms…
RanGTP in the nucleus
ABUNDANT IN NUCLEUS
RanGDP in the cytosol
ABUNDANT IN CYTOPLASM
Importin binds cargo
RanGEF
Guanine Nucleotide Exchange Factors swaps GDP out allowing GTP in
Bound to chromatin
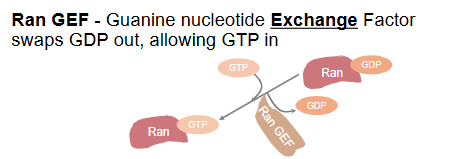
RanGAP
Activates hydrolysis of GTP to GDP
Bound to the outer pore
Nuclear Export of Proteins
Exportins bind to NES (nuclear export signals) of cargo
In presence of Ran-GTP
Exportins release:
NES-cargo
In presence of Ran-GDP
Ran-GDP and exportins are then returned to nucleus
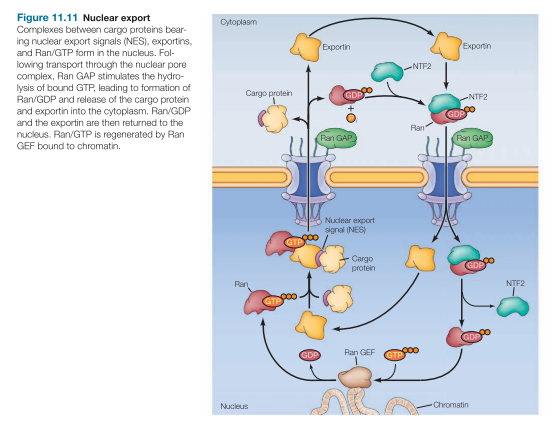

Ran-GTP would accumulate in the nucleus
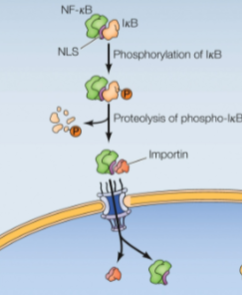
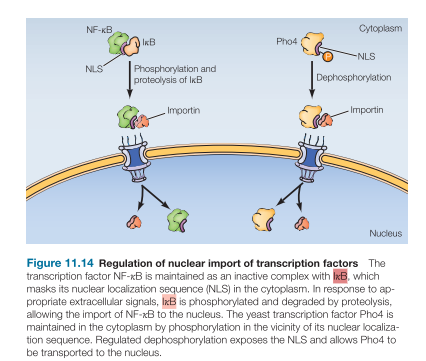
Regulation of Nuclear Import of Transcription Factors: Masking the NLS (not yeast)
Mask NLS by protein binding:
NF-kB transcription factor is bound by a protein (IkB) which masks NLS in the cytoplasm
Extracellular signals cause IkB phosphorylation recognized by an E3 ligase, causing ubiquitin-mediated proteolysis, allowing NF-kB nuclear import
Regulation of Nuclear Import of Transcription Factors: Masking the NLS (Yeast)
Yeast transcription factor Pho4 is kept in cytoplasm by phosphorylation near/at nuclear localization signal
Signal: phosphate depletion
Need for cellular phosphate, results in dephosphorylation, exposes the NLS, and Pho4 transported to nucleus
mRNA export
Independent of Ran
Once spliced and polyadenylated, mRNAs are bound by exporter complex proteins (quality control for fully proceeds mRNAs)
A helicase, on cytoplasmic face of nuclear pore complex, releases the mRNA from exporter complex
Helicase bound cytosolic face of NPC provides directionality

RNAs that function in nucleus either retained or moved out and back in:
snoRNAs for rRNA processing in nucleus - not exported
lncRNAs stay in the nucleus
snRNAs exported, associate w proteins, and reimported as RNPs for splicing
RNAs that function in cytoplasm:
tRNAs & miRNAs exported directly via specific exportins; participate in translation and RNA regulation cytoplasm
rRNAs are first bound by ribosomal proteins, in nucleolus, then exported as ribosome subunits for translation functions in cytoplasm
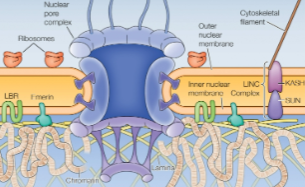
Nuclear Lamina
Lines the inner nuclear membrane
Structural support (links to cytoplasmic cytoskeleton)
Attachment points for chromatin (affects chromatin organization)
Participates in nuclear envelope breakdown and reassembly
Intermediate filament
Interact w both
Chromatin
Inner membrane proteins
Bind to protein complexes that bind to the cytoskeleton in the cytosol. The inner lamina provides a link bw chromatin and forces in cytoplasm.
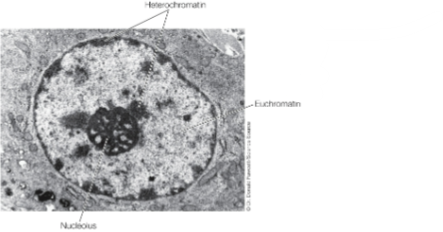
Subnuclear Organization
Heterochromatin (inactive): near nuclear membrane (via lamins interactions) & periphery of nucleolus
Euchromatin (active): preferentially localizes to the rest of the nucleus
Evidence for the location of active/inactive genes
EM of hetero vs euchromatin
FISH of chromosomal DNA/RNA
Chromatin IP/ Lamin Association
Heterochromatin in interphase nuclei
Euchromatin (active) is distributed throughout the nucleus
Heterochromatin (inactive) is associated w the nuclear envelope (nuclear lamina) and the nucleolus
Nuclear bodies
Membrane-less organelles, condensates
Domains within nucleus, maintained by interactions bw proteins or protein-RNA
Nucleolus
rRNA synthesis & processing, ribosome assembly (needed in large qty)
larger in metabolically active cells
Functions in rRNA synthesis, processing and ribosome assembly
Maintained by interactions between protein and RNA components
Contains 100s of copies of rRNA encoding genes
Subdomains form a multilayered biomolecular condensate within nucleolus
Multilayered biocondensate: Ribosome Assembly
Domains for:
rRNA gene transcription
rRNA processing
Ribosome subunit assembly
Ribosomal Proteins
Synthesized in cytosol
Imported
Assembled into 40S and 60S subunits
Exported as ribosomal subunits (ribosome subunits are not to scale, are 30x smaller than nuclear pores)
Nuclear envelope breaks down at mitosis to enable formation of 2 nuclei
Lamin phosphorylation: envelope fragmentation
Nuclear envelope assembly: dephosphorylated lamins bind to chromatin and bound to inner membrane, reassemble nuclear envelope
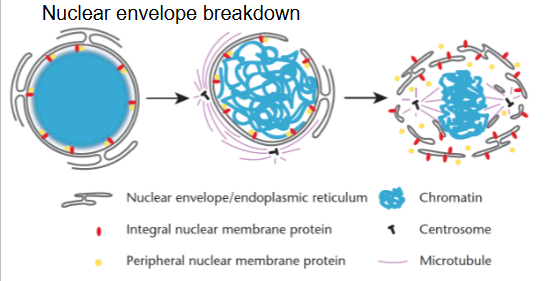
What happens to the nuclear proteins after cell division?
During cell division, the nuclear envelope breaks down, and the proteins within are scattered.
Cytosol contains
Many metabolic pathways
Protein synthesis
Cytoskeleton
Nucleus contains
Main genome
DNA synthesis
RNA synthesis and splicing
Ribosome assembly
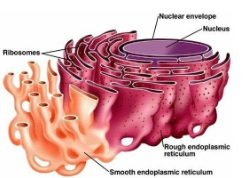
Rough ER Functions
Protein synthesis, modification, and transport to many organelles and plasma membrane
Smooth ER Functions
Lipid and steroid synthesis, detoxification
ER Structure
A continuous network of membrane-enclosed tubules and sacs (cisternae)
Nuclear envelope is part of the ER!
Typically largest organelle
Rough ER covered by ribosomes on its cytosolic surface
Key Players in ER Function:
Signal recognition particle and translocon
“As they emerge from the ribosome, signal sequences are recognized and bound by the signal recognition particle (SRP) consisting of six polypeptides and a small cytoplasmic RNA (SRP RNA). The SRP binds the ribosome as well as the signal sequence, inhibiting further translation and targeting the entire complex (the SRP, ribosome, mRNA, and growing polypeptide chain) to the rough ER by binding to the SRP receptor on the ER membrane (Figure 12.6). Binding to the receptor triggers the hydrolysis of GTP bound to the SRP, releasing the SRP from both the ribosome and the signal sequence of the growing polypeptide chain. The ribosome then binds to a protein translocation complex or translocon in the ER membrane, and the signal sequence is inserted into a membrane channel”
Chaperones (BiP), protein modification (S-S, lipid anchors)
“[…] chaperones that facilitate the folding of
polypeptide chains (see Chapter 10). The Hsp70 chaperone, BiP, is thought to
bind to the unfolded polypeptide chain as it crosses the membrane and then
mediate protein folding within the ER (Figure 12.14). Correctly assembled
proteins are released from BiP (and other chaperones) and are available for
transport to the Golgi apparatus”
Quality Control (ERAD, UPR)
ERAD - ER Associated Degradation ~ misfolded proteins are identified, returned from the ER to the cytosol, and degraded by the ubiquitin-proteasome system.
UPR - Unfolded protein response ~ activated if an excess of unfolded proteins accumulates in the ER
Activation of the UPR pathway leads to expansion of the ER and production of additional chaperones to meet the need for increased protein folding as well as a reduction in the amount of newly synthesized proteins entering the ER
![<ul><li><p>Signal recognition particle and translocon</p><ul><li><p>“As they emerge from the ribosome, signal sequences are recognized and bound by the signal recognition particle (SRP) consisting of six polypeptides and a small cytoplasmic RNA (SRP RNA). The SRP binds the ribosome as well as the signal sequence, inhibiting further translation and targeting the entire complex (the SRP, ribosome, mRNA, and growing polypeptide chain) to the rough ER by binding to the SRP receptor on the ER membrane (Figure 12.6). Binding to the receptor triggers the hydrolysis of GTP bound to the SRP, releasing the SRP from both the ribosome and the signal sequence of the growing polypeptide chain. The ribosome then binds to a protein translocation complex or translocon in the ER membrane, and the signal sequence is inserted into a membrane channel”</p></li></ul></li><li><p>Chaperones (BiP), protein modification (S-S, lipid anchors)</p><ul><li><p>“[…] chaperones that facilitate the folding of</p><p>polypeptide chains (see Chapter 10). The Hsp70 chaperone, BiP, is thought to</p><p>bind to the unfolded polypeptide chain as it crosses the membrane and then</p><p>mediate protein folding within the ER (Figure 12.14). Correctly assembled</p><p>proteins are released from BiP (and other chaperones) and are available for</p><p>transport to the Golgi apparatus”</p></li></ul></li><li><p>Quality Control (ERAD, UPR)</p><ul><li><p>ERAD - ER Associated Degradation ~ misfolded proteins are identified, returned from the ER to the cytosol, and degraded by the ubiquitin-proteasome system. </p></li><li><p>UPR - Unfolded protein response ~ activated if an excess of unfolded proteins accumulates in the ER</p><ul><li><p>Activation of the UPR pathway leads to expansion of the ER and production of additional chaperones to meet the need for increased protein folding as well as a reduction in the amount of newly synthesized proteins entering the ER </p></li></ul></li></ul></li></ul><p></p>](https://knowt-user-attachments.s3.amazonaws.com/60eb2b92-d833-498c-ac51-1253dd33effc.png)
Secretory Pathway
Rough ER → Golgi → secretory vesicles → cell