Pharmacology
1/166
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai | Chat |
|---|
No analytics yet
Send a link to your students to track their progress
167 Terms
Name the piece of legislation that covers:
Veterinary medicine
Veterinarians prescribing human medicine
Veterinary use of controlled drugs
Agricultural Compounds and Veterinary Medicines Act (ACVMA)
Medicines Acts (MA)
Misuse of Drugs Act (MDA)
12 Obligations veterinarians have when authorising RVM or PM
Choice and use of product is justified (with appropriate training + information to owner)
ONLY authorise RVMs after vet consultation (sufficient information)
If complaint is laid, actions other vets would take in the same situation is considered (decision must be consistent with peers)
Can authorise treatment without seeing animal (must have good knowledge of situation eg. regular farmer + knowledge of what diseases they commonly encounter + confidence that what the farmer describes is accurate + competent to administer themselves)
Must be bona fide client (animal is under your care)
Confirm client is competent at administering treatment (training)
Arranged provision of ongoing care for patient (in case of adverse drug reaction)
Documented authorisation (findings from clinical exam and details of RVM authorised) kept for at least 5 years
Can authorise for future supply for production animals (vet must have good knowledge of client, farm, and have ongoing recheck) + finite time and amount of drug + client must keep record of how much is used
Must provide written authorisation to client to obtain drug elsewhere (you are still responsible)
Critically important antibiotics - Alternatives? Drive to reduce
Controlled drugs: Record every sale or use + reconcile at least monthly
Authorising - Vet creates documented approval allowing client to (3):
Purchase a particular RVM to administer to particular animal in accordance with instructions of vet
Hold an RVM for anticipation of its use (eg. intracillin for mastitis)
In accordance with instructions of authorising vet
Dispensing vs. prescribing medicine
Dispensing - Preparing vet medicine for owner (eg. packing 2 dosages from commercial packaging to appropriately labelled alternative)
Prescription medicine - Human medicine that can only be sold under prescription
5 Features of a consultation (required for authorisation of RVMs)
Interview with client and exam animal
Collect + record sufficient information to ensure course of action is appropriate
Obtain consent to proposed course of action
MUST be responsible for the ONGOING health and welfare of the animal (eg. arranging after care when considering potential for adverse reaction)
Determine and provide appropriate level of advice and training of owner
What is “discretionary use” of medicine?
Use of medicine that is NOT listed on the label (eg. prescribing sheep medication for alpacas)
Must be able to justify discretionary use (reasonable decision by peers)
Describe the cascade for authorising/prescribing medicine
Is there an appropriate on-label (RVM authorised by vet with instructions following the label eg. correct species, dose, frequency, route)?
Is there an appropriate off-label (RVM used in a manner NOT specified in the conditions on registration for the product eg. incorrect species, different dose rate, frequency etc.)?
Is there a human PM (Under the Medicines Act)?
Compounding (specifically compounded by/on authority of that vet)
Overseas (talk to MPI to seek permission to import)
Registered vs. restricted vs. controlled veterinary medicines
Registered = All drugs must be registered with the ACVMA (ensures the benefits of the treatment will outweigh the adverse effects)
Contains compulsory information: species, condition to treat, dose rate, route, withholding periods (with sufficient trial data)
Restricted = Registered vet meds which pose some risks and use must be authorised by a vet with an APC (annual practicing certificate)
CAN be held by owner
Unrestricted - Anyone can purchase from supermarket (eg. Drenches, flea preparations, supplements, flea collars)
Controlled = Potential for addiction, and abuse (cannot use class A) with controlled drug register
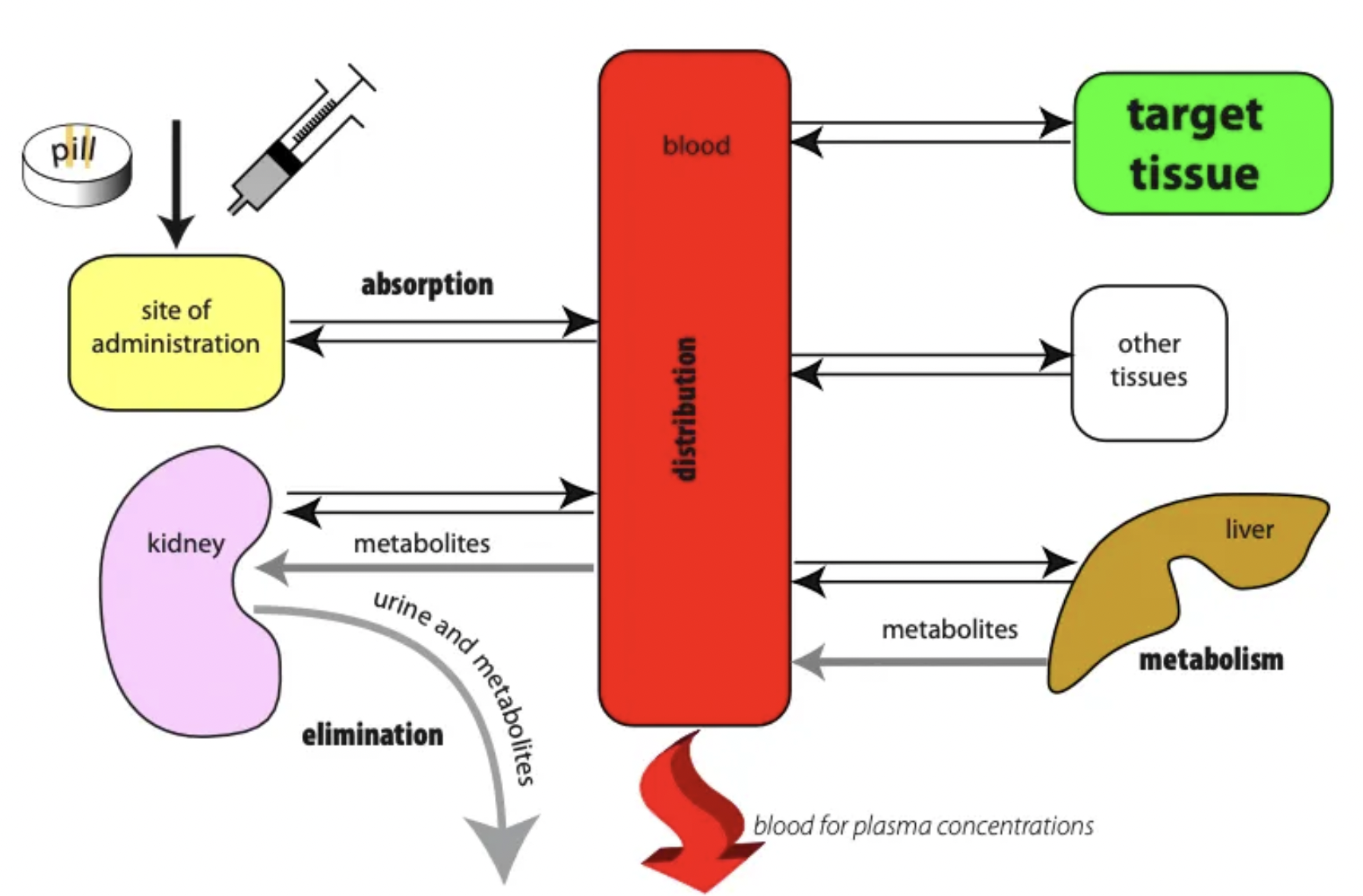
List the 4 pillars of pharmokinetics (+ definitions)
Absorption = Movement of drug into circulation
Distribution = Movement of drug from circulation to tissues
Metabolism = Body response to drug is to remove it by converting it to a water-soluble metabolite which is NOT toxic
Excretion = Elimination of water-soluble metabolite via faeces, urine, milk, sweat
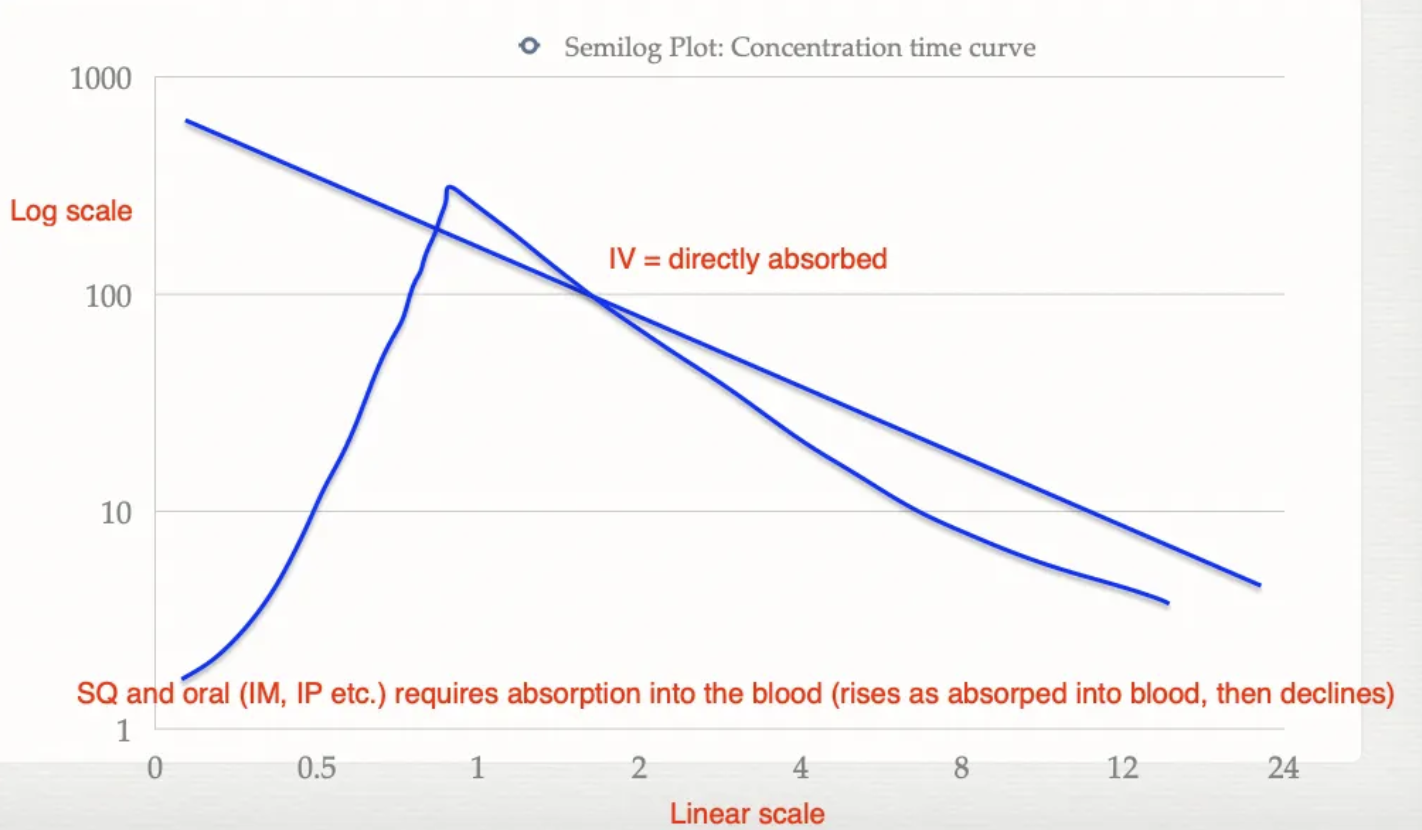
Describe the concentration of drugs in circulation overtime for IV vs. other routes of administration (eg. SC, PO, IM, IP)
IV = No absorption as directly into bloodstream → Gradual decreasing concentration (elimination)
Oral/SQ/IM/IP = Requires absorption and transport across the epithelium → Gradual increasing concentration (absorption) and then decreasing concentration (elimination)
List 3 routes of enteral administration (+ descriptions)
Oral = Absorbed through SI mucosa → Portal vein → Liver metabolism → Systemic
Sublingual = Absorption under tongue → Bypass liver = More effective
Rectal
List 7 routes of parenteral administration (+ cautions)
Intravenous (IV) = No absorption required
Caution: No insoluble salt suspensions or oils (must be completely soluble)
Intramuscular (IM)
Caution: Avoid semitendinosus → Potential injection in femoral artery and sciatic nerve
Use cranial thigh (quadriceps)
Subcutaneous (SC)
Intraperitoneal (IP) = Rats (IV difficult)
Caution: Massive absorption due to high SA and blood supply
Inhalational = Rapid absorption across alveolar membranes
Transdermal = Across skin
Caution: Slow absorption as natural barrier (faster in highly vascular dermis eg. MM)
Epidural/intrathecal
3 things required for drug absorption into bloodstream
Dissolution of drug
Movement of drug out of site of administration
Movement of drug into blood vessels = Cessation of absorption
Describe the pathway of absorption for oral drugs
Oral pills have coatings to pass through the acidic stomach in order to spare the drug
Absorbed across the small intestinal membrane
Cranial mesenteric vein → Hepatic portal vein
Liver metabolises drug (hence metabolites necessary for action)
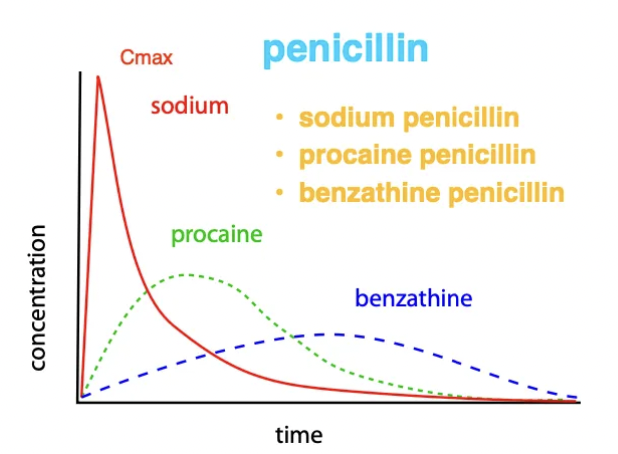
Describe how solubility of drugs influences the route of administration and the concentration overtime (eg. 3 forms of penicillin)
IV = Water-soluble ONLY → Rapid onset
IM/SC = Suspensions of insoluble salts/oils → Slow release
Example: Penicillin
Sodium penicillin IV = Water-soluble → Rapid absorption (highest Cmax) and rapid elimination
Procaine penicillin IM = Insoluble salt solution → Slower absorption (lower Cmax) and remains in body for longer
Benzathine penicillin IM = Insoluble → Extremely slow absorption and long duration of action (lowest Cmax → increase dose)
List 4 ways drugs can pass across the phospholipid membrane with examples (+ exception)
Passive diffusion = Small, highly lipid-soluble drugs (eg. fentanyl, diazepam, ethyl alcohol)
Facilitated diffusion = Large, highly lipid-soluble drugs (eg. corticosterone and drugs that must cross the BBB)
Active diffusion = Uncommon (eg. medicinal drugs, iron salts, fluorouracil)
Endocytosis = Large molecules which cannot enter by other means → Invagination of the cell membrane to form a vesicle around the drug (eg. oily preparations and cholesterol)
Exception: Penicillin = Water-soluble and requires specific transport molecules (excreted in urine as penicillin)
Drugs must be lipophilic to cross the phospholipid membrane to exert their effects
List 4 factors influencing absorption rate of PO drugs
Blood flow (shock → reduced absorption)
Contact time (vomiting/diarrhoea → reduced absorption)
Food (drug binds food and reduces availability for absorption)
Carrier-mediated transport (eg. P-glycoprotein)
List 4 factors influencing absorption rate of IM/SC drugs
Blood flow
Exercise → increased absorption of IM drug
Accidental intrafat injection → decreased absorption as poorer blood supply
SC → slow and variable (not as vascular as muscle)
Higher temperature → increased absorption rate
Inflammation (disrupted membrane → good absorption)
pH
Formulation (oil, insoluble salts, water-soluble etc.)
Use the Henderson-Hasselbalch logic to explain how drug pKa and environment pH affects the solubility of the drug
pH = pKa → 50% ionised / 50% unionised
pH < pKa → more protonated
pH > pKa → more unprotonated
Weak Acids: HA ⇌ H⁺ + A⁻
Low pH (acidic) environment = Shift → HA (unionised and lipid-soluble) → Cross cell membranes
High pH (alkaline) environment = Shift → A⁻ (ionised and water-soluble) → Trapped in compartment
Weak Bases: BH⁺ ⇌ H⁺ + B
Low pH (acidic) environment = Shifts → BH⁺ (ionised and water-soluble) → Trapped in compartment
High pH (alkaline) environment = Shifts → B (unionised and lipid-soluble) → Cross cell membranes
EXAMPLE: Describe absorption of aspirin in mouth vs. stomach vs. duodenum
Aspirin pKa = 4.4
Oral pH = 7.4
Stomach pH = 1
Duodenum pH = 6
Aspirin = Weak acid
Mouth pH > Aspirin pKa → More drug unprotonated and IONISED (A-) = Water-soluble and cannot enter through oral mucosa = ION TRAPPING
Stomach pH < Aspirin pKa → More drug protonated and UNIONISED (HA) = Lipid-soluble and absorbed through gastric mucosa
Duodenum pH > Aspirin pKa → Although more drug unprotonated and ionised, the higher SA of the SI means more aspirin is absorbed here than in the stomach
What is “ion trapping”?
Use erythromycin as an example when used IMM for mastitis
Ion trapping = Drug is initially lipid-soluble upon administration and is transported across the cell membrane. In the new site, the pH is different and it becomes water-soluble and therefore CANNOT move back
Example: Erythromycin = Weak base
Erythromycin given IMM into pH of 6.8 (acidic)
pH < pKa → Drug remains protonated (BH+) and ionised (water-soluble) and hence cannot enter plasma
Drug remains in milk where it is required
Define bioavailability
Fraction of drug that reaches systemic circulation (IV always 100%)
List 4 factors that distribution of a drug is affected by (+ examples)
Blood flow (higher blood flow → more distribution of drug to tissue)
Brain > Muscle > fat
Capillary permeability (more permeable → more distribution of drug to tissue)
BBB = Astrocytes tightly adhere endothelial cells together → Fewer drugs pass
No access to ionised drugs (eg. penicillin and aminoglycosides) EXCEPT with meningitis → Compromised BBB
Lipid-soluble drugs rapidly equilibrate and redistribute to the brain
Drug structure (size eg. peptides and proteins and lipid solubility)
Protein binding (higher PB → lower distribution)
Many drugs bound to albumin which keeps them in circulation, preventing distribution to tissues
Example: Thiopentone Na and phenylbutazone = High PB → Lower subsequent doses required as fewer proteins available for binding → More free drug available
Define volume of distribution (Vd) and its use
Definition: The theoretical volume that would be required to contain the total amount of drug in the body at the same concentration as in plasma
Higher Vd (eg. thiopentone) → higher distribution to all parts of body (extensive tissue binding)
Lower Vd (eg. aspirin) → stays in plasma for long periods so lower dose required to obtain necessary concentrations
Use: Used to calculate loading dose (higher Vd = higher loading dose)
Features of acidic vs. basic drugs (5)
Acidic drugs tend to: eg. Aspirin
Have higher oral bioavailability
Have higher hepatic clearance
Have higher protein binding
Have smaller Vd
Get absorbed better in stomach and duodenum
Basic drugs tend to: eg. Morphine
Have poorer protein binding
Have larger Vd
Have better CNS penetration
Be less selective
3 Outcomes of biotransformation
Prodrug → Active drug (eg. aspirin → salicylic acid)
Toxic → Non-toxic
Active drug → Inactive metabolite
Describe the 2 phases of drug metabolism (+ drug examples)
PHASE 1 = Reactive “handle” attached by cytochrome P450 enzyme (CYP450) via
Oxidation (hydroxylation, dealkylation, deamination) eg. dexmedetomidine
Reduction eg. warfarin
Hydrolysis eg. lignocaine
PHASE 2 = Conjugation with POLAR group (water-soluble molecule) via
Glucuronidation
Sulfation
Methylation
Acetylation
What is cytochrome P450? Why is it important?
Protein in hepatocytes which carry out phase 1 of drug metabolism (also in GIT, lungs, kidney and skin)
Importance: Important to know which CYP enzyme metabolises which drug (drug interactions)
List 5 factors influencing the CYP450 system (+ examples)
Enzyme Inducers = Drugs that increase synthesis and activity of CYP450 enzymes → Faster metabolism
eg. Phenobarbitone, alcohol, St John’s wort, griseofulvin
2nd dose of phenobarbitone remains in the system for LESS time → Increase dose rate to have the same effect
Enzyme Reducers = Drugs that reduce effect of CYP450
eg. Ketoconazole, tramadol, quinidine, grapefruit juice
Abnormal Phenotype = Abnormal CYPs which turn harmless compounds into toxins due to altered metabolism (faster/slower) of drugs
Liver Disease = Slower metabolism → Lower dose required
Neonatal/Geriatric = Lack of enzymes to metabolise drugs
List 6 compounds which drugs are conjugated with during phase 2 of drug metabolism (+ species differences)
Glucuronide (NOT cats) = glucuronidation
Sulphate (NOT pigs) = sulfation
Acetyl (NOT cats and dogs) = acetylation
Methyl = methylation
Glycine = methylation
Ornithine (ONLY birds)
Define “first pass metabolism”
Liver metabolises the drug FIRST before entering the systemic circulation to the target organ
Describe the mechanism and effect of enterohepatic recycling
Mechanism:
Conjugated drug excreted in bile (with glucuronic acid)
Unconjugated drug enters sinusoids to be excreted in the kidney
GIT bacteria remove conjugate for energy (sugar)
Drug available for resorption as no longer water-soluble
Absorbed back into bloodstream for re-use
Effects: Prolonged duration of action (esp. opioids) → Blips in drug concentration
3 Factors that influence renal elimination of drugs
GFR
Active excretion of water-soluble drugs (organic anion and cation transporters)
eg. Penicillin (given transporter competitor eg. TMP to make penicillin remain in body for longer)
Passive resorption of lipid-soluble drugs (pH important for ion-trapping)
Define withholding period and times for different species and products
Ruminants
Pigs/horses
Birds
Camelids
Rabbits
WHP: Time taken for drug to fall below the maximum residual limits (MRL) in nearly all animals → Defines period which must elapse between last treatment and use of animal as a food product
Ruminants: 91d (meat) and 35d (milk)
Pigs/Horses/Birds/Camelids/Rabbits: 63d (meat)
Birds: 10d (eggs)
List 6 drug targets (+ definitions)
Receptors = Protein molecules which bind specific ligands (drugs) and exert a response
Ion channels
Enzymes = Drugs compete with substrate for enzyme active site
Carrier molecule = Transportation of small molecules into and out of the cell
DNA (eg. antibiotics and chemotherapy drugs)
Non-specific (eg. osmotic diuretic and radioactive iodine)
Describe the structure of the drug molecule required to bind to extracellular vs. intracellular receptors
Extracellular Receptors - Drugs do NOT cross the cellular membrane → Large and water-soluble
Intracellular Receptors - Drugs must cross the cellular membrane to exert effect → Small and lipid-soluble
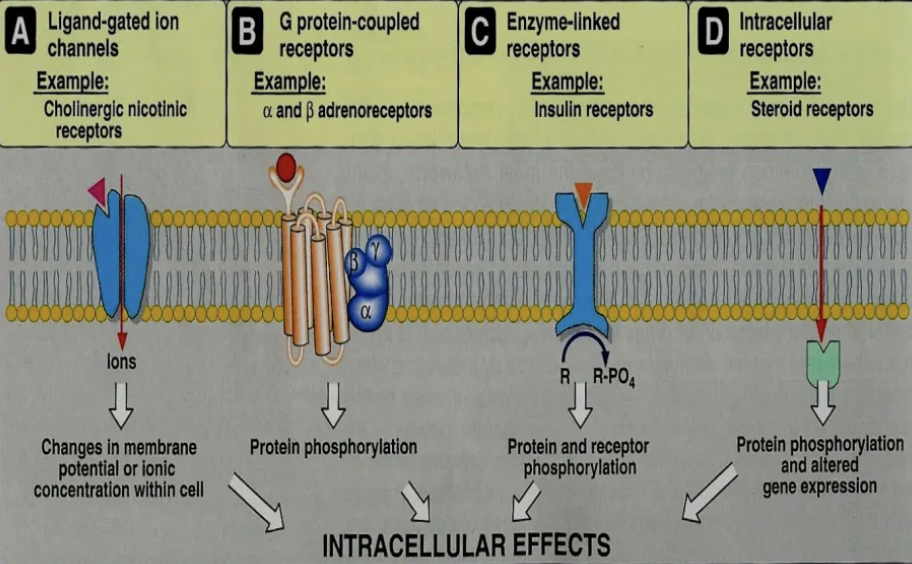
List 4 superfamilies of receptors
Onset of action/response
Intracellular/extracellular receptor
MoA
Example receptors
Ionotropic Receptors = Ligand-gated ion channels
Response: Rapid
Extracellular
MoA: Ligand binds receptor to open ion channel
Examples: Nicotinic ACh, AMPA, GABA receptors
Metabotropic Receptors = G protein coupled receptors
Response: Slower than ionotropic (s - mins)
Extracellular
MoA: Ligand binds receptor to change conformation of G-protein → Open channel or activate enzyme
Examples: Opioid, adrenergic, muscarinic ACh receptors
Tyrosine Kinase Coupled Receptors = Controlling cell growth
Response: Minutes to hours
Extracellular
MoA: Ligand (hormone) binds receptor to activate tyrosine kinase which phosphorylates the tyrosine residue on their substrates intracellularly → Activation of many intracellular signalling pathways
Examples: Hormones eg. insulin, thyroid, growth factor receptors
Nuclear Receptors = Receptors in nucleus or cytoplasm
Response: Hours to days BUT widespread response throughout body (longer duration of action)
Intracellular
MoA: Interferes with DNA to increase/decrease gene transcription and therefore, protein synthesis
Example: Corticosteroid receptors
How do must drugs influence ion channels? Provide an example
Most drugs block ion channels (NOT open) eg. lignocaine local anaesthetic → Blocks Na+ channels to prevent entry into nociceptive neurons
List 3 drugs which bind enzymes to exert their effects
Most antibiotics
Organophosphates: Block enzyme that breaks down ACh → Accumulation of ACh
NSAIDs: Bind cyclooxygenase enzyme necessary for conversion of arachidonic acid to prostaglandins
List 2 drugs which depend on binding to carrier molecules to exert their effects
Antidepressants = Block reuptake transporters → Prevents reuptake of serotonin (5-HT) and noradrenaline into presynaptic neuron → Increased neurotransmitter concentration in synaptic cleft → Increased ruing and intensity of post-synaptic effect
Ivermectin = Actively pumped out of the BBB by P-glycoprotein carrier molecule (small and lipophilic enough to passively cross the BBB)
Describe 3 ways that receptors can be complex (+ examples)
Same transmitter → Multiple receptor subtypes → Different effects
Example: Adrenaline/NA
α₁: vasoconstriction
α₂: ↓ neurotransmitter release (presynaptic)
β₁: ↑ heart rate & contractility
β₂: bronchodilation, vasodilation
Same receptor → Different effects in different tissues
Example: β₂ receptors
Bronchi → Bronchodilation
Uterus → Relaxation
Skeletal muscle vasculature → Vasodilation
Receptor regulation = Number depends of level of stimulation
Up-regulation (chronic antagonist exposure) → Supsensitivity when drug withdrawn
eg. Chronic β-blocker use → β-receptor up-regulation → rebound tachycardia if stopped suddenly
Down-regulation (chronic agonist exposure) → Tolerance
Chronic β-agonist use → β-receptor down-regulation → reduced bronchodilator response
Paradoxical pharmacology = Long-term drug exposure produces opposite effects to acute effects due to receptor regulation
What is tachyphylaxis? 3 Mechanisms
Definition: Rapid decrease in receptor response to a large initial dose of drug over a short period of time
Acute and reversible reduction in drug effect
Increasing drug dose will NOT change response as ALL receptors are blocked (must wait 1 week to give smaller dose)
Mechanisms: Desensitisation
Decreased synthesis of receptors
Increased synthesis of enzymes to deactivate existing receptors (eg. specific kinases which phosphorylate receptors)
Internalise receptors via endocytosis
What is tolerance? 3 Mechanisms
Definition: Chronic decrease in receptor response to repeated exposure of drug
Chronic and irreversible reduction in drug effect
Increasing drug dose WILL change cellular response
Mechanisms: Desensitisation
Decreased synthesis of receptors
Increased synthesis of enzyme to metabolise drug → Faster elimination of drug from body
Internalise receptors via endocytosis
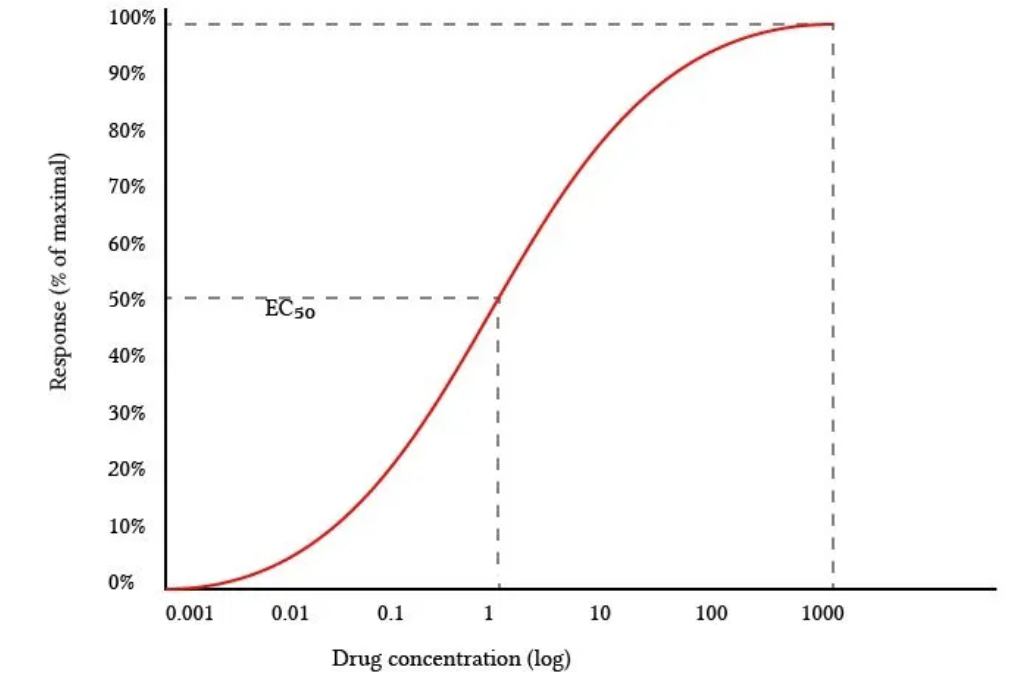
Dose response curve
Definition
Emax
EC50
Definition: Semi-log plot with log (drug concentration) on x-axis and % of maximum response on y-axis
Phase 1 = Small initial response
Phase 2 = Exponential response
Phase 3 = Drug occupies ALL receptors which become saturated (more drug → NO response)
Emax: Maximum response of drug
EC50: Effective concentration 50 = Potency of drug (i.e. concentration drug required to be 50% effective)
High specificity and drug binding afinity
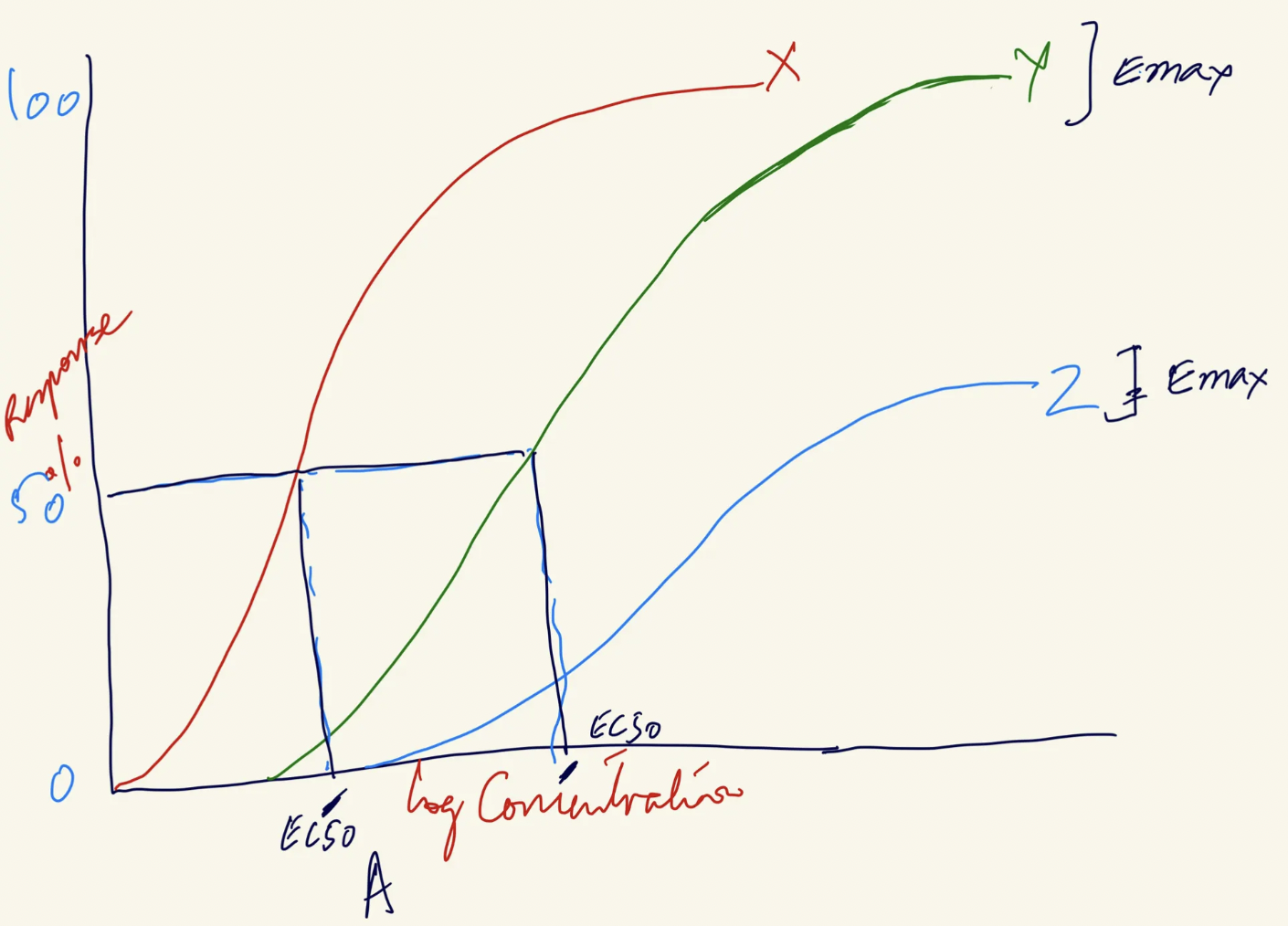
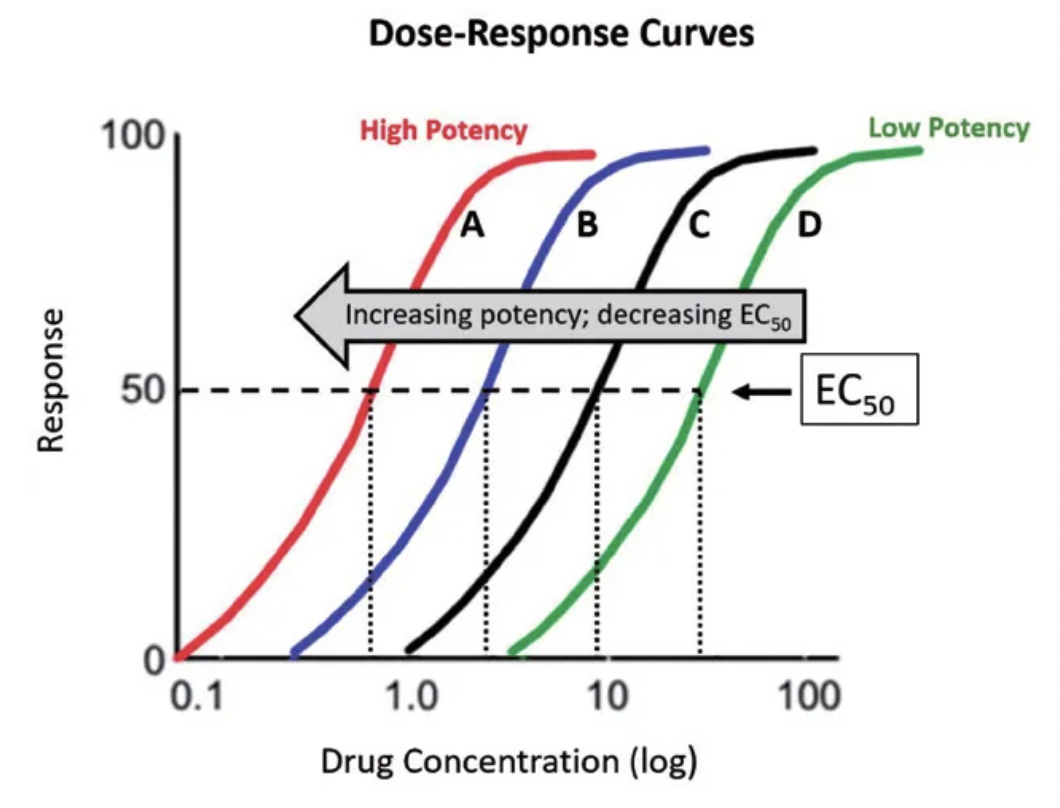
Which drug is most potent? Which drug is most effective? Why?
Most potent = Lowest EC50 → Left shift on x-axis
Drug X = Most potent
Drug Z = Least potent (highest EC50)
Most efficacious = Highest Emax → Drug exerts more response AFTER binding
Drug X and Y = Highest efficacy (highest Emax)
Drug Z = Lowest efficacy (lowest Emax)
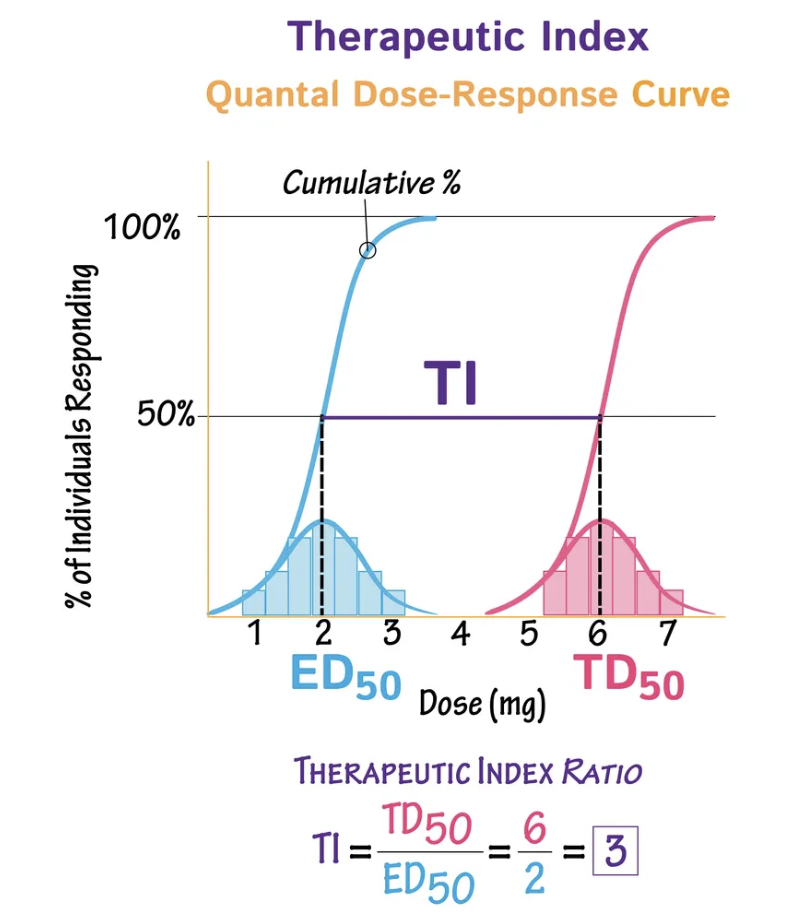
What is the “therapeutic index (TI)"?
Measure of how safe the drug is = Range of doses which are effective WITHOUT toxic side effects
TI = TD50 ÷ ED50
TD50 = Toxic dose for 50% of the population
ED50 = Effective dose for 50% of the population
ALWAYS choose drug with higher TI → Higher margin of safety
Types of Drug-Receptor Interactions
Full agonist
Partial agonist
Inverse agonist
Competitive antagonist
Non-competitive antagonist
Drug which interacts with a receptor to produce:
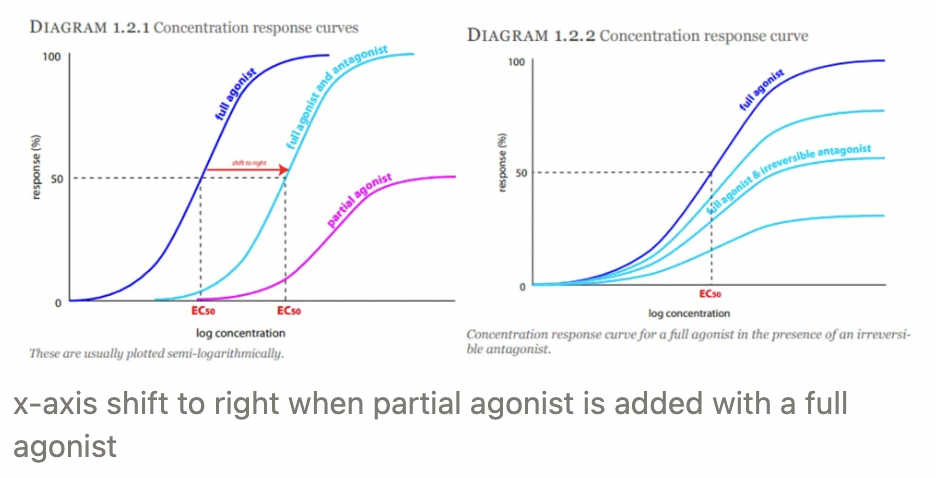
Full agonist = Maximum response → Achieve Emax
Partial agonist = Response LESS than Emax → Low efficacy
Acts as antagonist in presence of full agonist
X-axis shifts to the right when partial agonist added to full agonist
Inverse agonist = Negative/opposite response
Competitive antagonist = Irreversible/reversible competition with agonist for SAME binding site
Non-competitive antagonist = Binds to another site (allosteric site) to change conformation of agonist receptor to prevent agonist from binding
Pain vs. nociception
Pain = Unpleasant sensory and emotional experience associated with actual/potential tissue damage
Nociception = Transmission of pain signals to cerebral cortex (ONLY sensory component and NO emotional component)
Hyperalgesia vs. allodynia
Hyperalgesia = Increased sensitivity to a stimulus which is normally painful and may persist AFTER pain is treated
eg. Hurt toe → Extreme pain when touched again
Mechanism: Excitation of peripheral nerve endings stimulate other receptors to increase response of EP receptors
Allodynia = Pain caused by stimulus that is NON-painful
eg. Clothing or grooming causes pain
List 5 ways to assess pain
Behaviour = Unprovoked assessment of response to normal stimuli)
Response to analgesia = Change in behaviour, ANS etc.
Autonomic signs = HR and RR
Electroencephalogram (EEG)
Pain scores = Glasgow composite pain scale

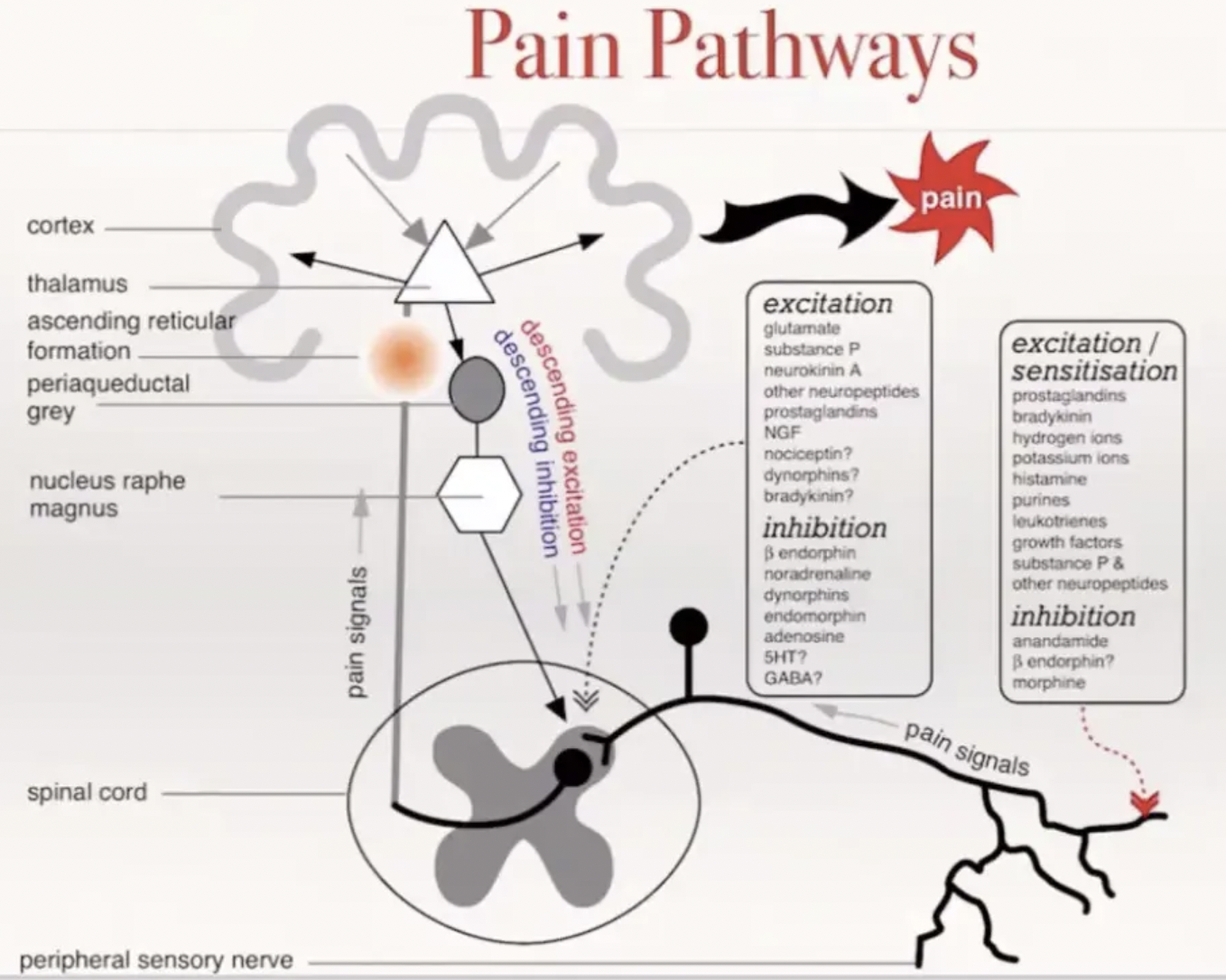
Describe the pain pathway
Initiation of pain signal = Excitation at the peripheral nerve endings
Tissue injury damages the cell membrane → Release of arachidonic acid
Arachidonic acid → PGE2 by COX enzymes
PGE2 binds EP receptors which open Na+ and Ca2+ channels
Also binds bradykinin receptors and acid-sensing ion channels which sensitise EP receptors
Influx of Na+ and Ca2+ → Membrane depolarisation
Action potential generated
Conduction of pain signal = Through A-delta and C fibres which synapse in the dorsal horn of the spinal cord (lamina I and II)
A-delta = Fast, sharp and well-localised pain
C fibres = Slow, dull and aching pain
Lamina II (substantia gelatinosa): Region of the dorsal horn in the spinal cord where modulation of pain signals in the 2nd-order neuron occur through release of excitatory/inhibitory neurotransmitters
Excitatory = Substance P and glutamate released by 1st-order neurons to activate 2nd-order neurons
Inhibitory = GABA, glycine, endogenous opioid, noradrenaline, serotonin → Inhibit substance P
Complex interaction of 1st-order, 2nd-order neurons and descending pathways which synapse gap between 1st- and 2nd-order neurons
TWO ascending tracts of spinal cord
Spinothalamic Tract = 2nd-order neuron decussates and ascends to the contralateral nuclei in the thalamus → Synapse with 3rd-order neuron → Somatosensory cortex → Localise pain
Projections into the periaqueductal grey matter to modulate pain via descending inhibitory pain pathways (opioid analgesia)
Spinoreticular Tract = 2nd-order neuron ascends to the contralateral reticular formation of the brainstem → Thalamus → Hypothalamus → Cortex → Emotional component of pain
Describe the gate control theory of pain
Aggressive touch/pressure on a painful area modulates the pain signal in the substantia gelatinosa → Decreased pain sensation
Rubbing/pressure → Activation of non-nociceptive fibres (Aβ-fibres = touch-sensitive) which synapse in the substantia gelatinosa (dorsal horn)
The Aβ-fibre activity recruits GABAnergic interneurons in the substantia gelatinosa
Interneurons release GABA inhibitory neurotransmitter
GABA neurotransmitters inhibit the C-fibre nociceptors and modulate the nociceptive pathway (prevent conduction of pain signal to the somatosensory cortex)
2 Types of chronic pain (+ mechanisms)
Long-standing or repeated noxious stimulus (injury, inflammation) →
Peripheral sensitisation = Increased pain perception from normally painful stimuli (hyperalgesia) due to:
Up-regulation of Na+ ion channels → Increased sensitivity of nociceptive fibres (C and A-delta fibres) → Lower threshold for activation
Release of neuropeptides (substance P and CGRP = calcitonin gene-related peptide) → Release of PGE2 and bradykinins
Central sensitisation = Pain becomes amplified, persistent, and independent of original injury (allodynia) due to:
Neuropeptides and glutamate activate NMDA receptors in the dorsal horn
NMDA (memory) receptor activation → Increased Ca2+ influx
Changes in gene expression and ion channel activity → Long-term increase in neuronal excitability
SAME sensory input, but increased response at the level of the CNS (increased excitability of neurons in the dorsal horn)
List 6 classes of analgesic drugs
Opioids
NSAIDs
Local anaesthetics
Alpha-2-agonists
NMDA antagonists (ketamine)
GABA agonist (gabapentin)
5 Desired effects of NSAIDs
Anti-inflammatory #1
Analgesic via anti-inflammatory effect (pain = cardinal sign of inflammation)
Inferior analgesic effects to opioids
Corticosteroids are anti-inflammatory ONLY → Suggests TWO anti-inflammatory mechanisms of action
Anti-pyretic
Anti-thrombotic = Reduce thromboxane concentration in circulation
Anti-endotoxic = Reduce effect of endotoxins (eg. LPS) released by G- bacteria
Antibiotics do NOT treat endotoxins (only bacteria itself)
Describe the MoA of NSAIDs
NSAIDs inhibit action of COX enzymes necessary to convert arachidonic acid (from cell membrane damage) into prostaglandins
Free arachidonic acid takes the lipoxygenase pathway to produce leukotrienes (instead of cyclooxygenase → PGE2)
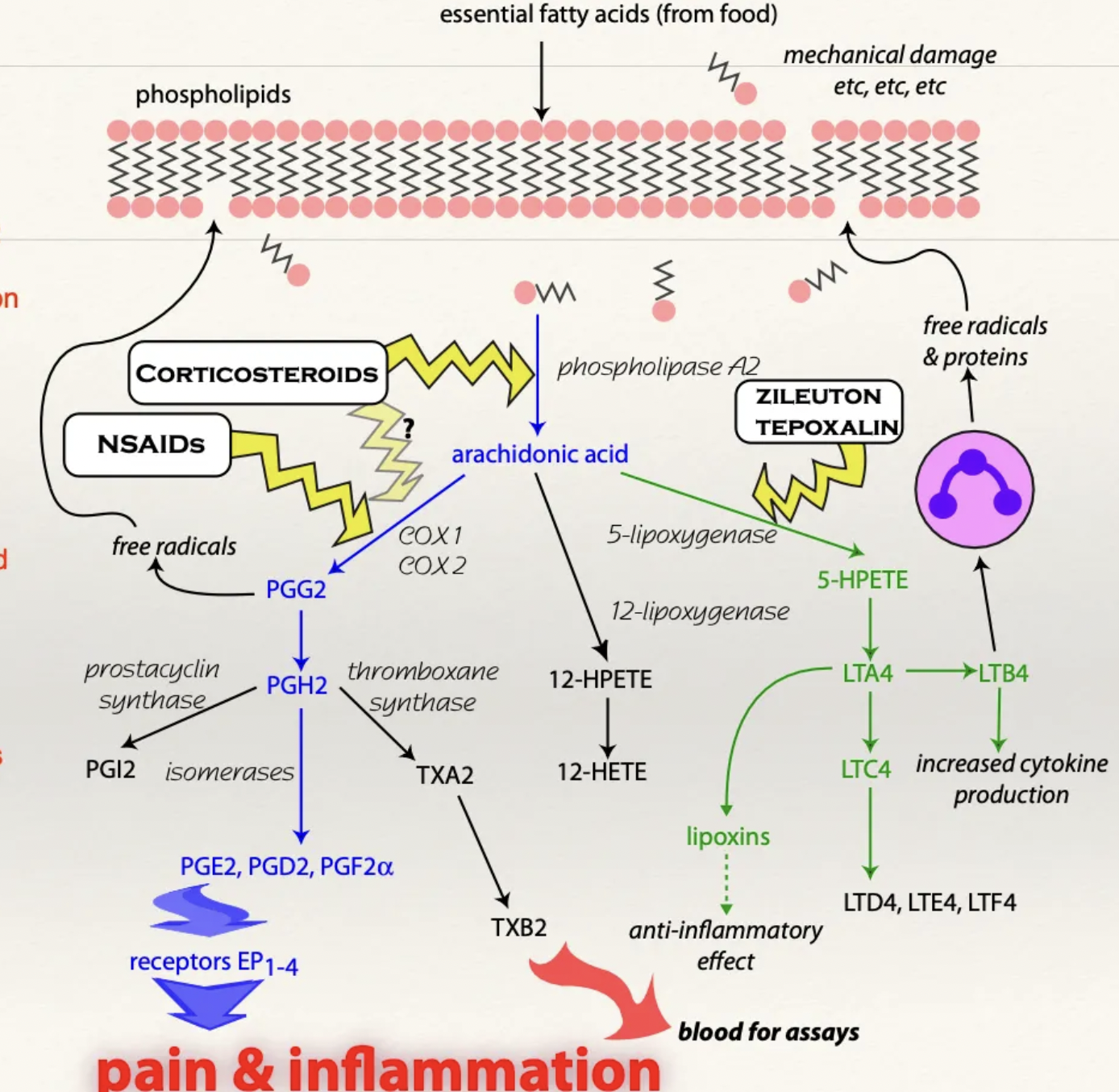
Function of COX1 vs. COX2 vs. COX3
COX1 = Assists with NORMAL physiological functions
Protect gastric mucosa
Maintain renal blood flow
Mediate production of thromboxane in platelets AND assists with platelet aggregation
COX2 = Inducible enzyme triggered by inflammation and injury
Produces PGI2 = Potent vasodilator
ALSO physiologically present in brain and kidneys
COX3 = Present in brain with unknown function
Why is meloxicam superior to aspirin?
Meloxicam has a higher affinity to COX2 enzymes → Superior analgesic and anti-inflammatory properties AND fewer side effects (less inhibition of physiological COX1)
List 7 side effects of NSAIDs (animals)
GASTRIC ULCERATION → Melaena, anorexia and haematemesis (coffee-grounds)
KIDNEY FAILURE
Vomiting/diarrhoea/inappetence (unknown MoA)
Increased bleeding time
Carprofen (Rimadyl) = Rare idiosyncratic hepatotoxicity (Labradors)
Phenylbutazone = Agranulocytosis → Increased risk of infection
Dermal reactions
List and describe the MoA of 2 side effects of NSAIDs on HUMANS
COXIBS (eg. firocoxib) = Heart failure (humans ONLY)
COXIB drugs only target COX2 → Inhibited release of PGI2 (potent vasodilator) → Vasoconstriction of coronary and brain arteries → Myocardial infarct and stroke
Asthma (humans ONLY)
Leukotrienes produced cause bronchoconstriction in patients with pre-existing lung pathology
Describe the mechanism of gastric ulceration caused by NSAIDs (or stress)
Prostaglandins play an important role in NORMAL physiological processes by binding receptors on gastric glands which protects the gastric mucosa from acidic stomach contents. NSAIDs inhibit PG synthesis →
Decreased mucosal blood flow
Decreased bicarbonate production
Decreased mucus secretion
Increased release of H+ from chief cells
ANY NSAID given for 5 - 7 days produces clinical signs associated with gastric ulcers
List 8 ways to reduce NSAID-induced gastric ulceration (when required for long-term therapy eg. OA)
Intermittent treatment (give for 5 days then wait)
Multimodal analgesia to reduce dose rates required
Misoprostol
Sucralfate
Antacid
Omeprazole
Atropine
Antihistamines (eg. ranitidine and cimetidine)
Describe the normal physiological effect of prostaglandins on the kidney
NSAIDs rarely cause AKI in healthy patients with normal BP
Decreased BP detected by JG cells which activates RAAS
Angiotensin II released causes generalised vasoconstriction (i.e. efferent arteriole constriction)
Efferent arteriole constriction results in increased GFR and stabilisation of pressure
PG acts as angiotensin II ANTAGONIST to cause vasodilation and relieve pressure in the efferent arteriole (maintains homeostasis)
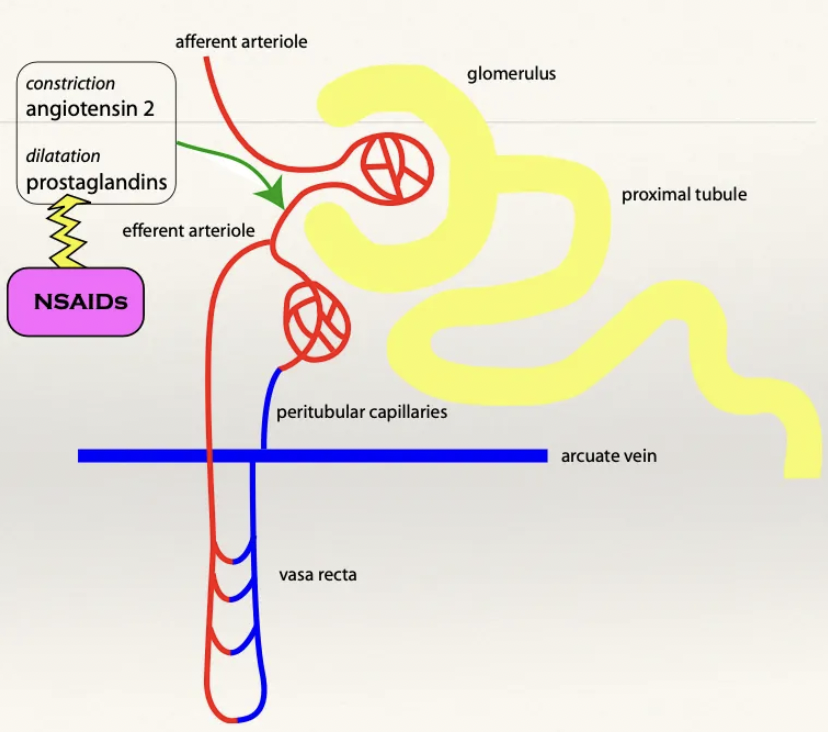
Describe the mechanism on AKI due to NSAIDs
Hypotensive patient administered NSAIDs → No PG to cause vasodilation
→ Permanent efferent arteriole constriction
Causes ischaemic necrosis and kidney failure (efferent arterioles supply the peritubular capillaries)
Avoid NSAIDs while under GA as it induces kidney failure due to hypotension (use opioids for analgesia instead)
List 5 indications of NSAIDs
Muscle damage
Mild pain
Osteoarthritis (intermittent use during flare-up periods)
Post-op pain
Colic
7 Contraindications of NSAIDs
Pre-existing GI complications (or consider omeprazole to reduce gastric ulceration risks)
Avoid COX2 inhibitors with history of myocardial infarction, angina, chronic CHF and strokes (inhibit PGI2)
Liver disease/hepatotoxic drugs (NSAIDs metabolised by liver)
Kidney disease/nephrotoxic drugs (eg. diuretics, ACE-i, aminoglycosides)
Patients with altered haemostasis (thromboxane is COX1-dependent)
Patients with low circulating volume (CHF, ascites, diuretics, dehydration, hypotension) → Exacerbate renal damage
Glucocorticoids
3 Specific NSAIDs which are contraindicated in different species
Aspirin contraindicated in cats and cattle
Cat half-life = 22hr
Cattle half-life = 25 minutes
Phenylbutazone contraindicated in cattle (long half-life NOT for production animals)
Naproxen and ibuprofen contraindicated in animals (long half-life due to enterohepatic recycling)
List the commonly used NSAIDs in cattle, horses, dogs and cats (+ COX enzyme affinity)
Cattle
Flunixin ((non-selective)
Ketoprofen (non-selective)
Tolfenamic acid (COX2)
Horse
Phenylbutazone (COX1 = Right dorsal colitis)
Flunixin (non-selective)
Ketoprofen (non-selective)
Tolfenamic acid (COX2)
Dogs/Cats
Meloxicam (COX2)
Carprofen (COX2)
Deracoxib/firocoxib (highly COX2)
4 Drug interactions with NSAIDs
Other drugs inhibiting prostaglandins (eg. EP4 inhibitors and glucocorticoids)
other drugs inhibiting renal blood flow (eg. diuretics)
Combination with highly protein-bound drugs → Increased risk of adverse reactions in patients with compromised hepatic function
Other drugs inhibiting CYP450 (eg. chloramphenicol, cimetidine, imidazole, antifungals)
2 Recommendations or NSAID use
Use short-term with lowest dose possible
Use in combination with opioids to produce superior analgesia with fewer side effects
What is neuropathic pain?
Maladaptive pain which originates from nerves (NOT tissue injury) eg. IVDD or chronic OA pain
List 6 drugs which help with neuropathic pain
Carbamazepine (anti-convulsant benzodiazepine) = Inhibits voltage-gated Na+ channels → Prevents sustained firing of neurons
Gabapentin/pregabalin = Inhibits voltage-gated Ca2+ channels → Disrupts NMDA receptors and excitatory systems
Methadone = NMDA receptor antagonist
Amantadine = NMDA receptor antagonist
Ketamine = NMDA receptor antagonist
Amitriptyline (TCA) = Anti-depressant for chronic pain to inhibit re-uptake of serotonin and NA → Increased descending inhibitory pathway activity
3 Advantages and 2 disadvantages of multimodal analgesia
Multimodal analgesia = Combination of ≥2 different drug classes
Advantages:
Superior analgesia (target different points on the pain pathway)
Reduced dose → Fewer side effects
Useful for longer term dosing regimes (eg. OA = Fentanyl patch with low dose NSAIDs)
Disadvantage: $$$
What are corticosteroids? What are the 2 classes?
Corticosteroids = Class of steroid hormones produced by the adrenal cortex
Classes:
Glucocorticoids = Produced by the zona fasciculata and zona reticularis
Cortisol #1 (cortisone in birds/rats)
Mineralocorticoids = Produced by the zona glomerulosa
Aldosterone #1
List 5 examples of glucocorticoids from least → most potent (+ duration of action)
Hydrocortisone = Short-acting (12hr)
Prednisone (dogs) → Prednisolone (horses/cats) = Intermediate-acting (12 - 36hr)
Triamcinolone (topical/intra-articular) = Intermediate-acting
Dexamethasone = Long-acting (48hr)
Bethametasone
Methylprednisolone acetate (insoluble salt) = Long-acting (3 - 5d)
Describe 5 levels of immune suppression
NSAIDs = Suppress immune response through anti-inflammatory effects
Low dose corticosteroids = Anti-inflammatory ± mild immunosuppression
Immunomodulators = Selective immune modulation (eg. oclacitinib)
High dose corticosteroids = Immunosuppression
Old anticancer drugs = ZERO immune response
Describe the mechanism of glucocorticoid binding to receptors
Glucocorticoid receptors are within almost EVERY cell → Widespread reaction
Intracellular receptor → Long onset of action but long duration
Mechanism:
Cortisol crosses the cell membrane and binds to a receptor to enter the nucleus
Altered gene transcription
→ Increased synthesis of lipocortin = Phospholipase A2 antagonist responsible for preventing synthesis of arachidonic acid
BOTH prostaglandins and leukotrienes are NOT synthesised
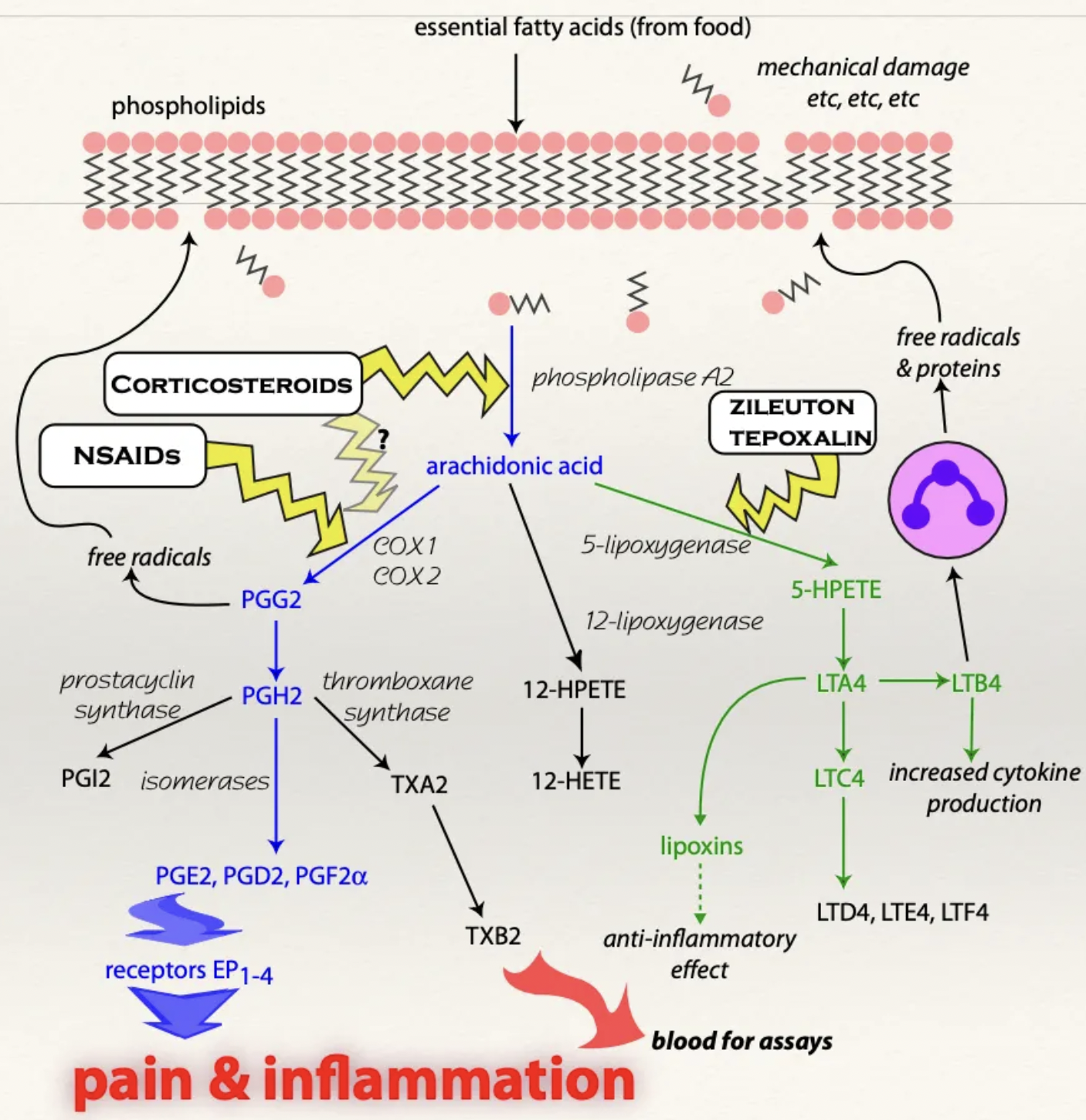
Do glucocorticoids have analgesic properties?
No (anti-inflammatory ONLY)
List 5 indications of corticosteroids
Reduce inflammation (eg. trauma or IBD)
Reduce allergies (eg. atopy)
Immunosuppression (eg. autoimmune disorders or immune system cancers)
Shock therapy (esp. refractory septic shock) → NOT indicated now
Parturition in cows (unethical)
How should glucocorticoids be used?
Lowest dose for shortest period possible (longer therapy → longer tapering-off period to restart the hypothalamic-pituitary-adrenal axis)
Induction dose for 1 - 2 days until no clinical signs of inflammation are observed
Transition to every other day treatment
Skip more and more days of treatment to taper drug off
10 Side effects of corticosteroids (+ MoA)
Corticosteroids are released during fight-flight response
Hyperglycaemia via protein catabolism, lipolysis and gluconeogenesis
Cortisol = Anti-insulin agent
PU/PD via ADH inhibition → UTI
Every glucocorticoid has a mineralocorticoid effect at high/long-term doses
Increased GFR (PU and generalised vasoconstriction due to lack of PG)
Osteoporosis via 2˚ hyperparathyroidism
Cortisol = Inhibited absorption of Ca2+ from duodenum and increased elimination from kidneys → Increased PTH → Increased osteoclastic activity
Immunosuppression via WBC apoptosis (except neutrophils) and reduced capillary permeability (reduced emigration of neutrophils and macrophages) → Increased risk of infection AND delayed wound healing
Euphoria/depression and polyphagia via increased dopamine and serotonin release
Thin and fragile skin via reduced fibroblast activity
Calcinosis cutis and alopecia due to increased PTH → Ca2+ deposited on skin
Muscle wasting via protein catabolism → Temporalis muscle wasting and pot belly
Infertility via reduced GnRH release from hypothalamus
Teratogenic
Inhibited ovulation and spermatogenesis
Induce abortion/parturition (promotes foetal lung maturity and surfactant production at the end of gestation)
GIT via gastric ulceration (inhibited PG), hepatic lipidosis, pancreatitis
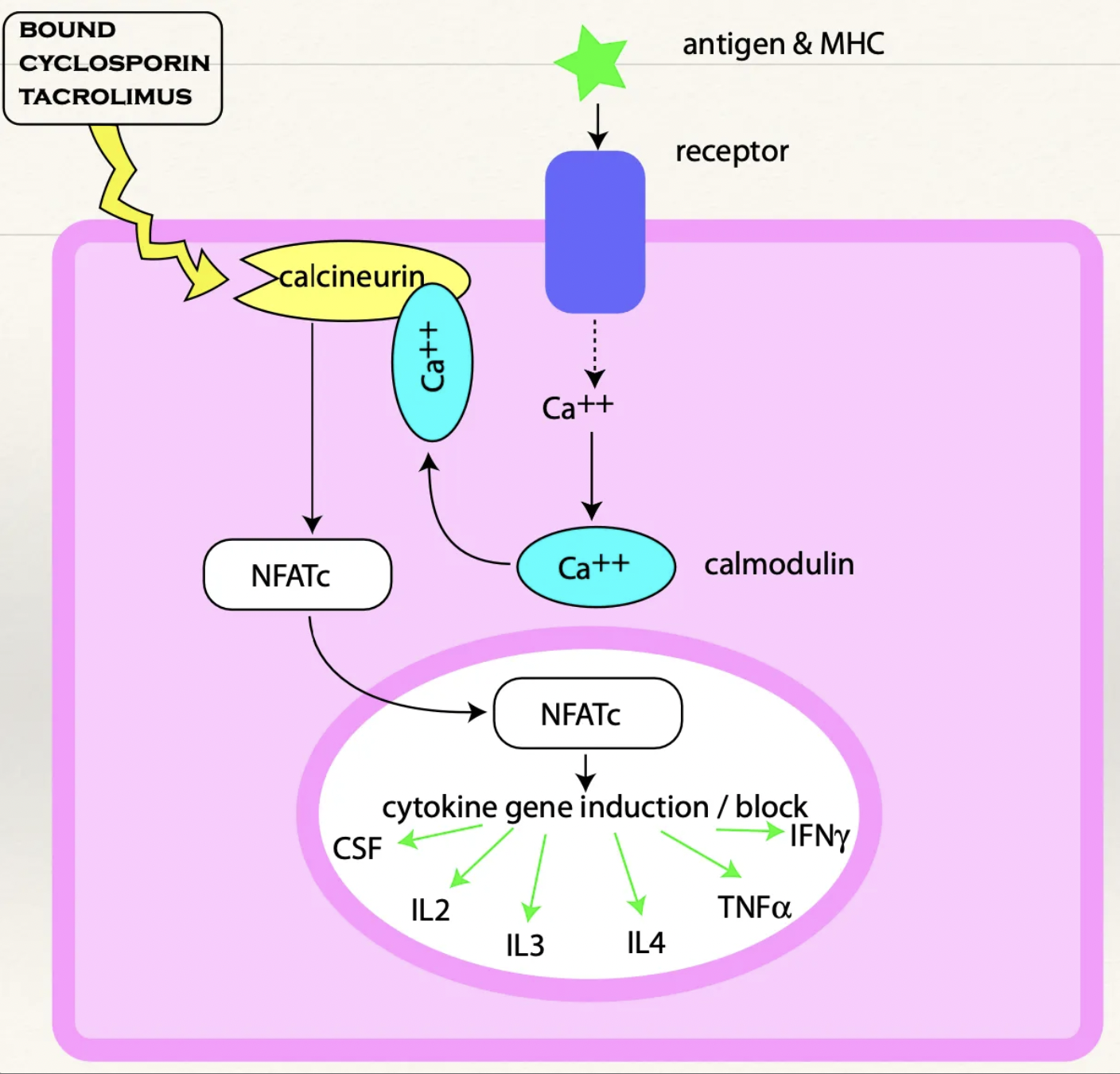
Tacrolimus
MoA
5 Indications
MoA: Binds immunophilin which inhibits calcineurin → NFATc is NOT transported to the nucleus → Prevents production of cytokines (eg. IL, TNF, IFN) → T-cell activation blocked which prevents clonal expansion and a cytotoxic response
Indications: Immunosuppression
Atopic dermatitis (eczema)
Keratoconjunctivitis sicca
Anal furunculosis
Adjunct to corticosteroids
Transplants
List 11 antibiotics groups (+ which of the 5 antibiotic classes they fit under)
Cell wall synthesis inhibitors
Beta-lactams (penicillins and cephalosporins)
Bacitracin
Protein synthesis inhibitors
Aminoglycosides
Tetracyclines
Fenicols
Macrolides
Lincosamides
Nucleic acid synthesis inhibitors
Potentiated sulphonamides
Fluoroquinolones
Nitroimidazoles
Cell membrane destruction
Polymyxin B
Others
List 6 mechanisms of antibiotic resistance
Inactivation of drug (eg. β-lactamases and plasmid-mediated chloramphenicol acetyl transferase)
Increased efflux pumps → Enhanced removal of antibiotic
Decreased cell membrane permeability to antibiotic
Alter binding site (eg. Methicillin-resistant staphylococcus aureus)
Increased target protein (eg. increased PABA production for TMP)
Target in protected environment
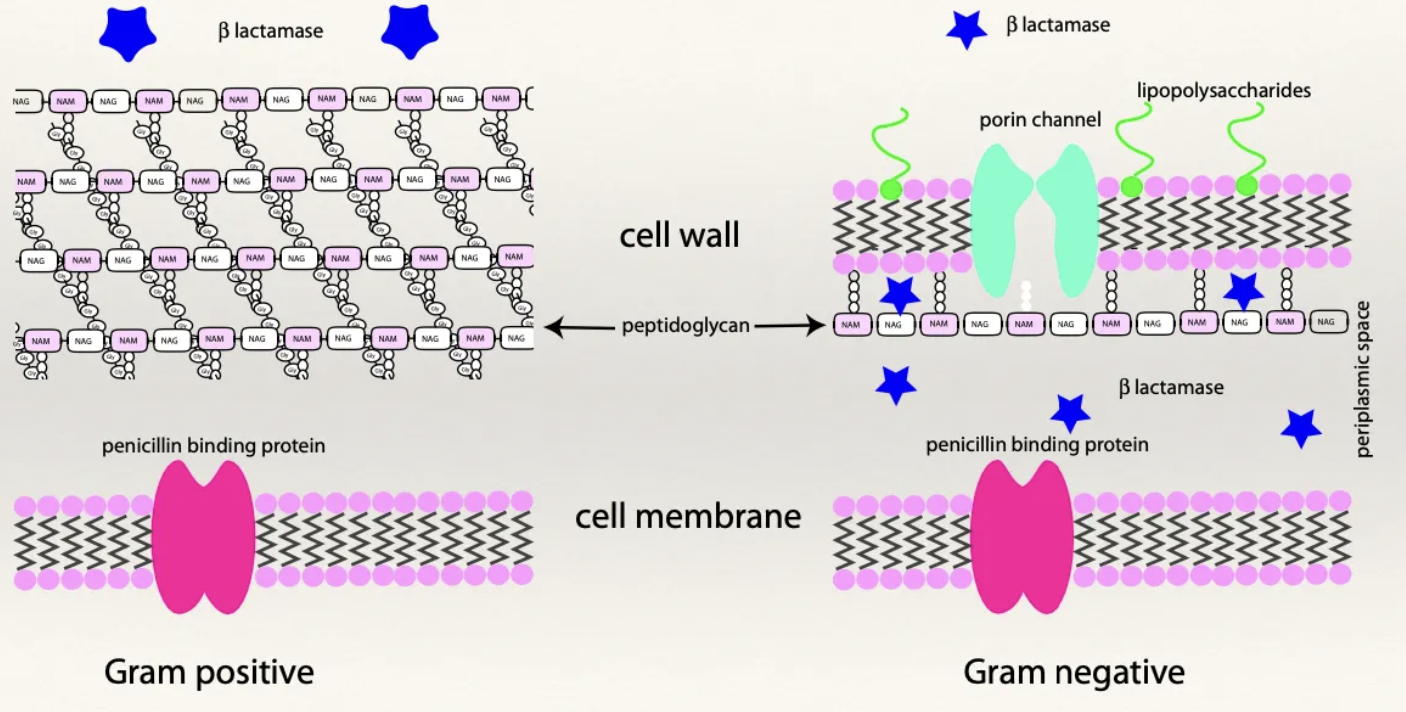
Describe the structure of G+ vs. G- cell walls
G+: Thick peptidoglycan wall and NO LPS layer
Alternating chains of N-acetylmuramic acid and N-acetylglucosamine which are joined together by peptide chains
No protein channels
Highly porous and easy for penicillin to pass through
G-: Thin peptidoglycan wall
Lipopolysaccharide (LPS) layer - Outer layer with LPS which are highly toxic to mammalian cells when broken down and released
Destruction of G- bacteria → Release of LPS causing endotoxaemia
NOT a porous cell wall (contains specific protein channels for nutrients and metabolic products to enter and exit the cell wall)
Penicillin must enter through protein channels to exert effect
Thin peptidoglycan wall
Periplasmic space - Separates the cell wall and the cellular membrane
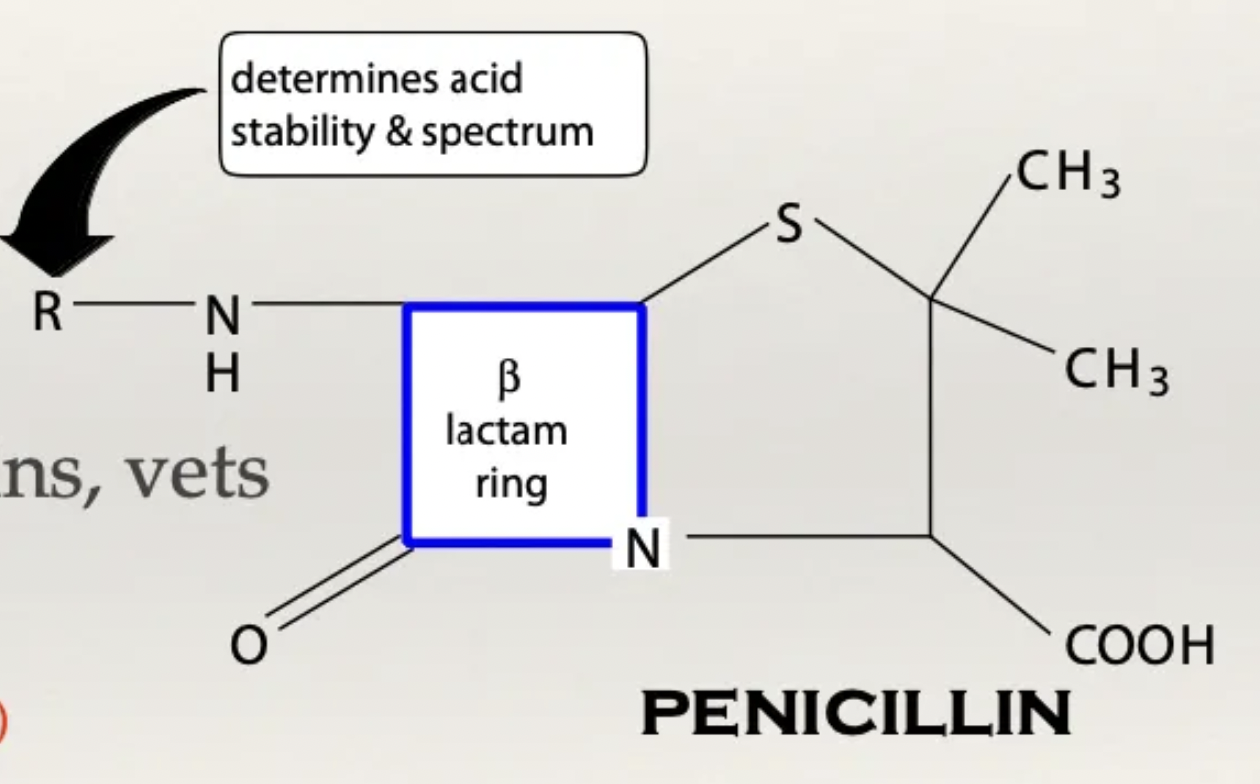
β-lactams: Structure
β-lactam ring = Important for antibiotic activity (broken ring → inactivated AB)
R chain = Determines spectrum of action and resistance
Large/complex R chain = Narrow spectrum (large R group cannot fit through G- protein channels) BUT highly resistant to β-lactamases (large R group protects the ring)
Small R chain = Broad spectrum (G-/G+) BUT less protection
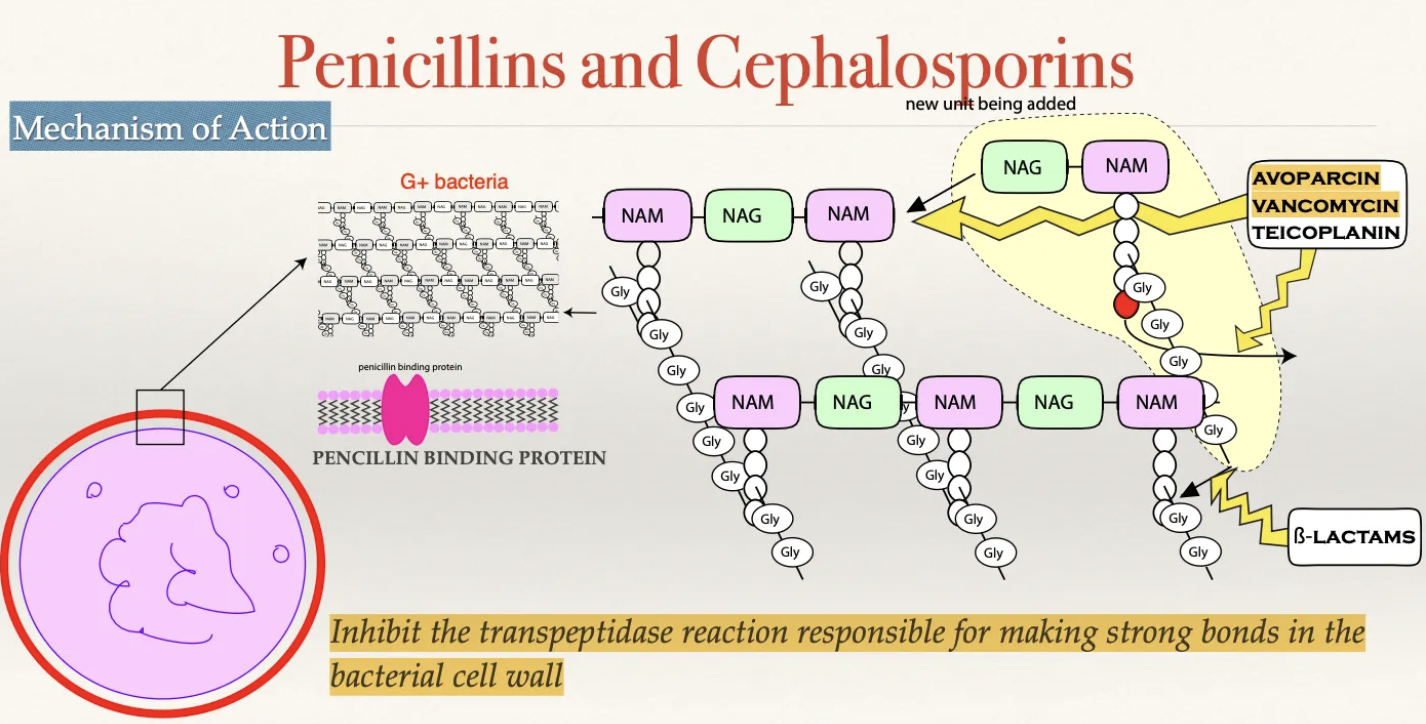
β-lactams: MoA
Drug MUST cross cell wall to enter the periplasmic space
G+: Porous cell wall and easy entry
G-: NOT porous cell wall (must enter through protein channels)
Drug covalently binds to penicillin binding protein in the cell membrane
PBP = Produces transpeptidase to assist in bonds between peptidoglycan chains of the cell wall via inter-bridging
Inhibited production of bacterial cell wall
Cell wall weakens causing the cell membrane to bulge and rupture
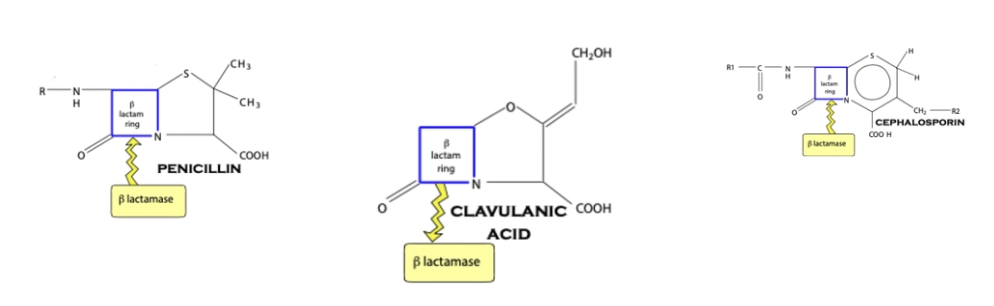
What is β-lactamase? How does it differ between G+ and G- bacteria?
Definition: Enzymes which destroy the β-lactam ring of penicillin and prevents it from working
G+: β-lactamases produced OUTSIDE of the bacteria → NOT confined to the periplasmic space
G-: β-lactamases trapped within the periplasmic space → Penicillin must penetrate through the cell wall to be inactivated
Clavulanic acid
Definition
Indication
Definition: β-lactamase inhibitor which increases activity of penicillins
Covalently bind with bacterial β-lactamase to inhibit destruction of the β-lactam ring
Indication: Given in conjunction with broad-spectrum penicillins against G- (not necessary for G+ as β-lactams are NOT confined to the periplasmic space)
List 4 examples of penicillins (+ spectrum of action)
Penicillin G (benzylpenicillin) = G+ (large R chain)
NOT PO
G- bacteria have LPS cell wall which prevents uptake of penicillin (small protein channels)
Na+ and K+ salts can be given IV (K+ concentrated resulting in disruption of membrane potentials)
Cloxacillin = Staphylococcus
Frequent component of IMM preparations in cattle
Amoxycillin/ampicillin ± clavulanic acid = G+/G- (small polar amino group)
Amoxycillin has -OH group to increase PO absorption
Ampicillin IM/SC ONLY (absorption impeded by food)
Ticarcillin/piperacillin = Extended spectrum (Pseudomonas)
Penicillins = Spectrum depends on R chains (G+ aerobes → G+ anaerobes → G-)
β-lactams: Pharmacokinetics
Absorption: Natural benzylpenicillin (G) must be given parenterally (ring destroyed by stomach pH)
Synthetic penicillins eg. ampicillin and amoxycillin can be given PO
Distribution: Poor (hydrophilic)
Ionised at blood pH (pKa ~2.7) → Restricted to the ECF (cannot cross cell membranes easily)
Cannot access BBB, eye or prostate
Metabolism: Minimal
Elimination: Urine (penicillin eliminated unchanged in urine)
Some synthetic penicillins accumulate in bile → Useful for GI infections
β-lactams: Duration of action
Na/K salt (IV) lasts 2hr
Procaine salts (IM) last 12 - 24hr
Procaine in oil lasts 48hr
Common on farm
Benzathine lasts ≥48hr
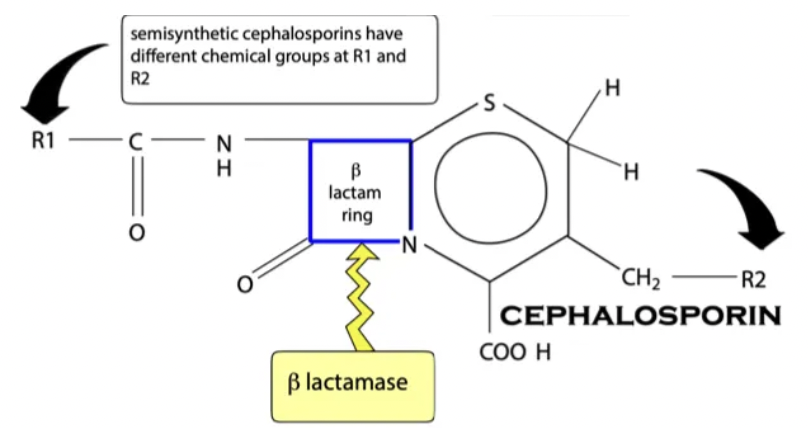
Cephalosporins
Structure
6 Examples
Caution
Structure: TWO R chains (protects β-lactam ring well → β-lactamase resistant)
Examples: Increasing spectrum of action
1st generation = Cephalexin, cefapirin, cephazoline (G+)
3rd generation = Ceftiofur, cefovecin (Convenia injection = 2w duration)
4th generation = Cefquinome
Caution: Avoid 3rd and 4th generation cephalosporins
β-lactams: 4 Adverse effects
Allergic reaction (rare in animals)
Electrolyte disturbances
Procaine reactions (esp. horses)
Antibiotic-associated diarrhoea (AAD) through damage of commensal GIT bacteria
Avoid in guinea pigs and hamsters
β-lactams: 5 Indications of use
Prophylaxis
UTI
Meningitis (BBB disrupted)
Skin infections
Respiratory infections
Aminoglycosides: MoA
Irreversibly binds to receptor protein of bacterial 30S ribosomal subunit → Inhibit protein synthesis
Distort codon arm and forces production of nonsense proteins
Aminoglycosides: Spectrum of action
G- aerobes ONLY
MUST have O2 transporters to enter the cell membrane
Useful synergism with β-lactams
Aminoglycosides: 4 Examples
Streptomycin
Gentamicin
Neomycin
Amikacin
Aminoglycosides: Pharmacokinetics
Absorption: ALWAYS give parenterally (no GIT absorption)
Distribution: Poor
Metabolism: Post-antibiotic effect = Bacterial growth suppression AFTER drug concentrations falling below MIC in the blood
Elimination: Kidneys (NEVER use in food animals as present in kidneys for months)
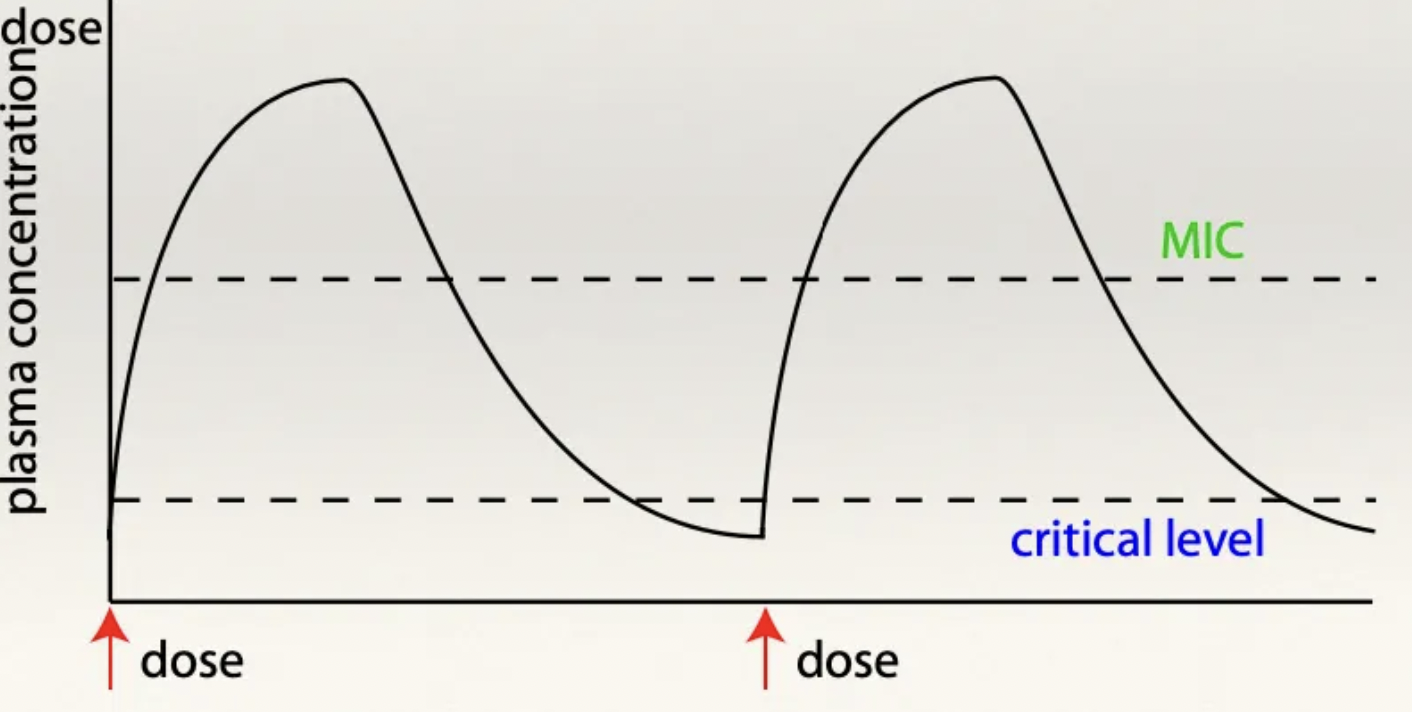
Aminoglycosides: 2 Adverse effects
Positively charged molecules attracted to negatively charged cell membranes of the:
Proximal convoluted tubules → Nephrotoxic
Cochlear of the ear → Ototoxic (kill hair cells)