biochem recombinant dna tech slides
1/63
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
64 Terms
difference between gene sequence and protein sequence:
gene sequence: a segment of DNA that contains the instructions for a protein. genes are made out of nucleotides.
protein sequence: is the specific order of amino acids that make up a protein. proteins are made out of amino acids.
Basically the gene's nucleotide sequence is transcribed and then translated into the protein's amino acid sequence, which determines the protein's structure and function.
In particular, once we know a protein sequence we can determine?
the gene sequence associated with that protein.
Knowing the gene sequence we can then go back to the?
vast databases currently available that include the genomes of nearly every organism we might be interested in.
Prokaryotic genes are especially accessible because?
Prokaryotes also stray a bit from the classic central dogma of molecular biology which says?
they rarely splice their mRNA so the gene fully encodes the protein without further processing of the mRNA.
one gene → one mRNA → one protein.
Some genes in prokaryotes are transcribed into polycistronic mRNAs?
This differs a little from the professor’s description of viruses earlier in the semester in which an mRNA would encode multiple proteins, but in this case, often a large protein is first translated then cleaved into?
in which one mRNA transcript contains the coding sequence for multiple genes, so multiple proteins are templated by that single mRNA.
smaller functional proteins by virus-encoded proteases.
The big advantage of knowing the sequence of a protein and therefore the gene encoding that protein is that?
These techniques allow us to?
we have access to recombinant DNA techniques.
tag proteins with purification tags that we can then use for affinity purification (GST, hexahistidine, MBP) or tags such as green fluorescent protein (GFP) that allow us to track proteins in cells.
In addition, we can use simple organisms like __ for the production of those proteins.
E. coli
While E. coli is the most commonly used organism, these techniques can also be applied using?
What differences does E.coli and these other types of cells?
host organisms like yeast as well as insect and mammalian cells.
these other cells tend to take more time when working with them but may have advantages. these cells are used when certain post-translational modifications of the produced proteins are required. Yields of proteins also tend to be much lower in these other types of cells.
To use recombinant expression of genes for the production of ? we typically incorporate?
protein.
the gene that we are interested in into a circular piece of DNA referred to as a plasmid.
Plasmid DNA occurs in __, especially in bacteria, and encode?
For example, plasmid DNA is often responsible for a phenomenon called? what does this phenomenon do?
nature
extrachromosomal genes that can benefit a particular organism.
horizontal gene transfer where certain strains of bacteria can share plasmids for things like antibiotic resistance.
In recombinant DNA technologies, the plasmid is referred to as a __. What do these things do?
vector.
These vectors are used to transform bacteria by introducing new properties via the plasmid.
What are the key components of a plasmid?
a promoter (a region of DNA located near a gene that signals where to begin the process of transcription)
transcription start site
His-tag coding sequence
multiple cloning site (MCS) (provides a specific region on the vector for the insertion of a new gene)
terminator (signals when transcription stops)
lacI coding sequence
origin of replication (a specific DNA sequence where the process of DNA replication begins)
bla coding sequence.
define restriction enzymes.
name 3 examples?
cut DNA at specific sequences (different enzymes cut slightly differently).
NdeI, XhoI and BamHI.
NdeI leaves a __.
two-base overhang.

BamHI leaves a __.
four-base overhang (also called sticky ends, but the professor’s preference is staggered ends).

To incorporate our gene into a vector we first?
amplify our gene of interest using the polymerase chain reaction (PCR). When we amplify the gene we can incorporate whatever sequences we want onto the end of our gene so we can incorporate the NdeI site and BamHI site.
Are Ndel and BamHI the only sites we can use?
We could use NdeI/XhoI, XhoI/BamHI and we could also use other sites like the NcoI site if we did not want the tag on our final protein.
To incorporate our gene into a vector we first amplify gene by PCR, then we?
We will then do a double digestion of our plasmid and our PCR amplified gene. Digestion of the vector with one or more restriction enzymes opens the vector making it linear. We analyze the digestion products by agarose gel electrophoresis.
Do we typically do double digest controls to ensure that the enzymes are working?
no we do single digest controls
single digest controls give us __ which differs from the undigested plasmid.
the ~5000 bp (base pair) linear product.
Why would we linearize the plasmid?
circular DNA supercoils tends to not run on a gel where we would expect for that size, so we must linearize the plasmid to get reliable sizes.
When we double digest the plasmid with both enzymes we should see a small shift in the?
t/f: While it is shown on the gel below that we could see the part that was cut out on the gel, this in practice is easy to see.
size of the plasmid since a portion has been removed.
FALSE. it is difficult to see.
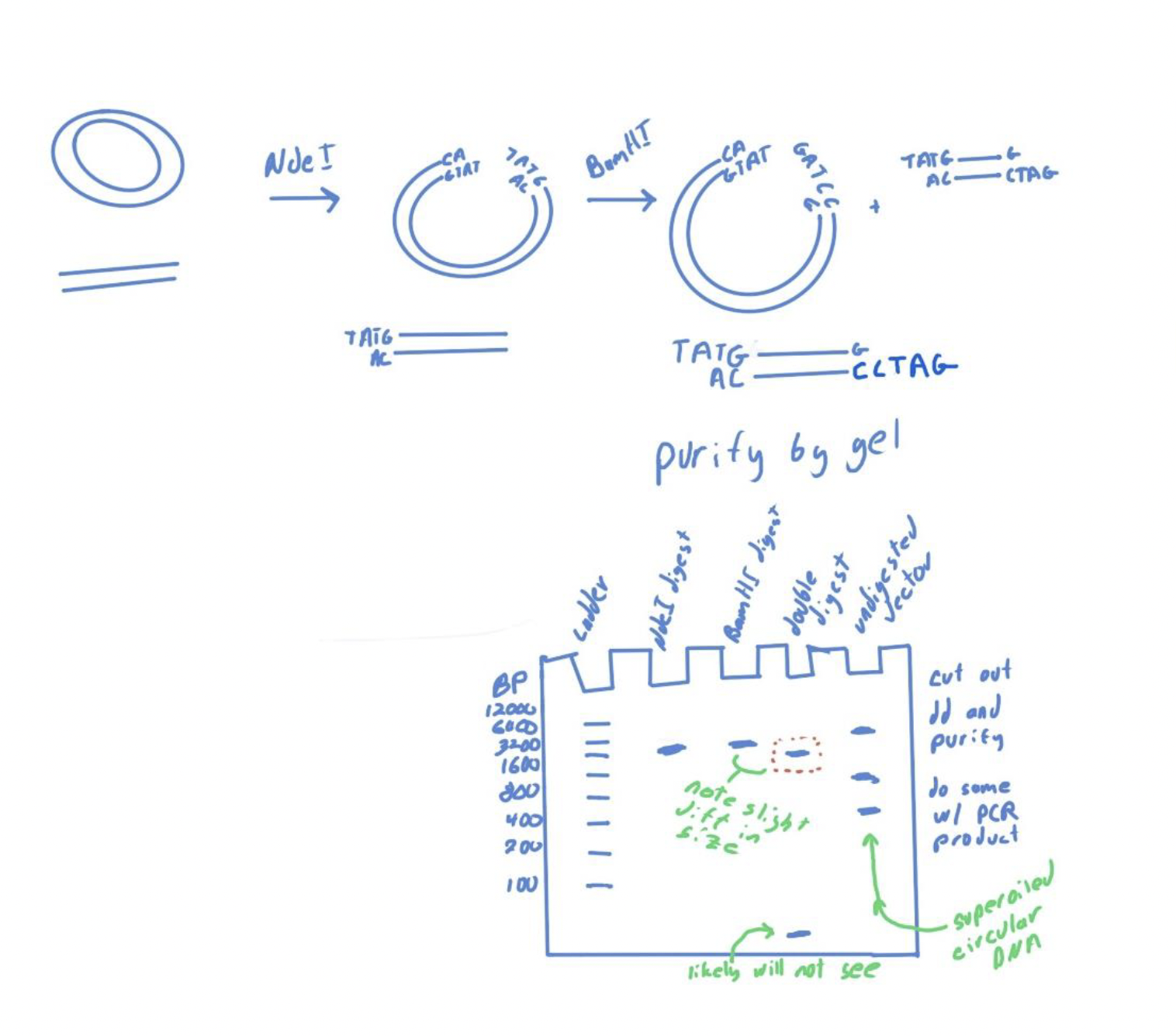
Why is it difficult to see the part of the plasmid that has been removed?
the reason that it is difficult is that we detect the DNA in the gel using stains like ethidium bromide which intercalates (inserts) into the DNA double helix.
The much larger fragment has many more sites for EtBr interaction relative to the smaller fragment which means that the smaller fragment will not be nearly as easily seen as the larger one.
dna gel electrophoresis differs from what we saw with proteins by?
we use an agarose gel which has much larger pores than the polyacrylamide gels in protein gel electrophoresis.
this agarose gel is better to use for the bigger nucleic acids which are bigger than proteins.
Why did we NOT use SDS for dna gel electrophoresis?
we did not need to add SDS to make the nucleic acids soluble and negatively charged. DNA is already negatively charged and readily soluble whether or not it is denatured.
To incorporate our gene into a vector we first amplify gene by PCR, then we perform gel electrophoresis then we?
After separating the double digestion of the gene and the vector on a gel, we then cut the bands out remove the gel and EtBr then use those pure materials for the next step.
We then paste our gene into the vector using an enzyme called a ligase. This gives us our new vector with our gene of interest inserted.
Ligase and ATP forms?
a phosphoester linkage between vector and insert.
Notice that before the MCS we see a sequence of nucleotides that encode __.
What is the significance of this sequence?
LVPRGS (lysine, valine, proline, arginine, glycine, serine)
is a site recognized by the protease enzyme thrombin.
What is Thrombin?
a protease enzyme that recognizes the LVPRGS amino acid sequence and cleaves the protein resulting from this vector after arginine residue.
Is there a relationship with Thrombin with tagging?
This cleaving site allows for the removal of the hexahistidine tag from the final protein.
This tag is encoded by the vector just before the thrombin site where you see His*tag in bold.
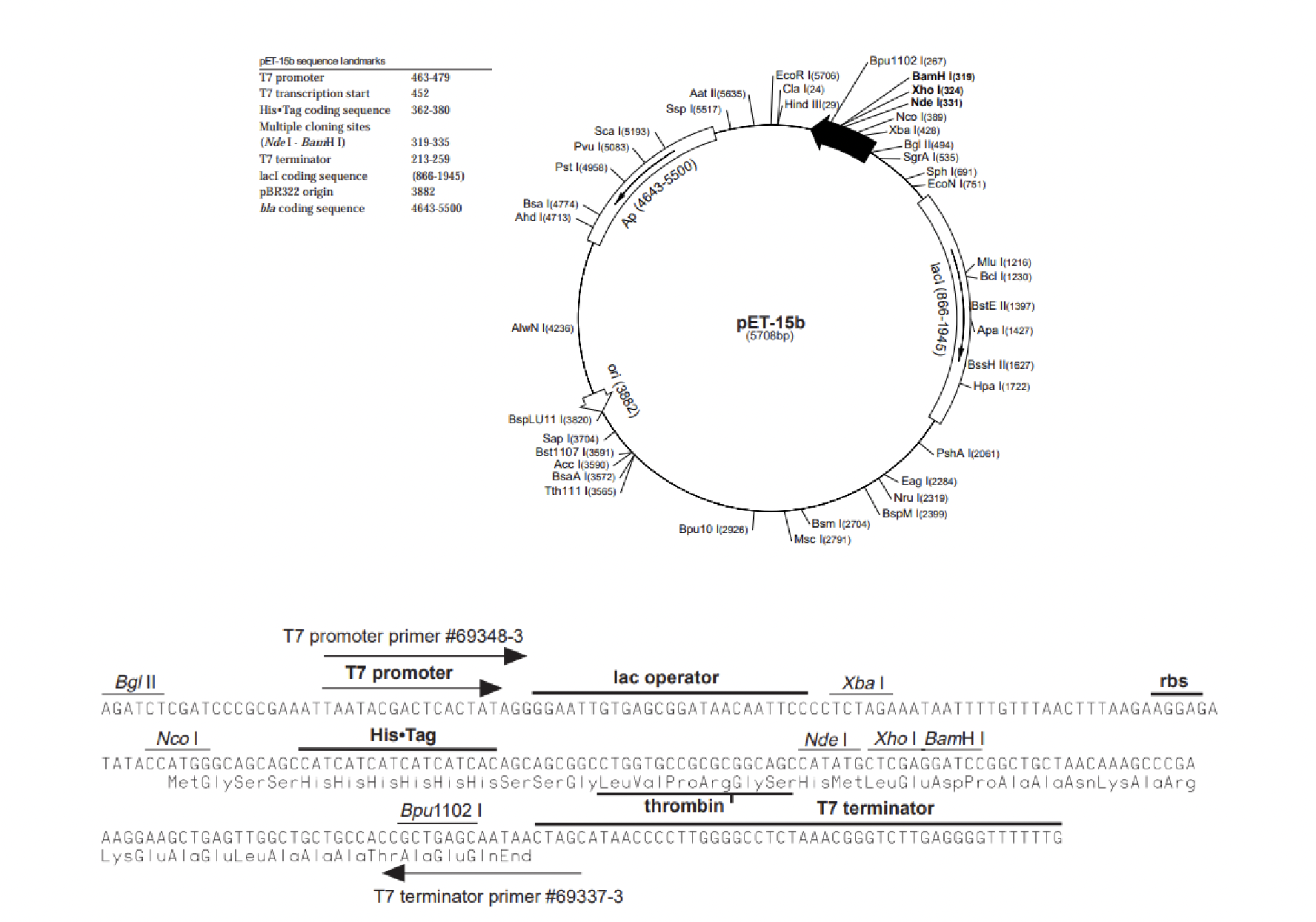
T/F: Is thrombin involved in blood clotting.
TRUE
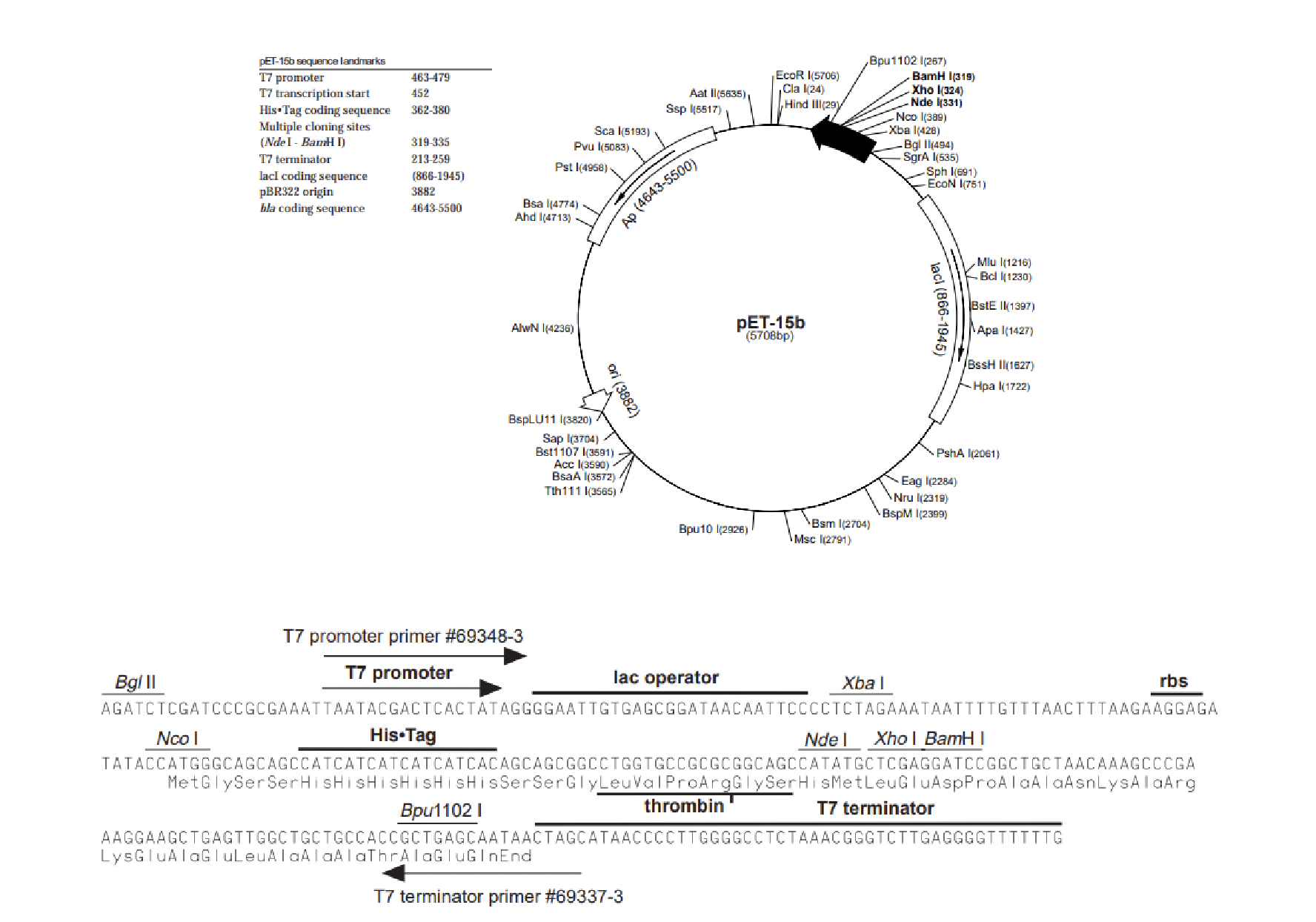
Prior to the His tag we see the promoter region which is a sequence of nucleotides that will?
recruit a DNA dependent RNA polymerase where transcription of this vector would start just after the promoter region and continue to the terminator.
What does the resulting mRNA have to do with translation?
The resulting mRNA would contain the rbs (ribosome binding site) region as well which is a sequence of nucleotides in the mRNA transcript that promote an interaction with the ribosome to initiate translation.
what is the origin of replication?
the start site for replication of the plasmid, and we usually only think about it when we want to transform an organism with multiple plasmids.
can we use the same origin of replication for each type of plasmid?
NO we have to use a different origin of replication for each type of plasmid otherwise daughter cells will not produce both.
The lacI coding sequence encodes the protein LacI (lactose repressor).
This protein was initially found in nature and was found to be responsible for?
sensitivity to lactose (a disaccharide that can serve as food for the organism). LacI is a repressor protein which means that it represses transcription of certain genes.
What happens when the Lacl protein binds to the lac operator region of the vector?
However, what if there is lactose present? Would this change anything?
When bound it physically blocks movement of the DNA dependent RNA polymerase and therefore blocks transcription.
However, if lactose is present it will bind to LacI, induce a conformational change in the protein that will then lead to dissociation of LacI from the operator and transcription can then occur. This allows for the control of gene expression from this plasmid.
In practice when we express genes from these vectors do we first want our cells to be focused on growing and dividing, or protein production?
When do we focus on protein production?
we first want our cells to be focused on growing and dividing, not protein production.
Once we have a culture full of cells (well into exponential phase of growth) we then induce protein production by adding lactose which switches the cells from focused primarily on growth to protein production.
Do we typically use lactose or do we use something else for protein production?
we use a molecule called isopropyl thiogalactoside (IPTG). IPTG is an analogue of lactose that is not consumed as food by the bacterium, and only induces protein production through its interaction with LacI.
does lactase have anything to do with lactate?
NO
why are lactose and IPTG considered analogues?
the American Chemical Society (ACS) style guide points out that when referring to a molecule that is similar to another or derivative of another we use the British spelling analogue.
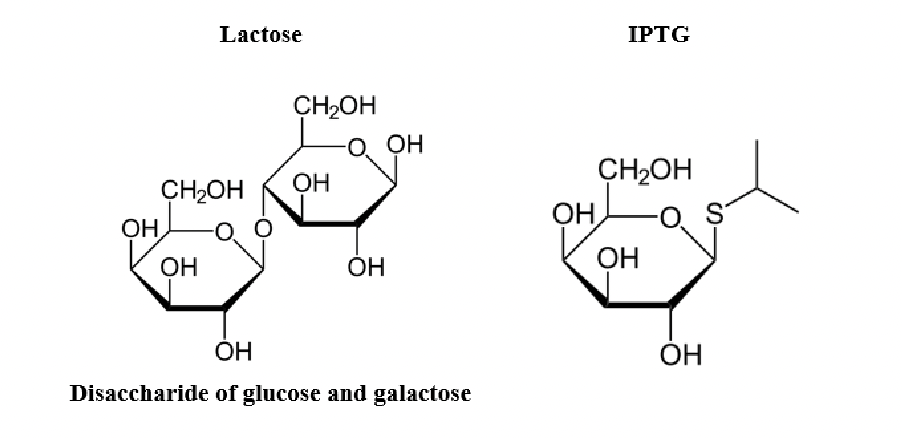
Bla is a gene that encodes a __. Note that once transformed into a host the vector the cells are always producing this beta lactamase and the LacI protein.
beta-lactamase protein.
We only control the production of the gene in the MCS. When were Beta lactamases proteins discovered?
very early on after the first use of antibiotics.
Basically if B-lactamase is produced in cells than what will not impact the bacteria?
antibiotics.
How does B-lactamase make the antibiotics ineffective?
The antibiotic normally __ which is an essential component of the bacterial cell wall.
The penicillin family of antibiotics contains a beta-lactam ring that is cleaved by the B-lactamase which makes these antibiotics ineffective.
inhibits the production of a molecule called peptidoglycan.
In the laboratory, the beta-lactam antibiotics that we typically use are ampicillin or carbenicillin (a slightly more shelf-stable form of ampicillin).
T/F: The bla gene therefore confers antibiotic resistance to these drugs to any cell that has taken up the vector.
TRUE
Why is B-lactamase considered a selectable marker?
The gene isolates bacteria that has been transformed with the vector. The bacteria that picks up the plasmid will survive.
Typically we prepare a vector with our gene of interest and transform it into cells using what two ways?
What will spreading these vector inserted cells spread on a selective media plate show?
Either electroporation (electric shock opens pores in the membranes that allow for uptake of the plasmid) or heat shock (increase temperature to 42 °C [107.6 F] in the presence of Ca2+ leads to opening of pores and uptake of plasmid).
We then spread these cells on selective media (media that contains antibiotic) and if the cells can grow it is because they took up the plasmid, and any cells that did not take up the plasmid die [recall these vectors have antibiotic resistance so now the bacteria will be resistant to the antibiotic because of this new resistance inserted].
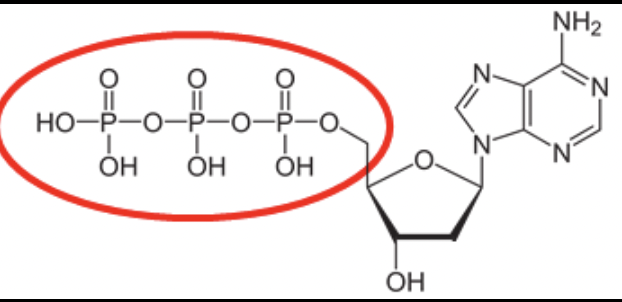
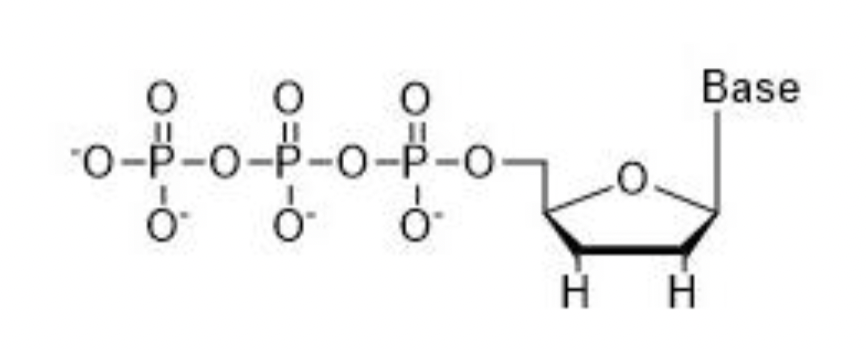
what is a dNTP?
deoxynucleotide triphosphates, which are the essential building blocks for synthesizing DNA. They consist of a deoxyribose sugar, a nitrogenous base (A, C, G, or T), and a triphosphate group.
What are the 3 core steps of PCR?
melt (melting dna at 95 degrees Celsius or 203 F)
annealing (temperature is lowered to 45 degrees Celsius allow short DNA primers [short sequence of dna or rna that is complementary to target) to bind to their complementary sequences on the single-stranded templates.)
extension (occurs at a higher temperature at 76 degrees Celsius where a heat-resistant taq DNA polymerase extends the primers by adding nucleotides, synthesizing new DNA strands.)
Why do we use taq polymerase?
We use taq polymerase because it can withstand the large temperature fluctuations required for PCR, prior to its use fresh enzyme had to be added for every cycle.
Typically PCR is performed in an instrument called a? What else does this instrument do?
Thermocycler that will change temperatures for each step for a specified amount of time. This is considerably more efficient than moving the sample between water baths set at different temperatures.
define introns and exons
introns: sequences of nucleotides that are not involved during the coding for proteins.
exons: a segment of DNA that contains the instructions for building a protein and is "expressed" or kept in the final messenger RNA (mRNA) copy. In a gene, exons are the parts that are spliced together after the introns (non-coding segments) are removed, forming the blueprint for a protein.
Why can we not use genomic DNA for PCR to obtain our gene of interest?
What is the solution?
Because eukaryotic genes often have introns that are spliced out of mRNA sequences we cannot use genomic DNA (the complete set of genetic material, or genome, of an organism, containing both its coding and non-coding regions) for PCR to obtain our gene of interest.
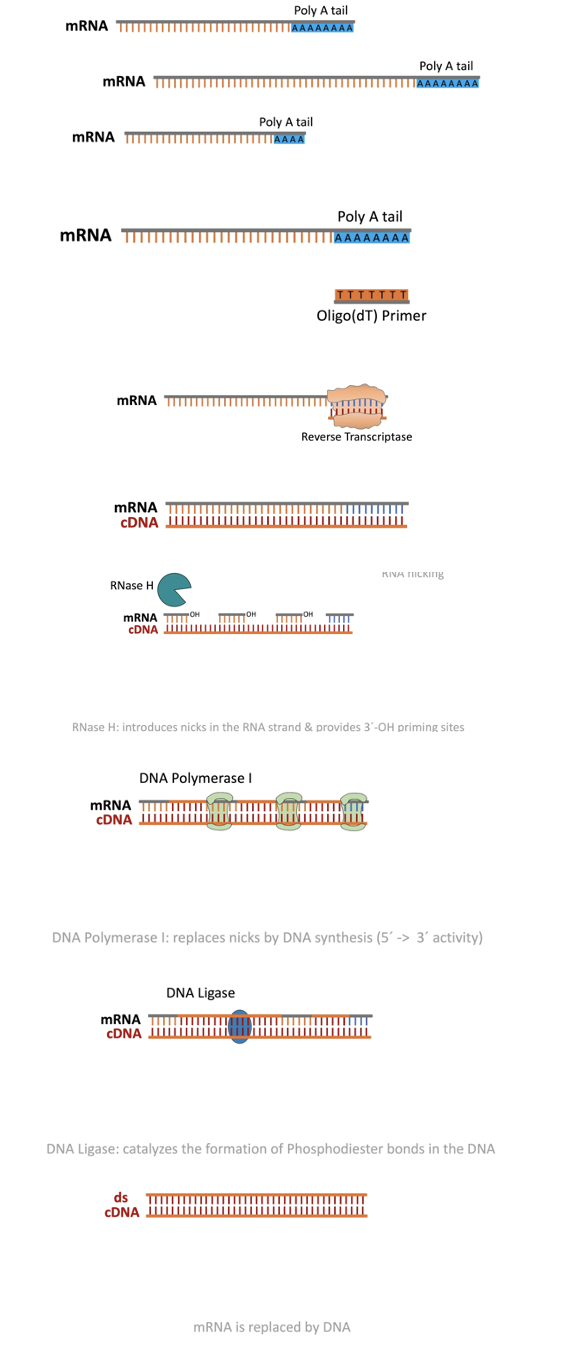
For this situation, we typically create cDNA libraries which stands for complementary DNA libraries.
What are cDNA libraries?
This method uses the enzyme reverse transcriptase which is a viral enzyme responsible for converting viral RNA genomes into DNA for host expression.
Here we take advantage of this enzyme and the fact that mRNA from eukaryotes have a poly A tail (string of more than 3 Adenine nucleotides) appended to the end of every mRNA transcript.
A primer containing a poly-T sequence (string of more than 3 Thymine nucleotides) anneals to mRNA isolated from a cell. Reverse transcriptase is then used to produce the complementary DNA sequence. mRNA is degraded by treatment with base or an RNase.
A known sequence is then ligated to the 3’ end of the mRNAs and a primer complementary to that sequence is then used for polymerization and formation of the cDNA library.
Purification tags are often incorporated into proteins via cDNA libraries and this is beneficial because?
it gives us access to simple to use affinity purification techniques that drastically simplify methods for protein purification.
We can also use these techniques to make fusion proteins where we combine multiple proteins into a single protein or chimeric proteins where we combine different parts of different proteins into one.
What are Fusion proteins especially useful for?
Name an example:
Fusion proteins are especially (which actually includes GST and MBP tagged proteins) useful for visualizing proteins in cells!
Green Fluorescent Protein, a protein first isolated from jellyfish and utilized by Roger Tsien the first time for tracking proteins in cells.
What is the makeup of Green Fluorescent Protein (GFP)?
How is fluorophore formed?
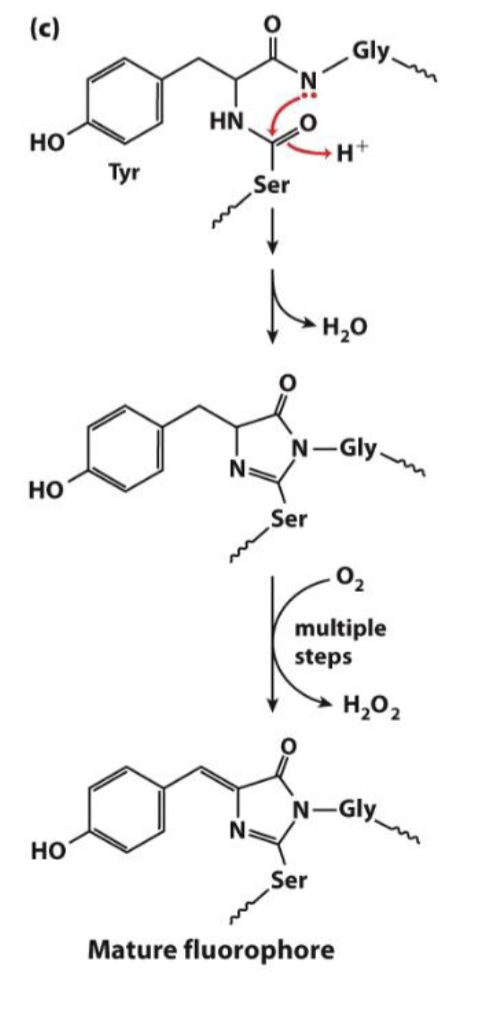
GFP is a beta-barrel protein where in the core of the protein is an SYG (serine, tyrosine, glycine) sequence.
This sequence forms the fluorophore through the attack of the G backbone nitrogen atom (normally a rare event considering the reactivity of amide nitrogen) with the S carbonyl forming a five-membered ring intermediate.
Oxidation then gives the conjugated fluorescent system shown in the figure below. Now there are many types of fluorescent proteins that emit at a variety of wavelengths allowing us to track multiple proteins at once in cells.
What was one of the clear precursors to PCR?
Are all components of PCR included in this?
Sanger sequencing.
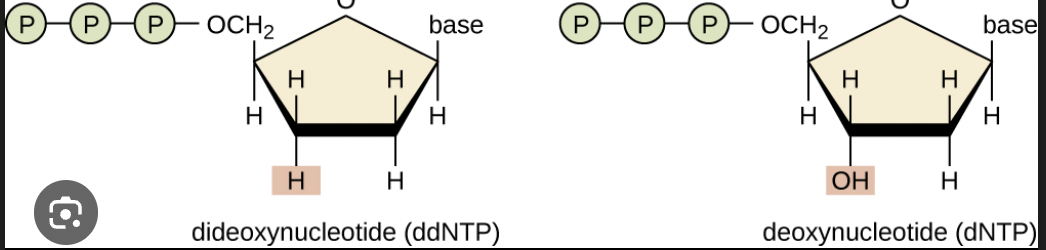
All of the components of PCR are included in Sanger sequencing along with a set of dideoxynucleotides (chain-elongating inhibitors of DNA polymerase).
Early methods to do Sanger sequencing took advantage of?
The ability to detect radioactive materials on film easily. Each dideoxynucleotide would have a radiolabeled base. Reactions would be set up where all PCR components would be added with just a single primer (not two) and one of the ddNTPs.
The ratio of the dNTP to ddNTP is typically around 1:100 meaning for every 100 dNTPs you will have one ddNTP.
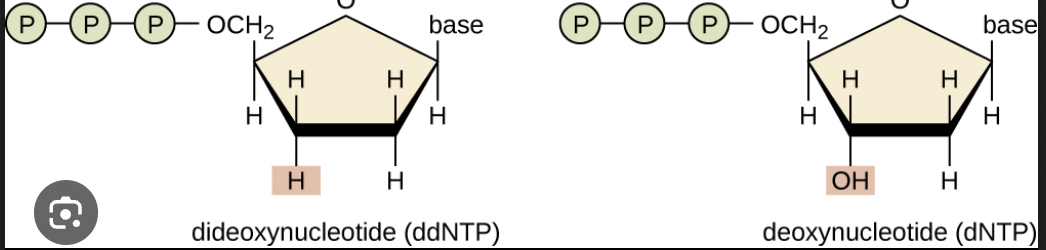
Is this a dNTP or ddNTP? why?
ddNTP because there is no 3’ OH group.
What is the significance of ddNTP (a structure that does not have a 3’ OH group) being incorporated?
Whenever this nucleotide is incorporated you terminate the extension of the nucleic acid.
At this ratio, you can ensure that at every step in the sequence, you will occasionally get chain termination and therefore a shortened sequence with the radioactive nucleotide incorporated.
Significance of dideoxynucleotides being typically conjugated to a fluorescent reporter group:
ddATP, ddGTP, ddCTP, and ddTTP each have a different fluorophore associated with them that emit a different color of light.
We can analyze these reactions with all ddNTPs present in one mixture separate by capillary electrophoresis which separates based on size and charge and detect the different colored fluorophores.
This image is an example of an electropherogram that would be obtained from modern Sanger sequencing.
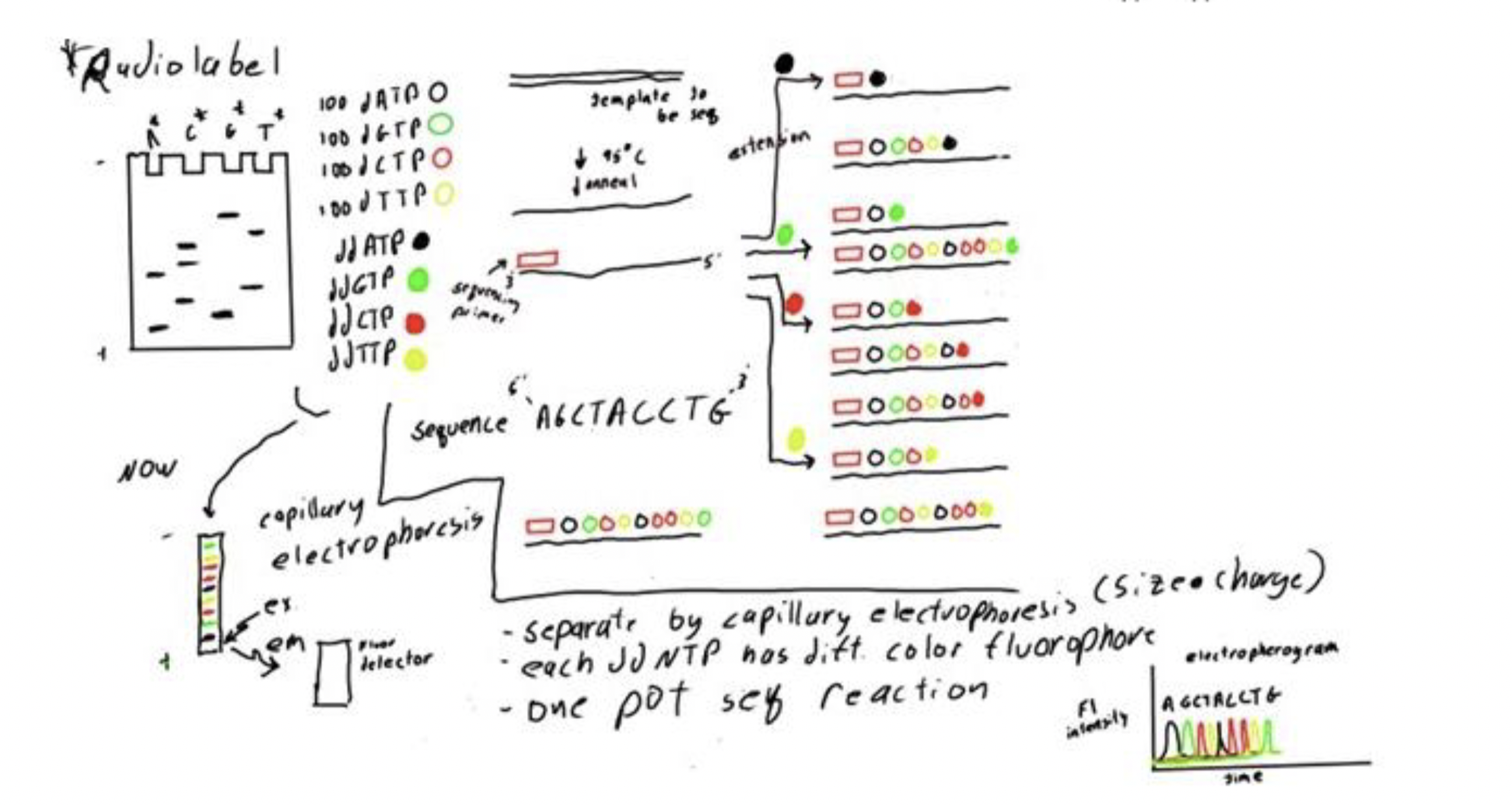
Is Sanger sequencing used for short sequences (1000-2000 base pairs) or for sequencing of entire genomes and other more complex applications?
What is used for the other?
used for short sequences (1000-2000) base pairs.
pyrosequencing and reversible terminator sequencing are used for sequencing of entire genomes and other more complex applications. These techniques are most relevant in bioinformatics.