NEURO - developmental and Hereditary Disorders
1/38
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
39 Terms
A newborn infant has a cystic swelling at the base of the spine that is covered with hyperpigmented skin and some coarse hair. Which of the following
is the most likely explanation?
a. Mongolian spot
b. Spina bifida occulta
c. Nevus flammeus
d. Meningocele
e. Encephalocele
MENINGOCELE i.e. evagination of the meninges (dura mater and pia arachnoid) around the cord or cauda equina through the defect in the spine. If meningeal and neural elements extrude together this is called a meningomyelocele. An encephalocele is a defect in the skull with extrusion of brain.
At age 5, a child is noted to have the loss of ankle jerks. At age 10, limb ataxia develops, followed by a peripheral neuropathy. During adolescence,
retinitis pigmentosa develops. Acanthocytosis is present. These are all characteristic of which of the following?
a. Multiple sclerosis (MS)
b. Sickle cell disease
c. Abetalipoproteinemia
d. Progressive multifocal leukoencephalopathy (PML)
e. HIV subacute encephalomyelitis
ABETA LIPOPROTEINEMIA (BASSEN-KORNWEIG SYNDROME).
Peripheral blood smear will show abnormally shaped RBCs (acanthrocytes - spiked or crenated) and plasma lipid profile will show low cholesterol and TGs. Autopsy shows posterior column and spinocerebellar tract degeneration.
Initial complaints = lost position sense. Deficits accumulate over the course of ears.
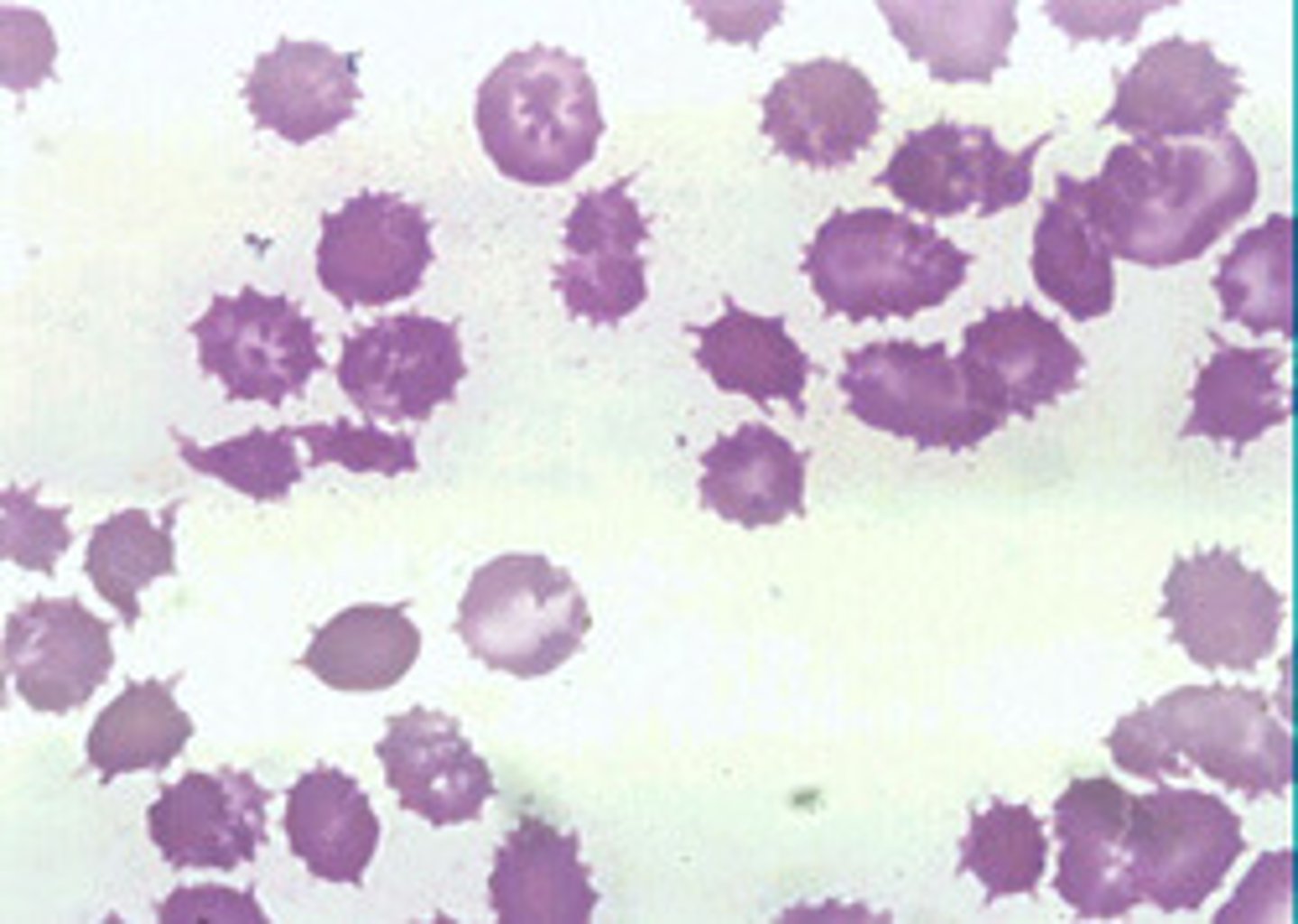
What supplement is helpful in ABETA LIPOPROTEINEMIA?
Vitamin E
What is retinitis pigmentosa? DDX?
Inherited degenerative eye disease (retinal dystrophy). Caused by abnormalities of the photoreceptors (rods and cones) or the retinal pigment epithelium (RPE) of the retina leading to progressive sight loss. Affected individuals may experience defective light to dark, dark to light adaptation or nyctalopia (night blindness), as the result of the degeneration of the peripheral visual field (known as tunnel vision). Sometimes, central vision is lost first causing the person to look sidelong at objects.
DDX = abetalipoproteineima, mitochondrial diseases, Bardet-Biedl syndrome, Laurence-Moon syndrome, Friedich's ataxia, Refum's disease.
In abetalipoproteinemia, VLDL, LDL largely absent in serum as a consequence of mutation of which gene?
a. Microsomal triglyceride transfer protein (MTP)
b. Huntingtin
c. Amyloid precursor protein
d. Dystrophin
e. Transfer RNA (tRNA)
MICROSOMAL TRIGLYCERIDE TRANSFER PROTEIN
Results in impaired VLDL formation and consequent decreased vitamin E delivery to the CNS and PNS. Patients also have disturbed fat absorption. It is the disturbed lipid that deforms the RBC cell wall (RBC production are normal). Fat is increased in the liver and patients exhibit lactose intolerence.
An assimilation of the atlas to the base of the skull (fusion) is a congenital abnormality associated with ...
Chiari malformation of the hindbrain.
A 5-year-old boy has mental retardation, homonymous hemianopsia, and hemiparesis. He had infantile spasm and still has epilepsy. Head CT
reveals calcifications in the cerebral cortex in a railroad track pattern. Which of the following does this child most likely have?
a. Glioblastoma multiforme
b. Oligodendroglioma
c. Acoustic schwannoma
d. Craniopharyngioma
e. Sturge-Weber syndrome
STURGE-WEBER SYNDROME
All of these disturbances will produce intracranial calcifications in some cases. The calcifications in
Sturge-Weber syndrome follow the gyral pattern of the cerebral cortex and consequently produce the railroad track pattern that is evident on plain x-ray of the skull. Calcium is deposited in the brain of the patient with Sturge-Weber syndrome, presumably because the abnormal vessels overlying the brain allow calcium, as well as iron, across the defective bloodbrain barrier. Craniopharyngioma and acoustic schwannoma produce calcifications, but these are obviously outside the cerebral cortex.
A 35-year-old woman has prenatal testing done which reveals that her child will have phenylketonuria (PKU). With PKU, serum may
exhibit dangerously high levels of
a. Creatine phosphokinase (CPK)
b. Nicotinamide
c. Phenylketone
d. Lactate dehydrogenase
e. Phenylalanine
PHENYLALANINE
Phenylketonuria is inherited as an autosomal recessive trait. It occurs in at least two forms. In one form, intolerance of phenylalanine is extreme, and dietary intake of that
amino acid must be restricted from birth. Alternatively, some persons have hyperphenylalaninemia without PKU. This latter group does not suffer the CNS damage seen with in utero exposure to high phenylalanine levels.
Such in utero exposure will occur if the mother is homozygous for PKU. If the mother is normal, infants with PKU are born with essentially normal nervous systems. Damage develops after birth in the susceptible group as
serum phenylalanine levels rise.
A 4 yo girl develops an intermittent red, scaly rash over her face, neck, hands, and legs. This is followed by developmental delay, emotional lability, and episodic cerebellar ataxia. She is diagnosed with Hartnup's disease. Her condition may respond to large supplementary doses of
a. Vitamin C
b. Nicotinamide
c. Thiamine
d. Pyridoxine
e. α tocopherol
NICOTINAMIDE
With Hartnup's disease there is intestinal malabsorption of tryptophan and other neutral amino acids. Tryptophan serves as a precursor for nicotinamide, but with more
than 400 mg of nicotinamide daily, the tryptophan malabsorption becomes less problematic. Inheritance appears to be autosomal recessive. Affected
children develop a scaly erythematous rash on the face similar to that seen with pellagra. The ataxia exhibited may be episodic.
Hepatosplenomegaly is most likely with
a. Tay-Sachs disease
b. Niemann-Pick disease
c. Alpers' disease
d. Subacute necrotizing encephalopathy
e. Wilson's disease (hepatolenticular degeneration)
Niemann-Pick disease is inherited as an autosomal recessive trait. By 9 months of age, patients with the infantile form usually have prominent hepatosplenomegaly. A deficiency
of sphingomyelinase in hepatocytes is diagnostic for the disease.
A 25-year-old woman with epilepsy is taking divalproex sodium during the first trimester of pregnancy. She is at slightly increased risk of having children with which of the following?
a. Holoprosencephaly
b. Defects of neural tube closure
c. Medulloblastoma
d. Agenesis of the corpus callosum
e. Kallmann syndrome
DEFECTS OF NEURAL TUBE CLOSURE
To what extent the antiepileptic divalproex sodium increases the risk of defects of neural tube closure, such as meningomyelocele, is debatable, but there is at least a
slight increase in the risk. The vitamin most clearly implicated in cases involving hypervitaminosis is vitamin A. Congenital malformations as a group are slightly increased in the offspring of women with epilepsy even if they are not taking antiepileptic drugs before or during pregnancy. The importance of folate supplementation in women with a
prior history of neural tube defect has been shown in several studies.
Agenesis of the corpus callosum
Chiari syndrome
Aicardi syndrome - agenesis of the corpus callosum is associated with retardation, epilepsy, vertebral anomalies, and chorioretinitis
With agenesis of the corpus callosum, MRI will reveal
a. Atrophy of the frontal lobes
b. Abnormally shaped lateral and third ventricles
c. Cerebellar aplasia
d. Schizencephaly
e. Encephaloclastic porencephaly
ABN SHAPED LATERAL AND THIRD VENTRICLES
On coronal sections of the brain, the lateral ventricles will have a typical batwing conformation if the patient has agenesis of the corpus callosum. The third ventricle may be
dilated and may open onto the surface of the brain. Patients with this congenital anomaly may be asymptomatic or may exhibit a variety of cognitive disorders.
A boy has the onset of difficulty walking at 16 mo. Reflexes are decreased. Over the course of several months, the patient becomes dysarthric and mental functioning decreases. Testing reveals that the patient has a deficiency of arylsulfatase A.
Which of the following does this patient most likely have?
How is the diagnosis made?
METACHROMATIC LEUKODYSTROPHY
Auto recessive enzymatic defect causing retardation, ataxia, spasticity, and sensory disturbances, but individual elements of this disorder may appear alone in less serious cases. The disease is usually symptomatic during infancy.
NERVE BIOPSY
Sulfatide granules may be evident in nerve tissue, as well as in tissue outside the nervous system, in persons with metachromatic leukodystrophy. The disease is usually fatal
within a few years of obvious symptoms. At autopsy, there may be evidence of dysmyelination or demyelination in the CNS, as well as in the peripheral nervous system.
A 15-year-old boy has moderate mental retardation, ADD, a long face, enlarged ears, and macroorchidism. Development has been steady but always at a delayed pace. The most likely cause for this patient's low intelligence is which of the following?
a. Turner syndrome
b. Klinefelter syndrome
c. Fragile X syndrome
d. Reye syndrome
e. Tuberous sclerosis
FRAGILE X
With the fragile X syndrome, the terminal elements of the long arm of the abnormal X chromosome appear stretched or broken away from the rest of the chromosome. Retardation usually becomes evident during childhood. Affected men have large ears, a high-arched palate, hypotelorism, and large testes. Autism also occurs
among affected men.
Women carrying chromosomes for Fragile X
a. Are invariably normal
b. Have mild retardation in about one-half of cases
c. Have high-arched palates and hypotelorism
d. Have hyperextensible joints
e. Have prominent thumbs
Mild retardation in 1/2 cases
Men with the fragile X syndrome have hyperextensible joints and prominent thumbs, but carrier women may appear quite normal. The abnormal chromosome may be detected in
fetal lymphocytes and fibroblasts, thereby allowing for prenatal screening. Epilepsy develops in many affected persons, but the seizures are usually easily controlled, unlike the case with other hereditary causes of epilepsy.
A 35-year-old man complains of stumbling and slurred speech. His problem started several mo ago and has progressed slowly but consistently.
On neurologic examination, he is found to have scanning speech, nystagmus, limb dysmetria, and kinetic tremor. His intellectual function is
normal.
The most appropriate initial investigation is
a. Lumbar puncture
b. Serum drug screen
c. Routine urinalysis
d. Posterior fossa myelogram
e. Precontrast CT scan
PRECONTRAST CT SCAN
This man has signs of cerebellar dysfunction. That the deficit has been slowly progressive and is not associated with cognitive dysfunction makes it especially likely that a
structural lesion in the posterior fossa is responsible for the deficit. Because the lesion need not disturb the external shape of the cerebellum, a posterior fossa myelogram will not necessarily yield an answer. The CT scan will show if there is an intraparenchymal or extraparenchymal lesion.
Admission studies include a hematocrit of 55% and a routine UA w/ excess protein and some RBCs. The initial physical examination reveals an enlarged liver and spleen. Additional physical findings will most likely include
a. A Kayser-Fleischer ring around the cornea
b. Hypopigmented (ash leaf) spots on the trunk
c. Telangiectasias in the fundi on retinal examination
d. Bilateral hearing loss
e. Generalized hyporeflexia
TELANGIECTASIS IN FUNDI ON RETINAL EXAM
Erythrocytosis with cerebellar signs, microscopic hematuria, and hepatosplenomegaly suggests von Hippel-Lindau syndrome. This auto dom disorder is characterized by polycystic liver disease, polycystic kidney disease, retinal angiomas (telangiectasia), and cerebellar tumors. Other abnormalities that occur with this syndrome include adenomas in many organs. Hemangiomas may be evident in the bones, adrenals, and ovaries. Hemangioblastomas may develop in the spinal cord or brainstem, as well as in the cerebellum.
A postcontrast CT scan reveals a cyst and two smaller masses in the left cerebellar hemisphere. Your recommendation?
SURGICAL RESECTION
The cystic lesion and the other cerebellar lesions are most likely hemangioblastomas. They often bleed and produce potentially lethal intracranial hematomas. Radiation therapy and needle biopsies would increase the risk of bleeding. Rather than spontaneously involuting, these lesions generally enlarge and become more unstable as time passes. Intracerebellar hemorrhage is increasingly likely as time passes.
Within 6 years of his initial visit, the patient returns with a pathologic fracture of his spine. Biopsy reveals metastatic cancer. The source of the tumor is most likely the
a. Cerebral hemisphere
b. Cerebellar hemisphere
c. Liver
d. Kidney
e. Spleen
KIDNEY
Von Hippel-Lindau syndrome is associated with a high incidence of renal carcinomas. These malignant renal tumors usually develop years after the cerebellar hemangioblastomas, liver disease, or polycystic renal disease becomes symptomatic. People surviving intracranial hemorrhages caused by the intracerebellar hemangioblastomas often succumb to metastatic renal carcinoma. Treating the intracranial lesions does nothing to reduce the risk of metastatic renal cancer.
An infant has a head CT performed because of a large head and failure to thrive. The diagnosis of hydrocephalus is made. Congenital hydrocephalus may develop as a consequence of which
first-trimester maternal disorder?
a. Complicated migraine
b. Viral infection
c. Pseudotumor cerebri
d. Chorea gravidarum
e. Intervertebral disk herniation
VIRAL INFECTION
A maternal infection with mumps or rubella virus may produce aqueductal stenosis and, as a consequence, hydrocephalus. The aqueduct of Sylvius connects the third ventricle to the fourth ventricle. The lateral and third
ventricles enlarge as the choroid plexus produces fluid that cannot migrate to the subarachnoid space to be reabsorbed.
Uncorrected congenital hydrocephalus will usually produce which
of the following?
a. Dolichocephaly
b. Brachycephaly
c. Holoprosencephaly
d. Macrocephaly
e. Microcephaly
Macrocephaly
Congenital hydrocephalus usually requires shunting to avoid progressive enlargement of the head and thinning of the brain mantle.
A 6-month-old child is noted to have head lag, tongue fasciculations, and bilateral abducens palsies. MRI scan reveals a type 2 Chiari malformation. Which of the following defects would this child be likely to have?
a. A renal cyst
b. Pulmonary atelectasis
c. Spina bifida
d. Holoprosencephaly
e. A hepatic cyst
SPINA BIFIDA
Spina bifida may be extreme in some of the children affected by the Arnold-Chiari (type 2 Chiari) malformation. A myelomeningocele may be present at the level of the spina bifida. Spinal cord tissues may extend into this mass and lie just under the skin covering the neural tube defect. Children with obvious spinal defects usually have persistent problems with leg movements and bladder and bowel control.
A 7-year-old boy is taken by his parents to see a dermatologist. They have noticed nodules on his face and are concerned. The dermatologist
tells them that their child has adenoma sebaceum.
Adenoma sebaceum of the face is especially common with which disease?
TUBERCULOUS SCLEROSIS
Adenoma sebaceum occurs in about 90% of patients with tuberous sclerosis. A depigmented lesion, called a shagreen patch, occurs in only about 20% of these patients. Adenoma
sebaceum usually becomes apparent over the malar eminences of the face between 2 and 5 years of age and may evolve into difficult-to-treat angiofibromas of the skin.
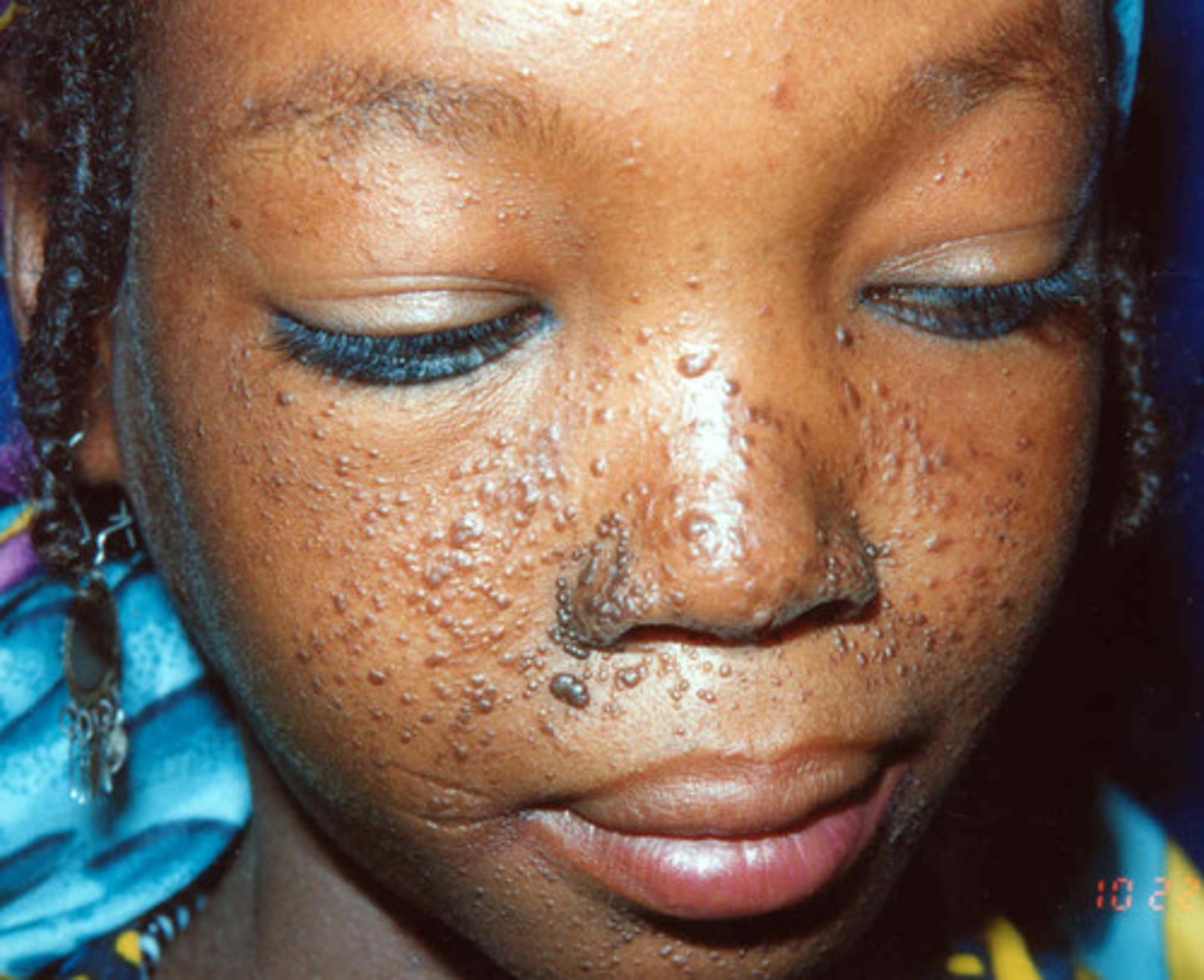
Tuberous sclerosis is inherited in
a. A sex-linked recessive pattern
b. An autosomal dominant pattern
c. An autosomal recessive pattern
d. A pattern most consistent with newly arising mutations
e. A pattern suggesting a mitochondrial gene defect
AUTO DOM
Although the inheritance pattern of tuberous sclerosis is autosomal dominant, the penetrance is variable.
A severely impaired child may be born to a negligibly affected parent. Despite the consensus that inheritance is autosomal dominant, estimates of spontaneous mutations in affected persons are as high as 70%.
Retinal problems with this tuberous sclerosis:
a. Include retinal phakomas
b. Include retinitis pigmentosa
c. Include retinal telangiectasias
d. Include retinoblastomas
e. Are generally not part of the disease
PHKOMAS
Retinal phakomas, which require no treatment, are a principal criterion for making the diagnosis of tuberous
sclerosis. Along with adenoma sebaceum and periventricular tubers, they are virtually pathognomonic. Other findings that are typical of tuberous sclerosis include ash leaf spots, shagreen patches, CNS calcifications, renal tumors, cardiac rhabdomyomas, and seizure disorders.
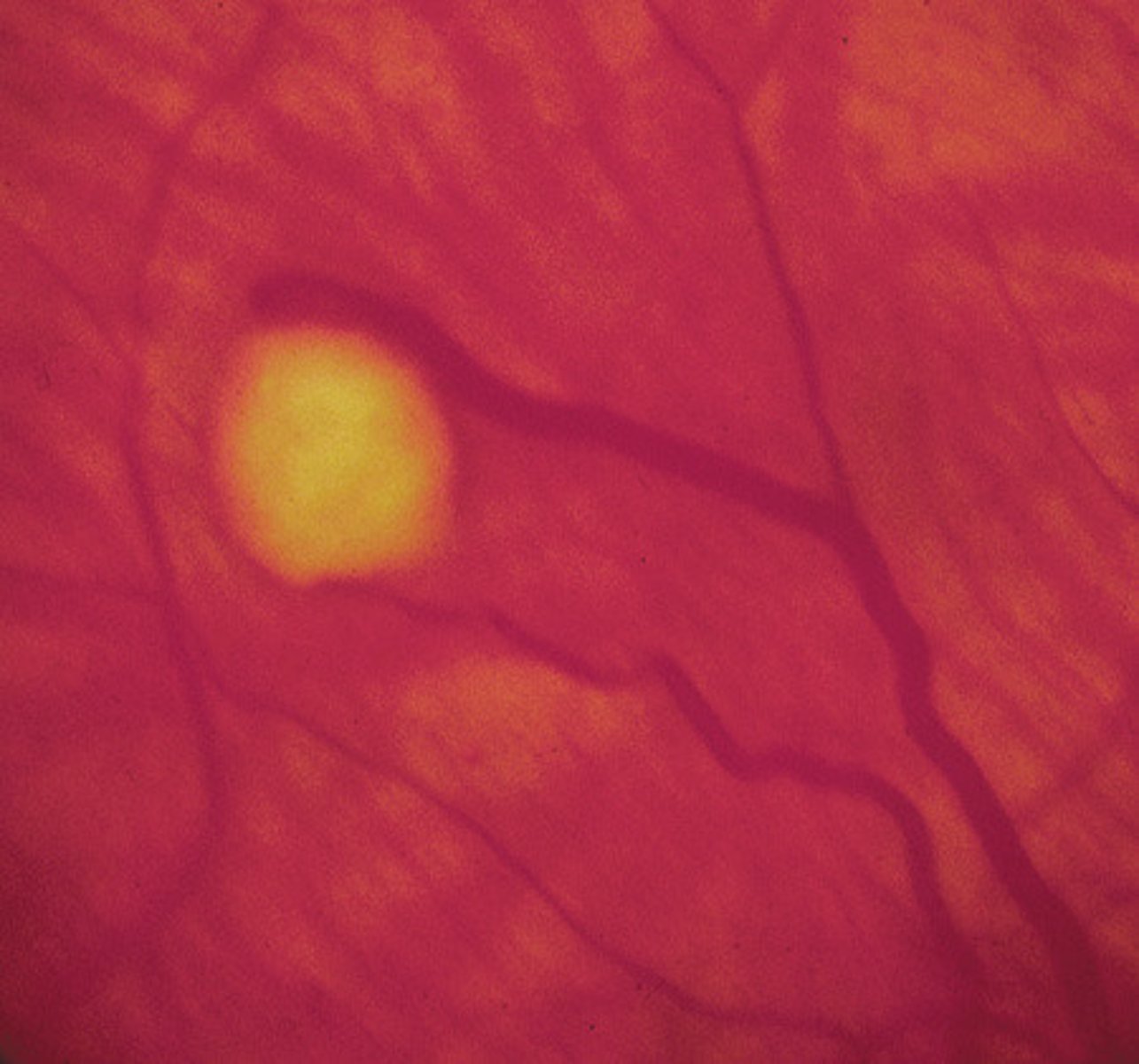
Calcifications evident on the skull x-ray or CT scan of a patient with this disease usually represent
a. Calcified subependymal glial nodules
b. Calcified meningeal adhesions
c. Meningeal psammoma bodies
d. Calcified astrocytomas
e. Calcified granulomas
CALCIFIED SUBEPENDYMAL GLIAL NODULES
By 5 years of age, more than half of patients with tuberous sclerosis will have subependymal glial nodules that have calcified. These nodules usually do not become malignant,
but they may enlarge sufficiently to produce an obstructive hydrocephalus.
A 50-year-old man complaining of dizziness is found to have a cyst occupying 50% of his posterior fossa and incomplete fusion of the cerebellar
elements inferiorly. There is no evidence of an obstructive hydrocephalus. His longevity can be estimated to be
a. Less than 3 months
b. Less than 1 year
c. Less than 5 years
d. Less than 10 years
e. Unaffected by this finding
UNAFFECTED BY THIS FINDING
That the cerebellar elements are not fused in the midline suggests an asymptomatic Dandy-Walker malformation. This congenital disorder of brain formation may become symptomatic soon after birth if an obstructive hydrocephalus develops as one facet of the anomaly. In the absence of an obstructive hydrocephalus, the patient may remain asymptomatic throughout life.
The treatment of choice for children with infantile spasms as part of tuberous sclerosis is
a. Carbamazepine (Tegretol)
b. Phenobarbital
c. Phenytoin (Dilantin)
d. Divalproex sodium (Depakote)
e. Adrenocorticotropic hormone (ACTH)
ADRENOCORTICOTROPIC HORMONE (ACTH)
Usually given as a gel intramuscularly to control infantile spasms in children with tuberous sclerosis; 40 to 80 mg is divided into two doses. Treatment continues until the infantile spasms abate or the EEG pattern of hypsarrhythmia resolves. This usually requires 6 to 8 weeks of treatment.
The ACTH should not be stopped abruptly.
A 9-year-old boy has been generally healthy. However, his parents are concerned that his many areas of hyperpigmented skin may have some significance. They have been told that these are café au lait spots. Café au lait spots are commonly found on patients with
a. Tuberous sclerosis
b. Neurofibromatosis
c. MS
d. Sturge-Weber syndrome
e. Ataxia telangiectasia
Café au lait spots in patients with neurofibromatosis are usually larger than a few centimeters and occur in several locations in individual patients. Some have ragged
edges and are called coast of Maine spots. They occur with both type 1 and type 2 neurofibromatosis, but are much more common with type 1.
The newborn infant with motor neuron disease is likely to exhibit
a. Seizures
b. Hypotonia
c. Hypsarrhythmia
d. Moro reflexes
e. Spina bifida
HYPOTONIA
The child with congenital weakness, hypotonia, and muscle atrophy may have Werdnig- Hoffmann disease, a congenital motor neuron disease. This is an especially lethal form of motor neuron disease and may limit the child's life expectancy to weeks or months. A similar pattern of disease that appears in older children is less lethal and is called Kugelberg-Welander disease. These types of motor neuron diseases are also known as spinal muscular atrophies (SMAs). Anterior horn cell disease is presumed to be a pivotal feature.
Many children with Tay-Sachs disease develop blindness before they die, with retinal accumulation of gangliosides that produces
CHERRY RED SPOT
More than 90% of children with Tay-Sachs disease develop cherry red spots on the retina. The red spot at the fovea develops as retinal ganglion cells become distended with
glycolipid. There are no ganglion cell bodies overlying the fovea, and so the red color of the vascular choroid is apparent in this region but obscured by more opaque glycolipid-engorged cells over the remainder
of the retina.
The parents of a 10-year-old boy bring their child in to see you. The child has been diagnosed with cerebral palsy, and the parents do not really
understand what this means. As part of your explanation, you tell them that cerebral palsy is a static encephalopathy because
a. Deficits do not appear after birth
b. The injury to the brain does not progress
c. Affected persons fail to reach any developmental milestones on time
d. Affected persons have resting tremors
e. The EEG exhibits a disorganized background rhythm
THE INJURY TO THE BRAIN DOES NOT PROGRESS
A static encephalopathy is one in which brain damage has been arrested but neurologic problems persist. Establishing that the brain lesion is not progressive may require extensive testing. A young child with a static motor disorder is said to have CP. Neurodegenerative diseases with slow or stepwise progressions may appear to be static encephalopathies over the course of months, but prove to be progressive encephalopathies over the course of years. The brain lesion with CP is static, but the deficits associated with CP may evolve as the child matures.
A 6-year-old child is brought to the neurologist because of developmental delay. Her morphological features are typical and chromosome analysis confirms a diagnosis of Down syndrome (trisomy
The brain of this patient is expected to be
a. Smaller than normal for age and body size
b. Larger than normal for age and body size
c. Abnormally long in anteroposterior measurements
d. Hydrocephalic
e. Excessively convoluted
SMLLER THAN NORMAL FOR AGE/BODY SIZE
The brain of the patient with Down syndrome (trisomy 21) is typically foreshortened. The gyral pattern is simplified, and the frontal lobes are small. The occipital lobes may be
slanted, and the overall shape of the skull is abnormal.
Porencephaly usually develops as a consequence of
a. Fetal alcohol syndrome
b. Vascular or other destructive injuries to the fetal brain
c. Trisomy 13
d. Trisomy 21
e. Dandy-Walker syndrome
VASCULAR OR OTHER DESTRUCTIVE INJURIES TO THE FETAL BRAIN
In utero damage to the fetal brain may be evident at birth as large cysts in the brain. The presence of one or more of these intracerebral cysts is called porencephaly. Some
pathologists believe that schizencephaly, a related abnormality in which brain segmentation is abnormal, is caused by similar phenomena, which include incidents such as strokes and viral encephalitides in the fetal brain.
What percentage of patients with tuberous sclerosis have mental retardation?
a. 1
b. 10
c. 25
d. 65
e. 99
Of the 65% of patients with tuberous sclerosis who are retarded, half are severely retarded. *Seizures are
invariably associated with retardation*. About 20% of patients with tuberous sclerosis develop the Lennox-Gastaut syndrome with persistent seizures and significant mental retardation. These children usually have a mixed seizure disorder, whereas those without Lennox-Gastaut syndrome
most often have complex partial seizures.
A child is born to a 19-year-old woman who has had two to eight drinks per day throughout her pregnancy. What is the major pathologic
effect of alcohol on the central nervous system of the developing fetus?
a. Cerebral ischemia
b. Periventricular hemorrhage
c. Macrocephaly
d. Impaired neuronal migration
e. Holoprosencephaly
IMPAIRED NEURONAL MIGRATION
Although the mechanism of alcohol's effect on the developing brain is not entirely clear, it appears that alcohol acts primarily to impair neuronal migration. This may result in formation of heterotopias (collections of cortical neurons in abnormal locations), cortical disorganization, and malformations of the cerebellum and brainstem. Mental retardation, learning disabilities, hyperactivity, and microcephaly, not macrocephaly, are the common clinical neurologic consequences of fetal alcohol syndrome.
A 37-year-old man has an MRI performed by his primary care doctor because of a long history of headaches. It is notable only for the finding
of a type 1 Chiari malformation. He is sent to a neurologist for further evaluation.
A type 1 Chiari malformation usually becomes symptomatic as which of the following in adults?
a. Epilepsy
b. Hydrocephalus
c. Ataxia
d. Dementia
e. Psychosis
ATAXIA
Both type 1 and type 2 Chiari malformations are primarily abnormalities of hindbrain development. With
the type 1, or adult, abnormality, the cerebellar tonsils extend below the foramen magnum. Affected persons do not usually become symptomatic until they are adults, and then the symptoms are largely referable to the cerebellum. With the type 2 malformation, cerebellar anatomy is usually
much more deranged, and the cerebellar vermis lies well below the foramen magnum. Type 2 malformations most often become symptomatic at birth or during infancy and may produce hydrocephalus with retardation.
A 25-year-old mother develops an illness during pregnancy. A diagnosis of cytomegalovirus (CMV) infection is made by serology. Prenatal
CMV infections may produce which retinal disturbance?
a. Chorioretinitis
b. Cherry red spot
c. Microaneurysms
d. Hypervascularity
e. Hemorrhage
CHORIORETINITIS
Microaneurysms and hypervascularity are typically seen with diabetic retinopathy rather than developmental disease. Hemorrhages in the retina would be more typical of
hypertensive encephalopathy or a coagulopathy. Neurologic problems that develop in the infant with a prenatal CMV infection include retardation, microcephaly, seizures, and hearing deficits. The virus often causes chorioretinitis,
optic atrophy, and architectural changes throughout the brain.