L6: Characterizing genetic diversity
1/35
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
36 Terms
how to identify genes of interest?
causal genes are gene that is responsible for variation in a phenotypic trait or a disease of interest
at meiosis, crossing overs swap - between - chromosomes, without recombination, all positions in a chromosome would be in -
recombination breaks the linkage between - in a chromosome
recombination mixes up - and creates new - combinations
regions. homologous, linkage
genomic positions
genetic variants, allelic
crossing over generates new combinations of the -
regions common to all progeny with the - phenotype will contain the -
parental chromosomes
mutant, causal gene
qualitative traits
discrete classes
can be categorical
quantitative traits
continuous variation
no discrete classes
measurable
qualitative traits are typically -
meaning what?
monogenic
gene at one locus controls trait
quantitative traits are typically -
meaning what
polygenic
combined effects of many loci
to map the phenotypic effects of a qualitative trait, track segregation of only -
one gene/locus
when mapping quantitative variation, track segregation of -
QTL mapping
several genes/loci
QTL mapping
each genetic region/variant is tested for associations with the phenotypic trait of interest
the likelihood of that marker/variant being associated with a gene controlling that trait is plotted across the genome
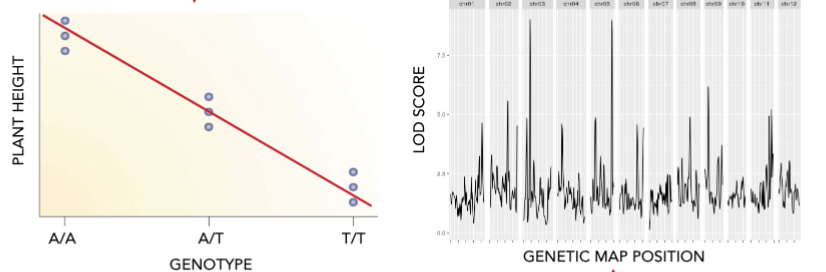
what is needed for QTL mapping?
segregating population
a group of related individuals obtained by crossing two or more parents, which shows variation for a trait of interest
genotype information
genetic markers differentiating the parental genotypes across the whole genome
phenotype information
measurements of trait of interest, as accurate and quantitative as possible
genetic mapping relies on
recombination breaking the association between alleles in nearby regions of the genome
the more recombination events you have, the higher the likelihood to break linkage between nearby genes
can narrow the candidate region further, meaning higher mapping resolution
therefore fewer possible candidate genes to choose from
how many recombination events?
on average, only 1-2 recombination per chromosome per generation
limited number of recombinations in each individual
recombination rates increase with chromosome number, not genome size
F2 characteristics
2 parents
“quick” to produce
limited recombination
heterozygous regions
one-and-done
RIL
2 parents
require multiple generations
more recombination
homozygous individuals
immortalized lines
MAGIC populations
multi-parent advanced generation inter-cross
many parents = more alleles
more recombination
homozygous, immortalized lines
very slow/work-intensive to produce
reference genome allows
placing and comparing of sequence reads from different individualshow
how to compare genetic diversity with short reads?
for genotyping, throughput and cost of sequencing is more important than read length
2nd gen seq produces large amounts of short reads for cheap
short reads are great to identify genetic variants between individuals
ideally comparing them to a reference assembly
limited to
small variants like SNPs or indel
non-repetitive region
how is read QC done?
trimming
mapping to reference genome
removing duplicates, indel realignment, base recalibration
variant detection
variant filtering
trimming
to remove low-quality bases and/or adapters
if inserts are smaller than 150bp, adapters will be sequenced as well
mapping to reference genome + removing duplicates
reads from each individuals are associated to the corresponding region on the reference assembly
reads from repetitive sequences will map to multiple regions of the genome, which are then discarded
variant calling/detection
positions that are variable (polymorphisms) are identified and collected in a variant call format (VCF)
variant filtering
assessing and improving the quality of raw sequencing reads, using that information to filter out or refine low-confidence variant calls generated from those reads
whole genome sequencing WGS
provides very dense, genome-wide information on genetic differences between individuals
cheap
stumped by repetitive regions or variants larger than a few base pairs
WGS is not same as genome assembly
WGS = producing sequencing reads from the whole genome
genome assembly = reconstructing the sequence and organization of the whole genome
don’t need to know every single variant to assemble a genome
genome-wide patterns of differentiation
high levels of linkage
limited number of crossing overs in an F2 population, so we don’t need to know the genotype of the individuals at every single nucleotide
recombination vs marker density for genetic mapping
recombination is a limiting factor in determining QTL mapping accuracy
even perfect knowledge of genotypes would not pinpoint the causal gene
not sequencing the whole genome allows
save time and computational resources
save money
focus on regions that could be more interesting to you
reduced representation in regards to gene space
RNAseq
potentially all genes, no previous knowledge required
but affected by expression levels and RNA is delicate
hybridization seq
selected regions, gene space and less seq needed
but seq knowledge required and its an initial investment
reduced-representation in regards to genotype by sequencing
WGS sequences the whole genome
GBS sequences only a subset of genomic regions
restriction enzymes recognize and cleave short DNA patterns
short reads genotyping focuses on -
SNPs
small but mighty
missense/nonsense
affects splicing
disrupt/modify promoter sequence
disrupt/add miRNA target sites
how to study structural variants?
short reads are poor at detecting structural variants
long reads allows identification of larger structural variants
getting phenotypic information from genotypic information
environment needs to be controlled to make sure only genetic factors are identified
genotypic information is increasingly easy/cheap thanks to new sequencing technologies
phenotypic information needs to be as accurate as possible
molecular phenotypes
phenotypes don’t need to be visible
physiological states like temperature, transpiration, water content
chemical or metabolite levels
expression levels eQTL
methylation levels mQTL
repeating QTL experiments in different conditions can identify genes controlling responses to -
environmental stimuli
what determines the success of a mapping experiment?
power = ability to detect genotype-phenotype associations
more power = genes with smaller effects can be detected
size of population, more robust assct, more recomb
complexity of trait, highly polygenic traits require more power
heritability of trait, how much variation is due to genetics vs environment
phenotype accuracy, sloppy phenotyping will mess up everything
genotyping density, enough genotype information