Lecture 9: Ionotropic and G-coupled protein receptors
1/23
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
24 Terms
where are ionootropic receptors found
neuromuscular junctions and postsynaptic membranes
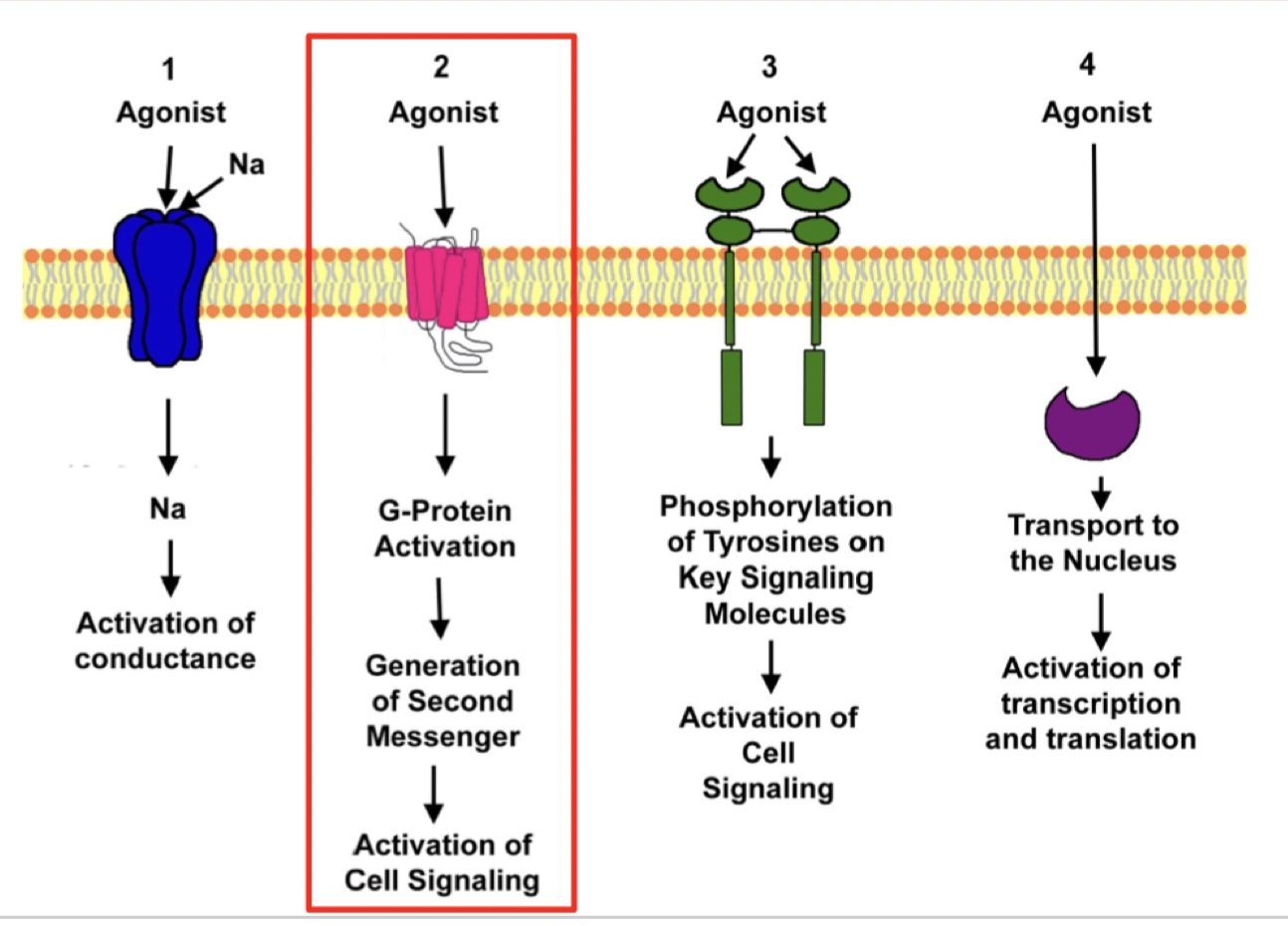
what are ionotropic receptors
ligand-gated ion channels that switch between closed and open states upon ligand binding
mediate rapid synaptic transmission by directly controlling the flow of ions across cell membranes in response to the binding of a ligand (ex. neurotransmitter)
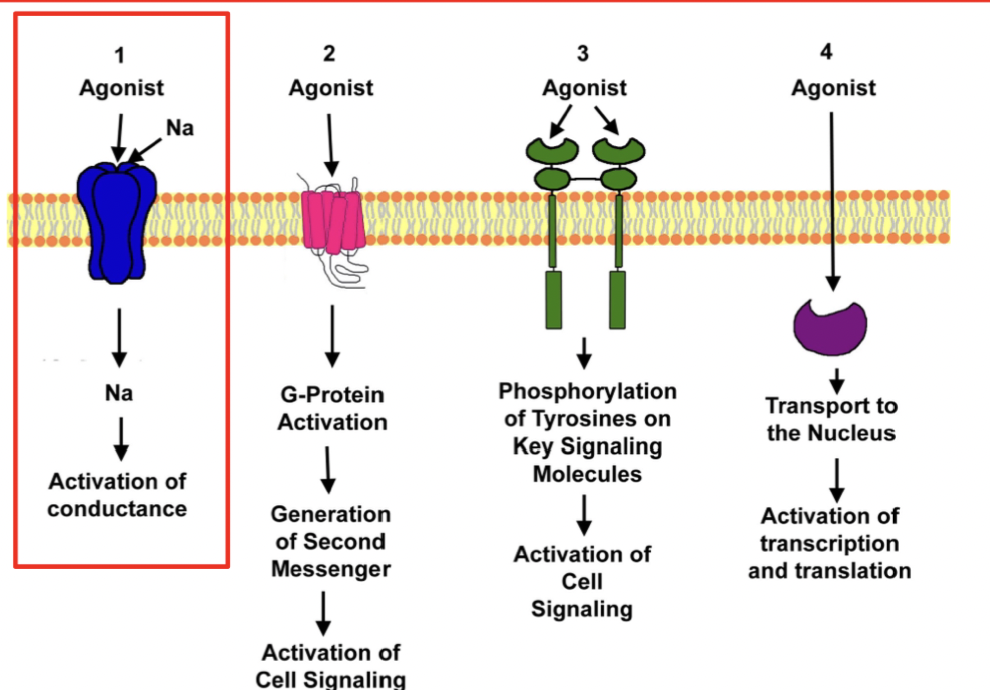
what is the mechanism of ionotropic receptor
The receptor (ex. acetylcholine receptor, GABA receptor, NMDA receptor) binds to a specific ligand
binding induces a conformational change, opening the pore.
Specific ions (ex. Na⁺, K⁺, Cl⁻, or Ca²⁺) flow down their electrochemical gradient, altering the membrane potential
*Channels are activated directly by bound ligand
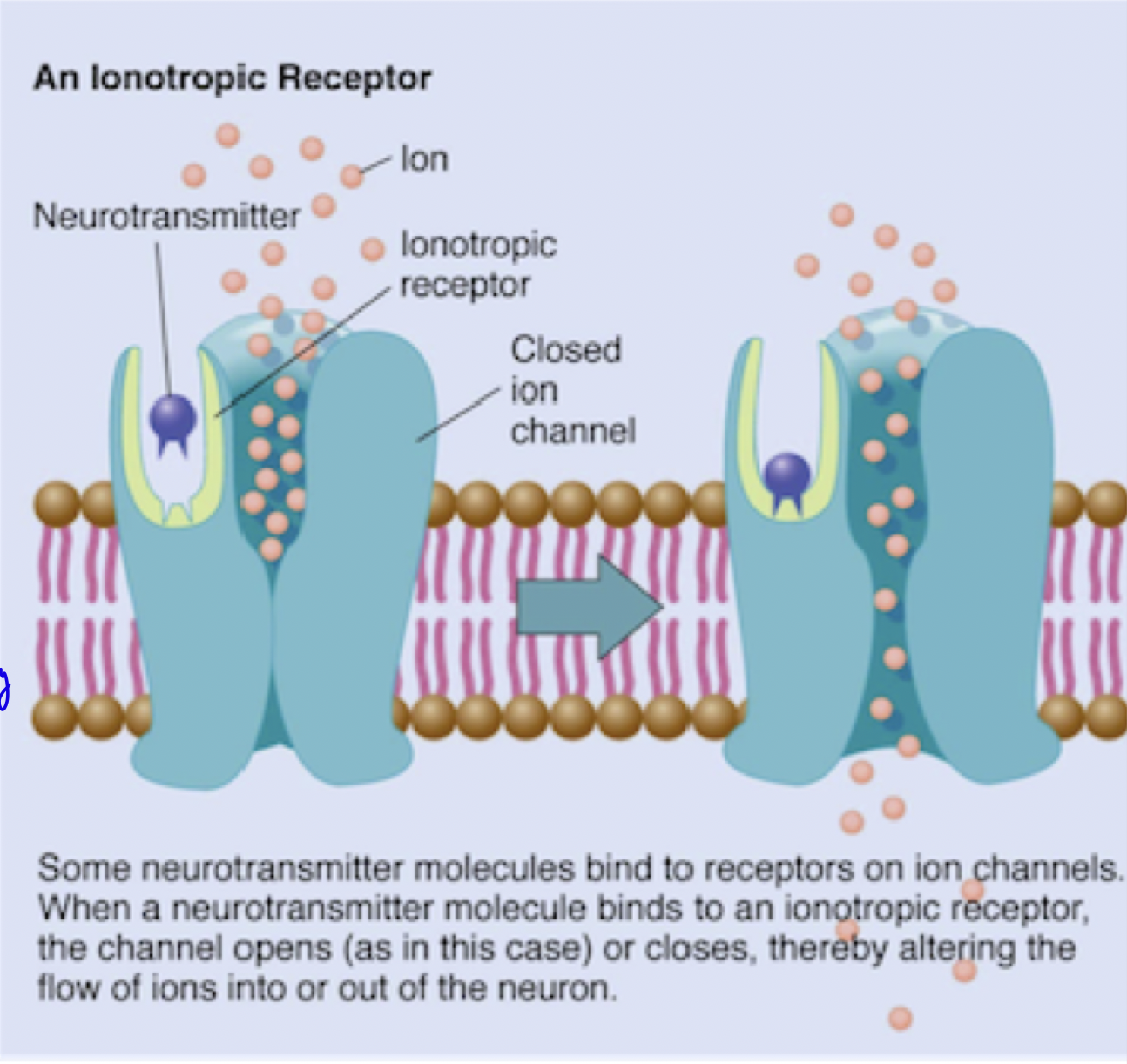
Membrane potential → Role of voltage dependent Na+ channels, and Ca2+ channels in muscle contraction?
resting inner nerve muscle membrane is -70mV → negative membrane potential
influx of positive ions (after ligand binding opens channel) depolarizes cell → this permits propagation of AP along axon.
AP activates (opens) intracellular Ca2+ channels, resulting in muscle contraction
eventually, the efflux of Na+ or influx of Ca2+ re-polarizes the membrane (back to resting negative membrane potential)
Therefore, Facilitate fast, direct signaling at synapses. Play a role in processes like excitation (e.g., dpolarization by Na⁺ influx) and inhibition (e.g., re-polarization by Cl⁻ influx).
how are Ca2+ channels classified
based on mechanisms of action and ligand, on physiological roles, and on inhibition by different toxins
different types found in different tissues
how does Ca2+ channels impact blood pressure normally?
Normally agonist binds, conformational change opening Ca2+ channel
Rush of Ca2+ which binds calmodulin
Downstream signalling: Activation myosin light chain kinase (MLCK), leads to increase smooth muscle contraction, reducing vasodilation, increase in blood pressure
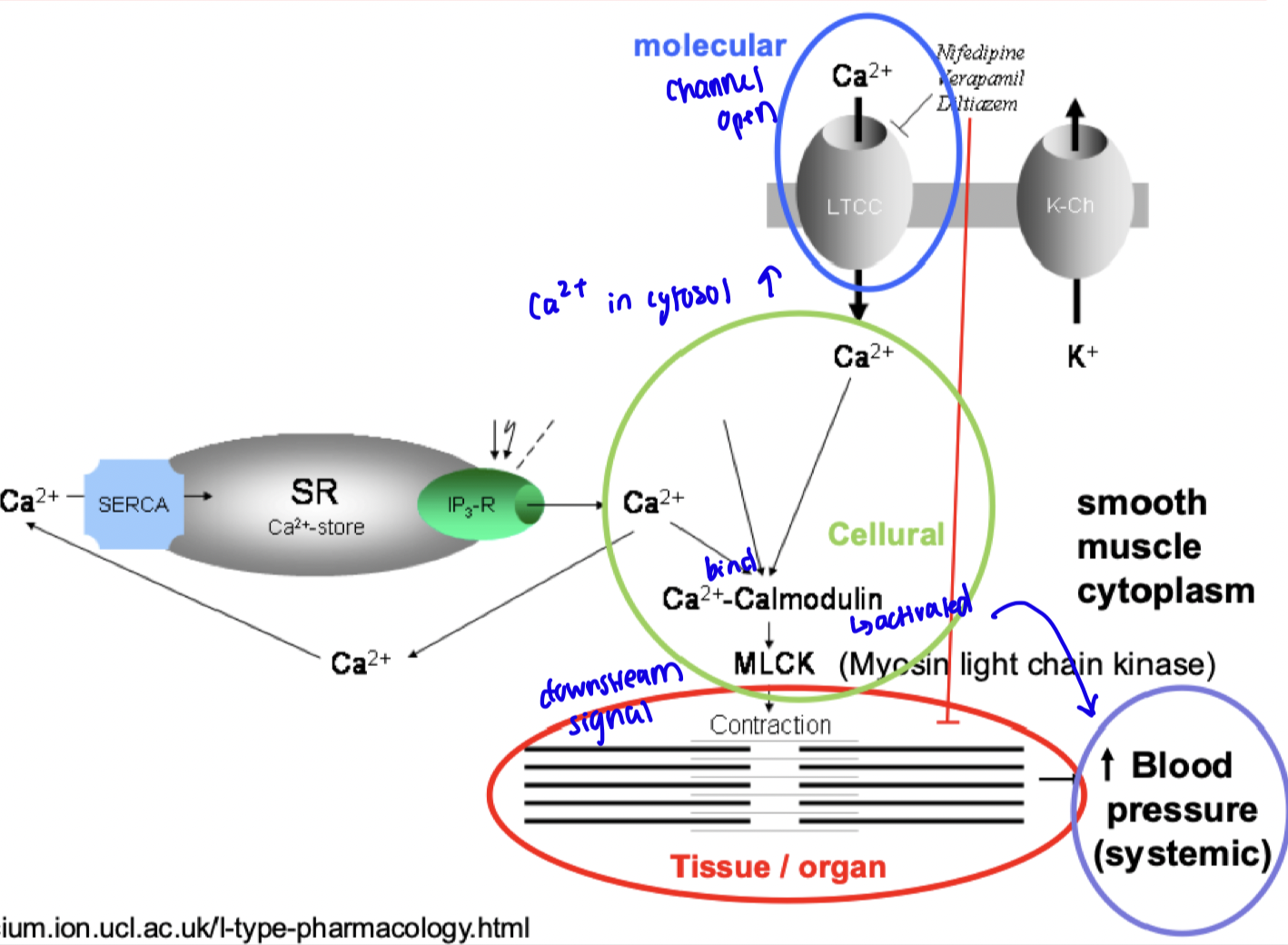
how is Ca2+ channel affected with a drug Amiodarone?
when drug antagonist binds and block receptor activation, intracellular calcium reduced.
Therefore decreases the activation of calcium-calmodulin and downstream pathways, ultimately reducing smooth muscle contraction favouring vasodilation and reducing blood pressure.
Impact of K+ channel with drug Amiodarone?
Drug blocks voltage-gated K+ channels, therefore no K+ going outside (more positive inside).
This slows re-polarization (more negative), therefore prolongation of the action potential duration (APD) and the effective refractory period (ERP).
**recall, it is the depolarization that propagates AP.
Impact: reduces the likelihood of reentrant arrhythmias by preventing premature excitations.
TRPV1 (transient receptor potential cation channel subfamily V member 1
Na+ and Ca2+ channel receptor
Activated (opens) by extra/intracellular ligand binding
When activated, TRPV1 allows the influx of Ca²⁺ and Na⁺ ions into cells
This depolarizes the neuron, triggering action potentials that signal to the central nervous system.
Detects and regulates body temp. Senses scalding heat, acid and pain. Therefore antagonists involved in pain relief!!
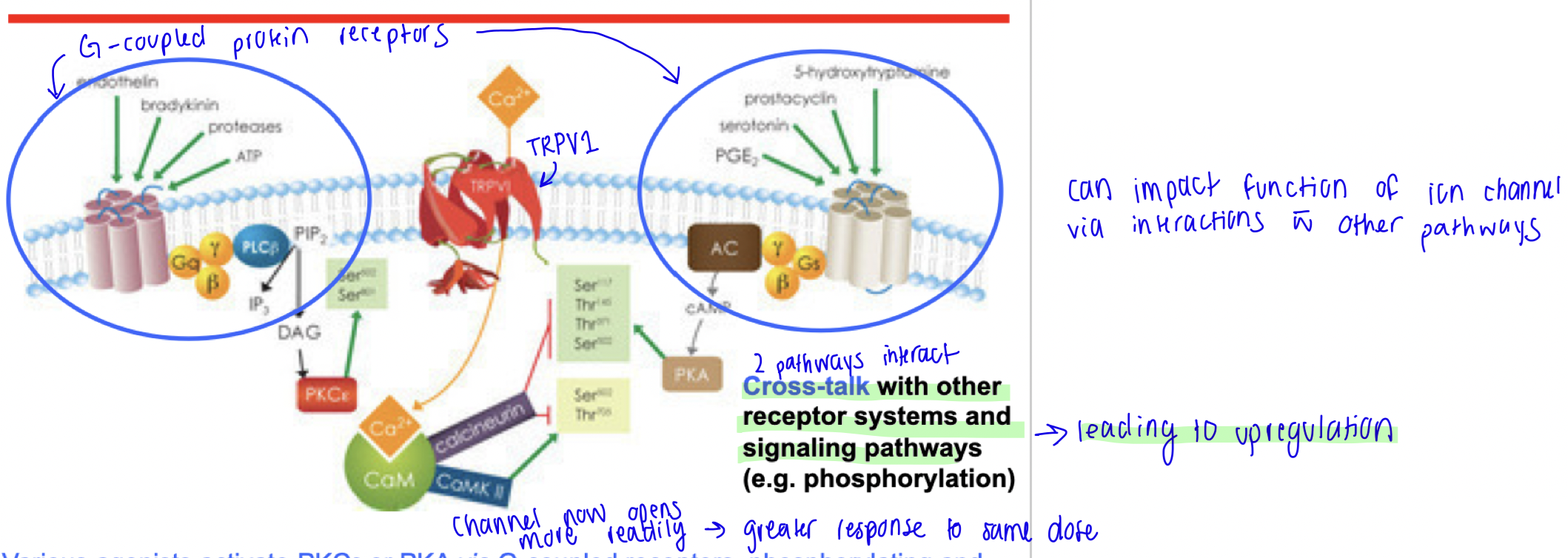
Cross talk
Interactions with different signalling pathways
Cross talk up-regulation
PHOSPHORYLATOIN - SENSITIZING
Upon activation, G-coupled protein receptors activate kinases which phosphorylates other proteins (including TRPV1)
This phosphorylates and sensitizes TRPV1 to acid, heat, or chemical agonists → channel now opens more regularly, resulting in greater response to the same dose.
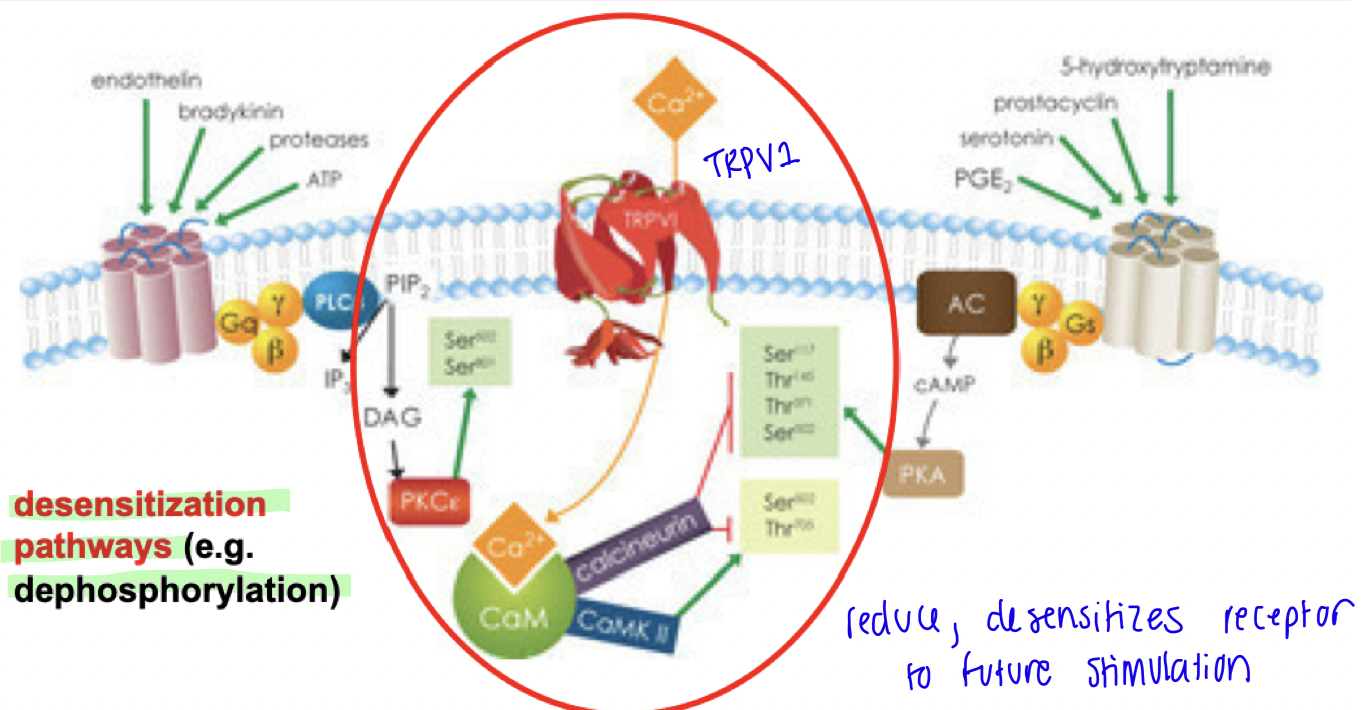
Cross talk down-regulation
DE-PHOSPHORYLATION - DESENSITIZES
If in presence of constant activation (ie. high dose / repetitive dose of agonists, blocking metabolism endogenous agonist), transient influx of Ca2+ which activates downstream target calmodulin, leads to phosphatase activity that de-phosphorylates and desensitizes TRPV1.
This down-regulates receptor sensitivity → next time agonist binds it will not create same response as before (decreased response to same dose)
how can xenobiotics mimic endogenous ligands?
Xenobiotics structurally resemble the endogenous ligand and bind to its receptor, activating the same signalling pathways
Agonists: xenobiotics that fully or partially activate the receptor, mimicking the endogenous ligand
how can xenobiotics block endogenous ligands?
ANTAGONISTS
Xenobiotics prevent the endogenous ligand from binding (competitive) or activating its receptor (non-competitive), inhibiting the natural signalling pathway
how can xenobiotics modify endogenous ligands?
Allosteric modulator - bind at a site other than the endogenous ligand's binding site to enhance (positive modulator) or reduce (negative modulator) the effect of the ligand
how do high efficacy agonists impact channel lifetime in ionotropic receptors?
activates large proportion of receptors at a time
β (opening constant) > α (closing constant)
favours open state
Prolonged ion flow and stronger activation of the receptor
how do low efficacy agonists impact channel lifetime in ionotropic receptors?
activates few receptors at at time
α (closing constant) > β (opening constant)
favours closed state
Weaker activation of the receptor compared to high-efficacy agonists
how do antagonists impact channel lifetime in ionotropic receptors?
Antagonists occupy receptor, conformatinal change, but do not activate
β (opening constant) = 0
therefore channel lifetime remains at a closed state → no ion flow
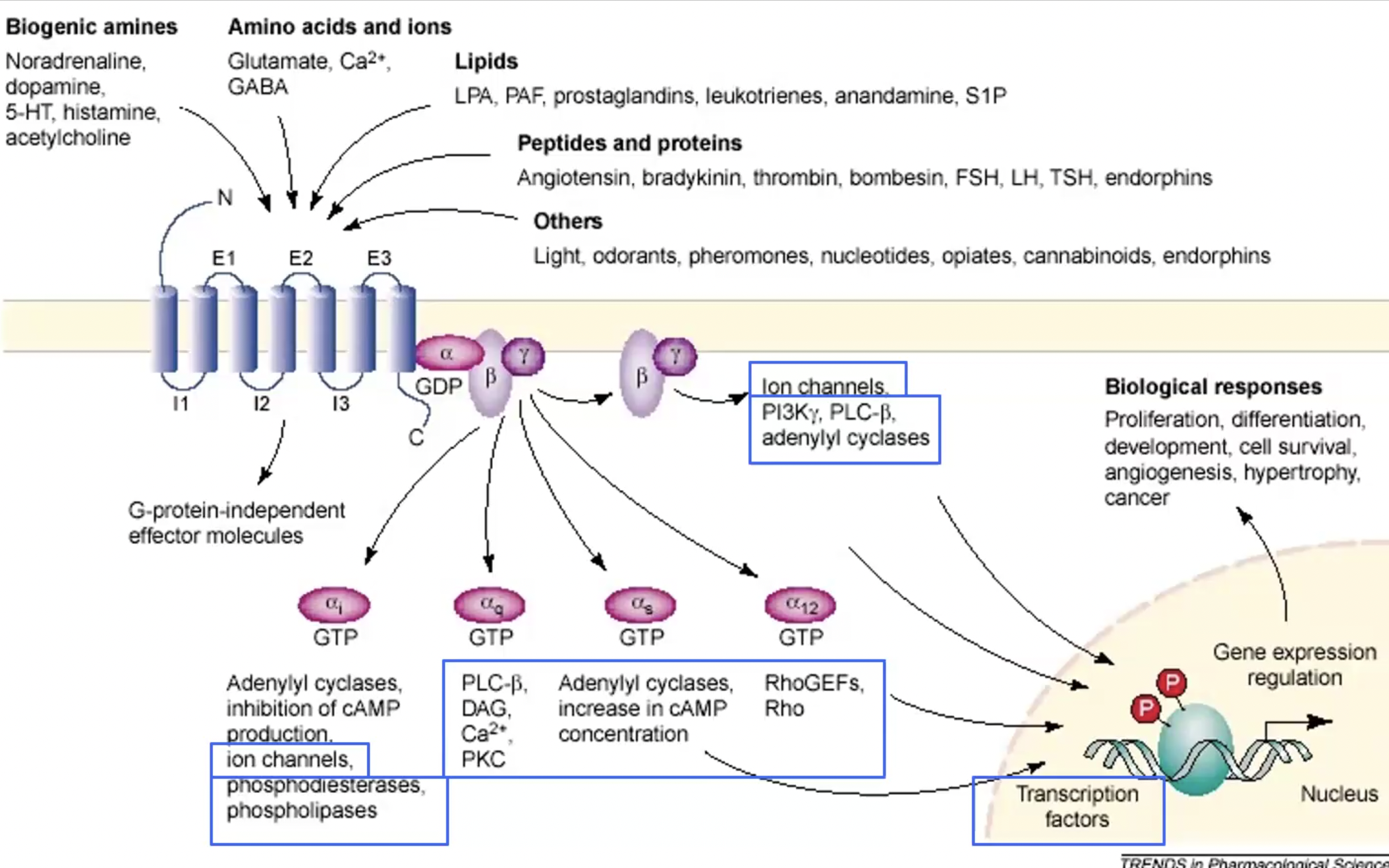
Mechanism of G-Protein Coupled Receptors (GPCR)?
Metabotropic receptors
Ligand binds
intracellular portion changes conformation
Recruits G protein (3 domains α, β, γ). They become activated (GDP → GTP)
G proteins move on to activate second messengers/effector molecules downstream (ex. cAMP, DAG)
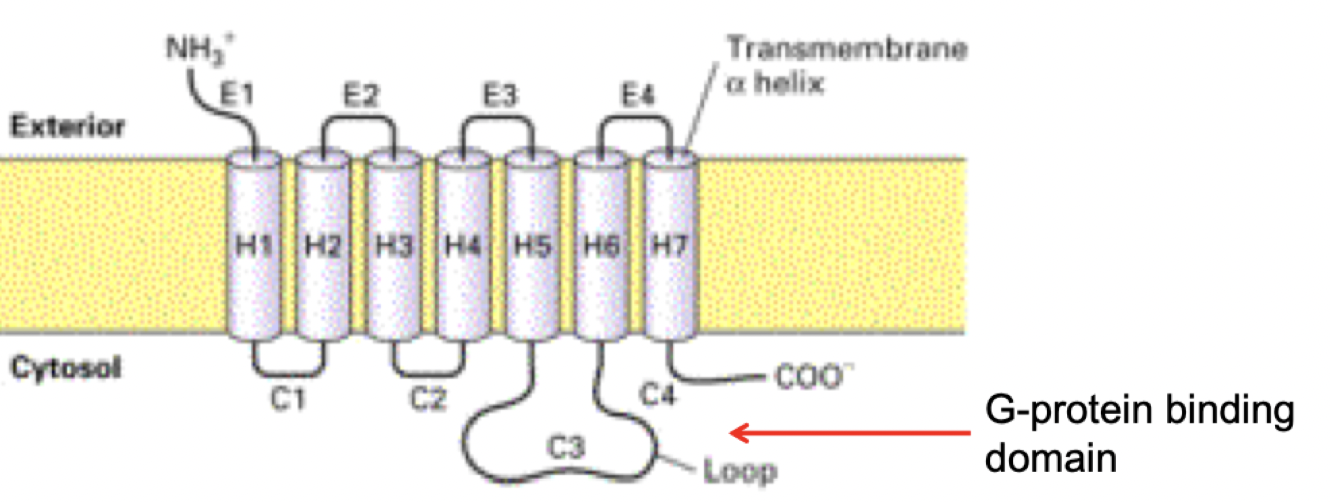
GPCR basic structure
400-500 aa length
7 transmembrane segments (alpha helices) → conservative portion that situates in plasma membrane
Extracellular domain with more variability in terms of binding ligand
Once agonist binds, intracellular part changes conformation, and gains affinity for binding partners (recruiting G protein)
Variation in GPCR
Different ligands bind different GPCRs activate different G proteins, which activate different effectors (particularly variability in α subunit)
Inhibitory and stimulatory receptors can have same target effector (ex. Gs stimulates and Gi inhibits adenylate cyclase)
Crosstalk in GPCR
ex. Cross-talk between receptors and signalling pathways can lead to rapid and longterm effects, and activate genes.
ex. g coupled proteins (like TRPV1) can regulate ion channels, also the pathways g coupled proteins activate can regulate ion channels.
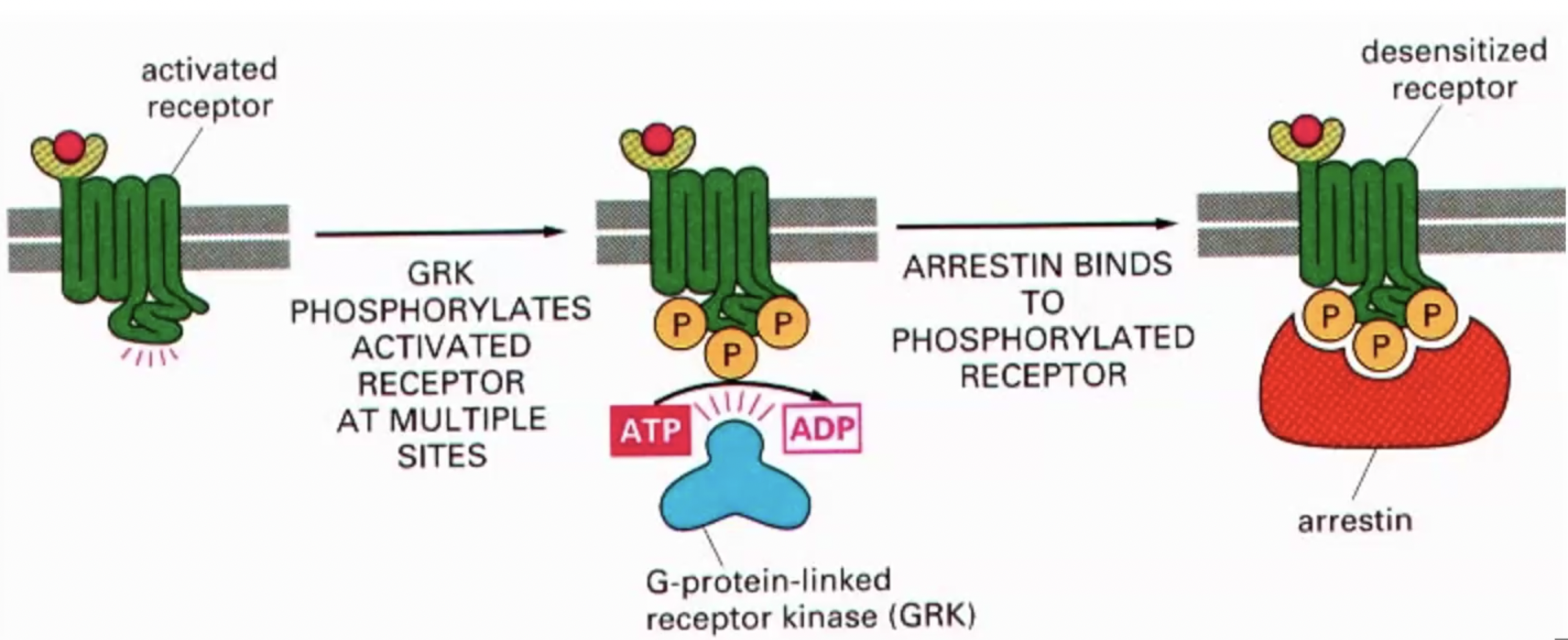
GPCR down regulation - MASKING
Temporary reduction in GPCR activity (desensitization) due to uncoupling from G proteins, without altering receptor numbers at the membrane.
Receptor binds agonist, changes conformation, recruits g-protein. With prolonged exposure to agonist, conformation gains affinity for g-protein-linked receptor kinase that phosphorylates intracellular side receptor at many sites, changing conformation which 1. makes less effective at activating g-proteins, 2. phosphorylation makes gain affinity for arrestin. Arrestin blocks G protein from inducing signalling
*masking = blocks further activation, reduces signal
*Reversible once the ligand is removed or the receptor is de-phosphorylated.
GPCR down regulation - INTERNALIZATION
GPCRs are removed from the cell surface and taken into the cell via endocytosis. This reduces density of receptors on the surface
Continuing on from masking, if constant presence of agonist still, arrestin receptor complex gains affinity for another enzyme that deposits clatherin into membrane allowing receptors and saturated proteins to be signalling for endocytosis
Clatherin brings receptors inside vesicle and degraded - no longer available to extracellular ligands.
Next dose there is fewer signalling as receptors as internalized (less units of activation). It required MORE substance to generate same effect.