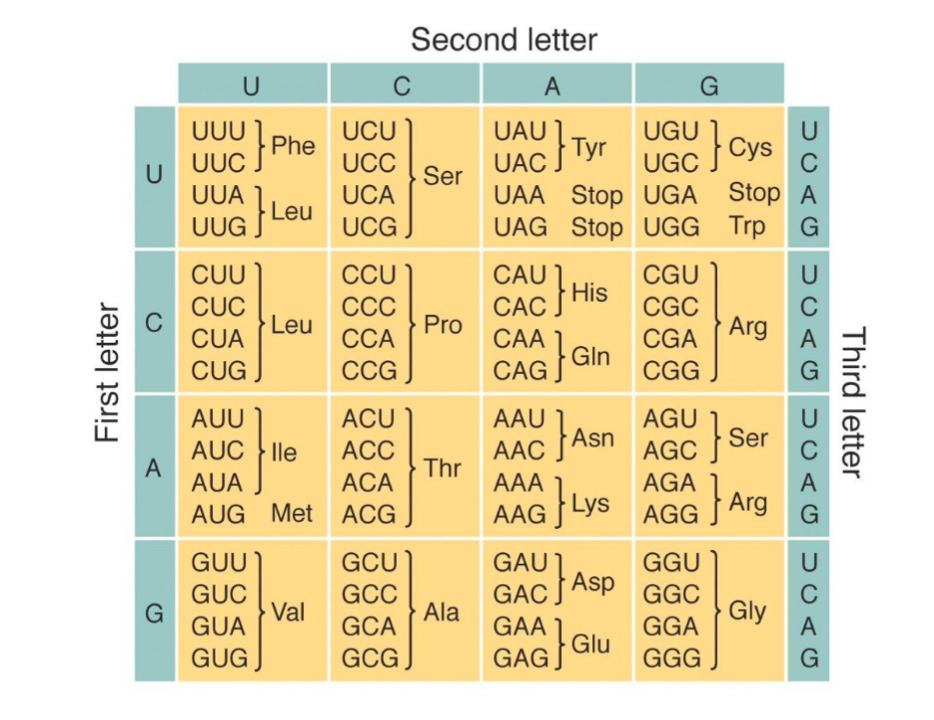
Synthetic Biology I & II
1/36
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
37 Terms
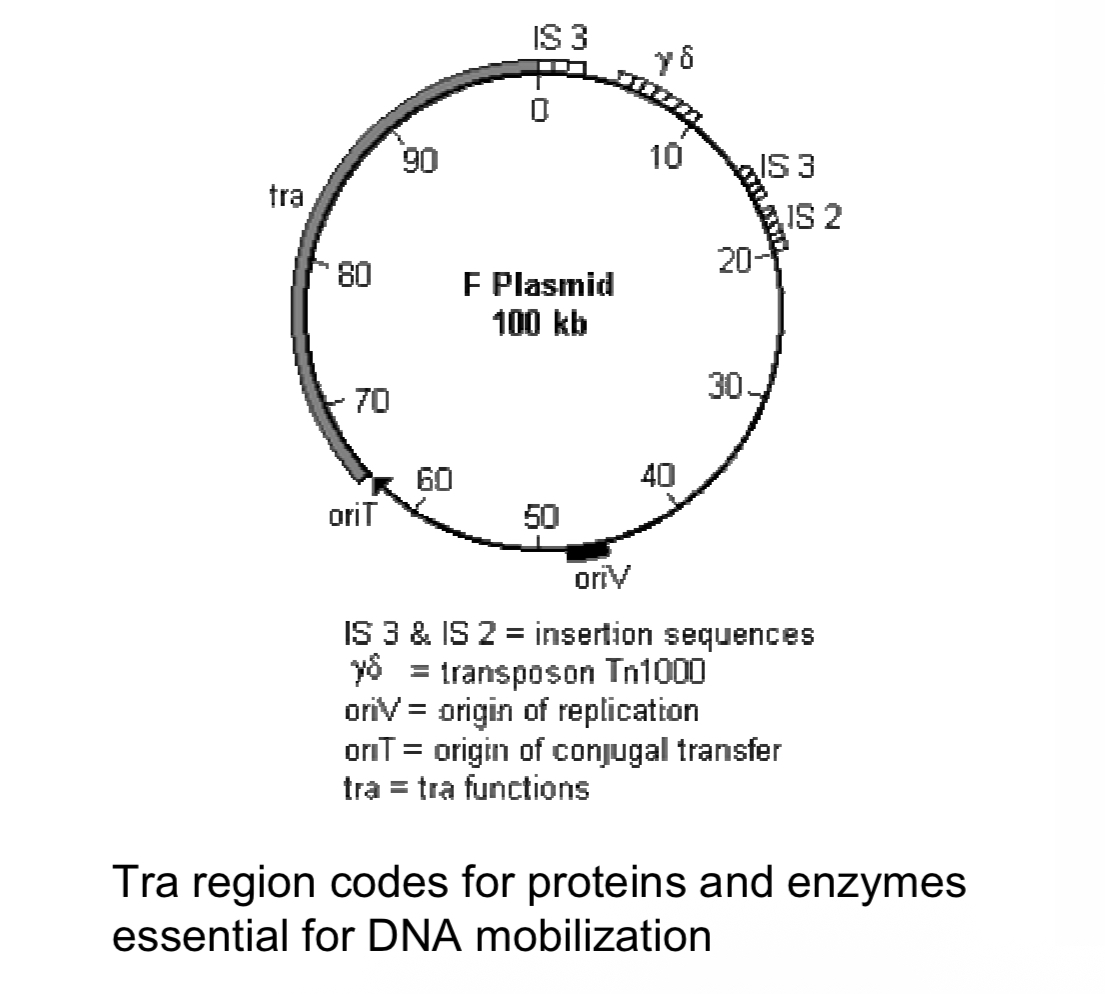
F (fertility) factor
plasmid that can be mobilized from F+ male bacteria and F- female bacteria. BAC
The F factor controls its own replication.
It has two origins of replication:
oriV is the origin for bidirectional replication;
oriS is the origin for unidirectional replication.
The F factor also has genes that regulate DNA synthesis so that its copy number is kept at a low level; and, genes that regulate the partition into the daughter cells after E. coli divides.
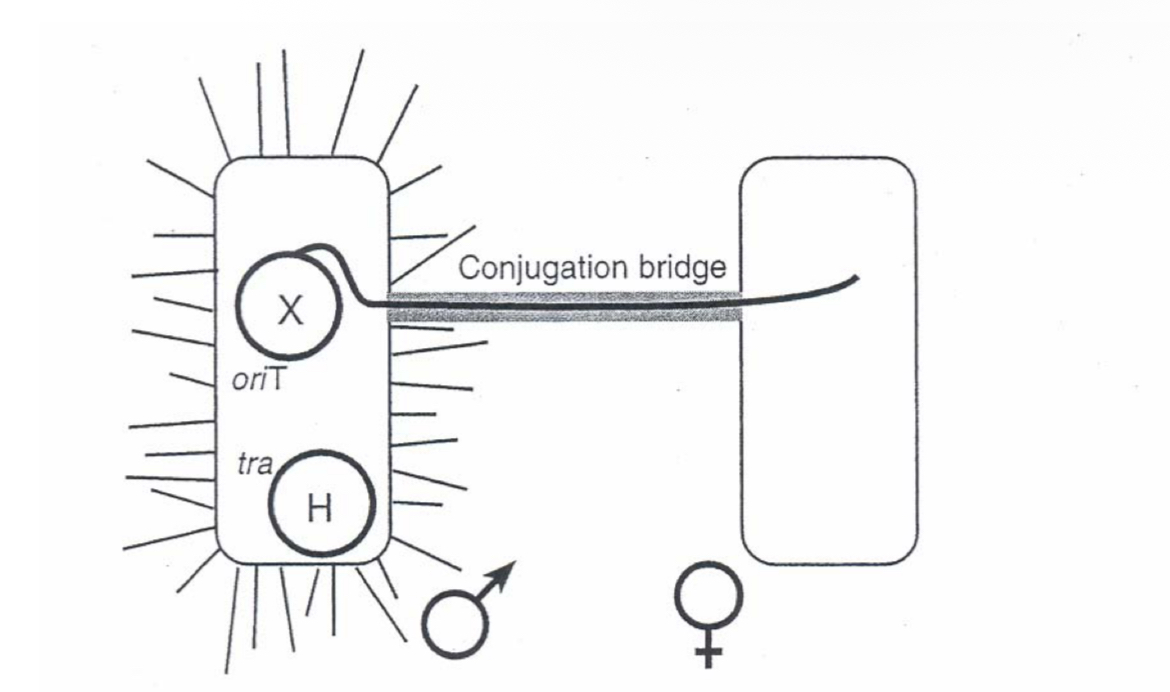
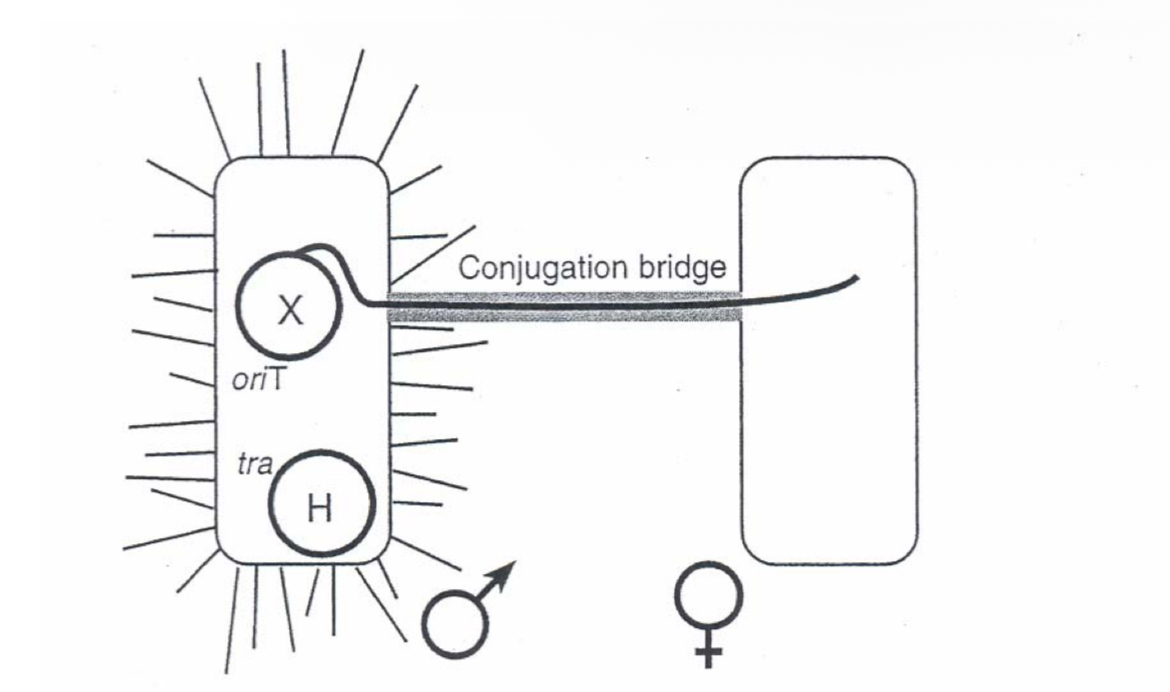
Conjugation
gene transfer from one to another bacterial cell
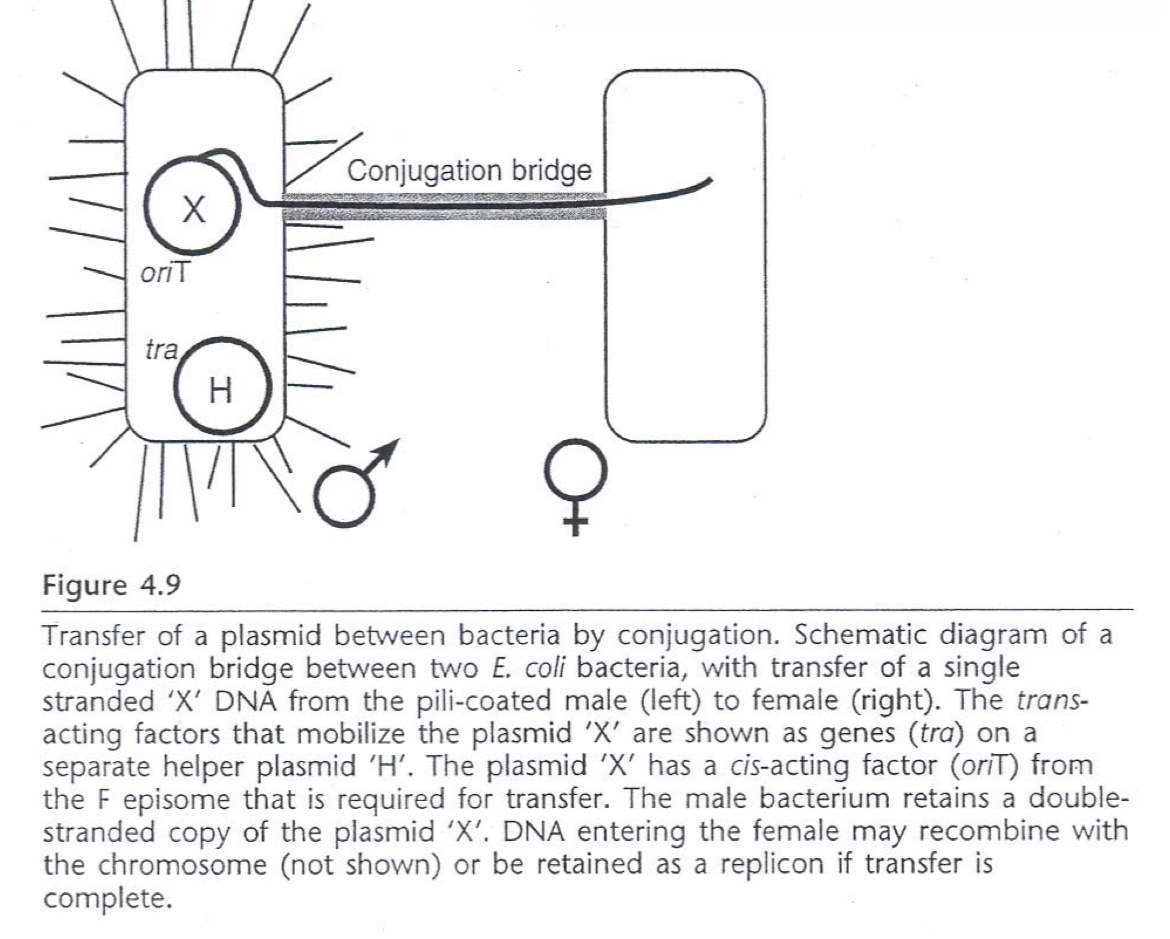
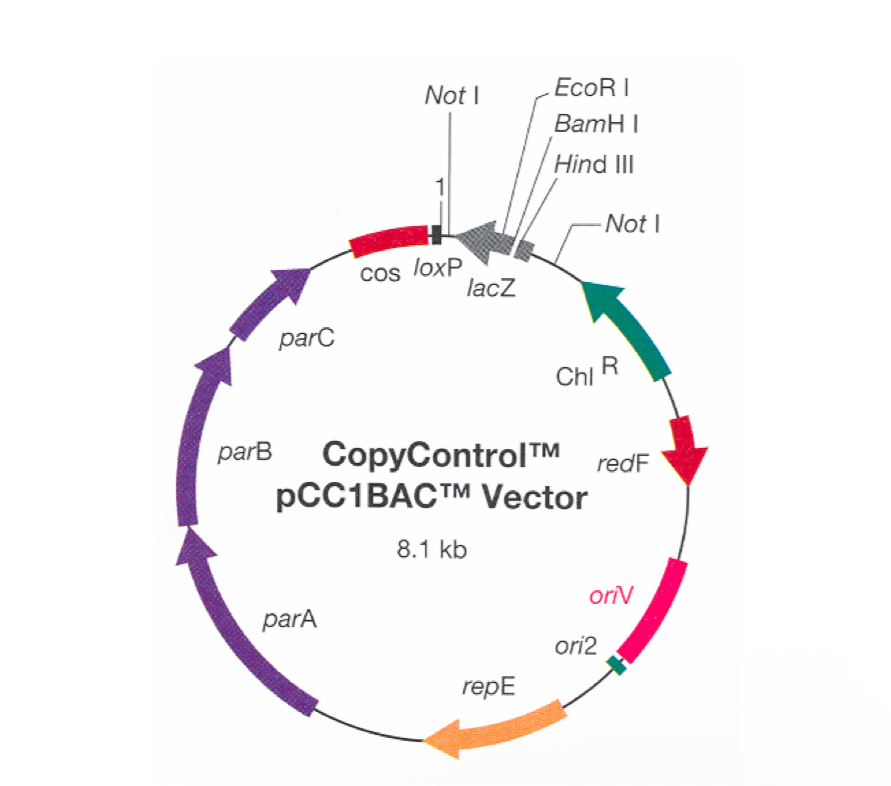
BAC (Bacterial Artificial Chromosomes) Modifications?
Added second replication origin; oriV.
Generates multiple copies of the BAC if TRF gene is present
TRF= trans acting factor.

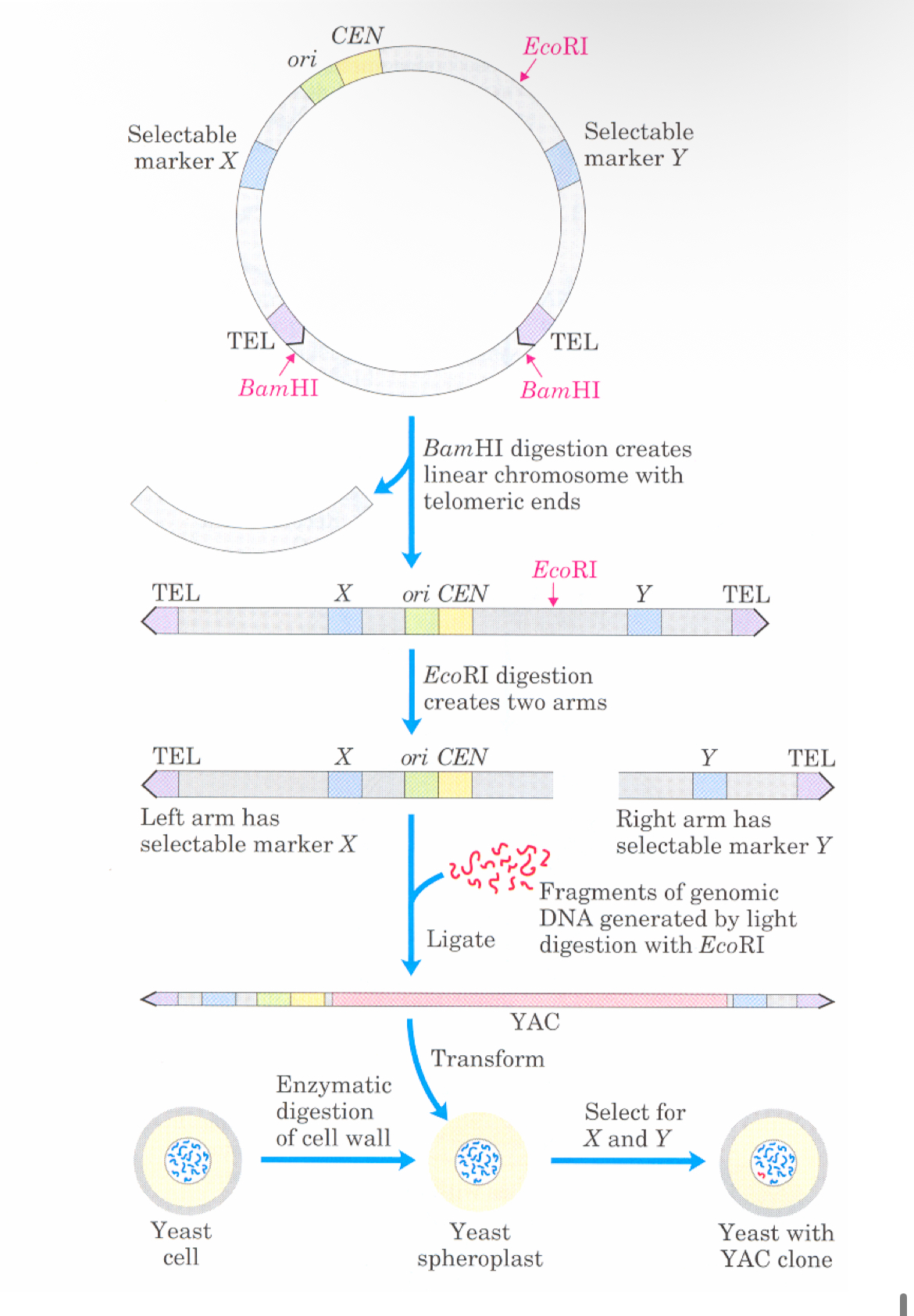
YAC (Yeast Artificial Chromosomes)

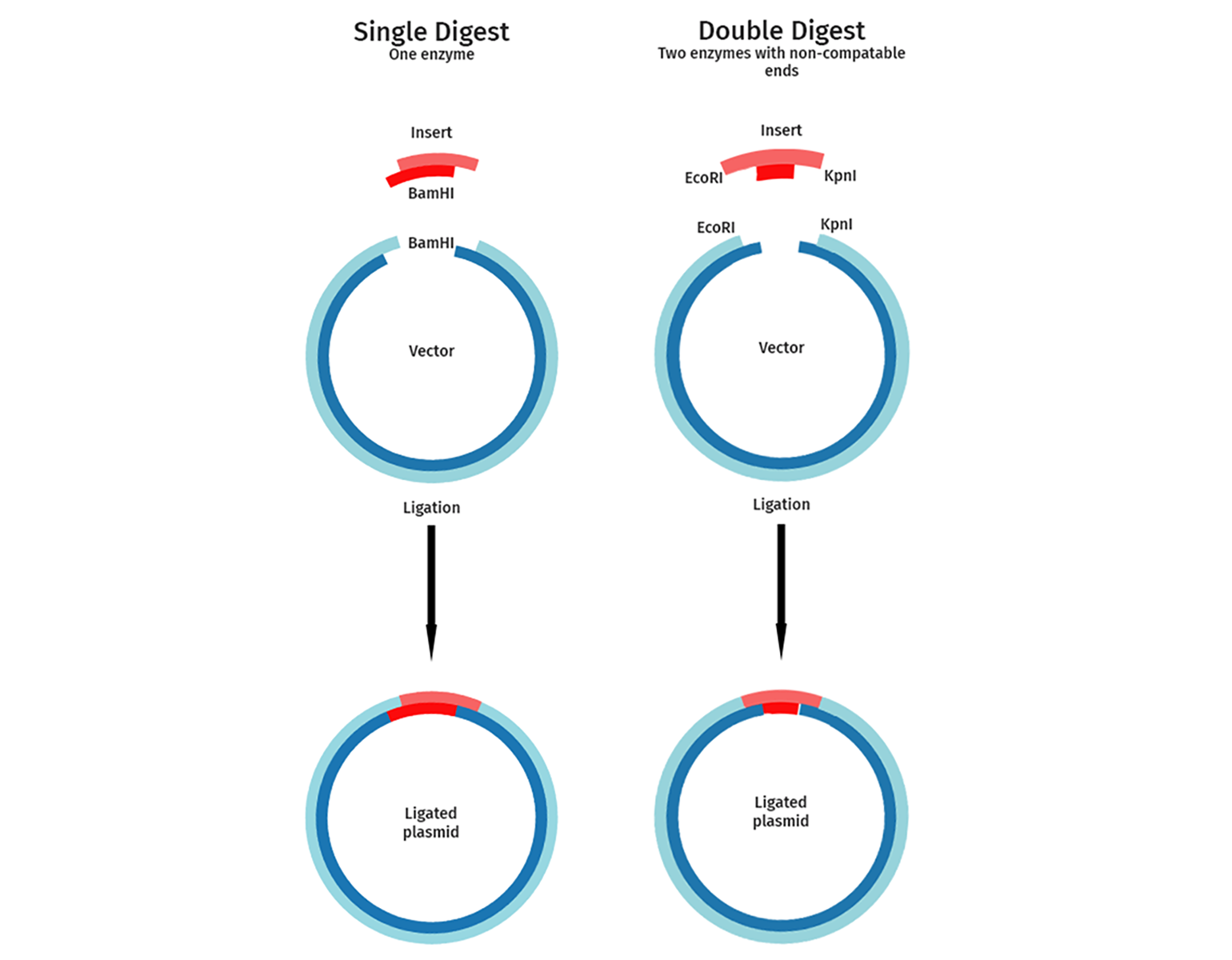
Single Digest vs Double Digest
Single vs. double digest refers to using one versus two restriction enzymes to cut DNA;
single digests cut DNA at one type of site, creating fragments
double digests use two enzymes simultaneously (or sequentially), generating smaller fragments and often creating directional cloning sites, allowing for specific insert orientation and preventing vector religation
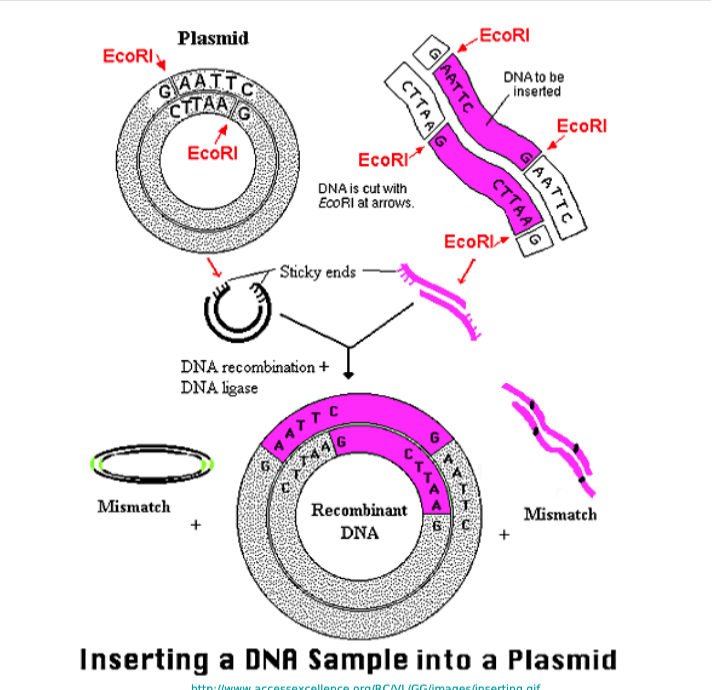
Single Digest Plasmids
Only cut by ONE TYPE of Restriction Enzyme.
typically uses blunt ends, but CAN have sticky ends.
can be cut however many times
Double-Digested Plasmids
cut by TWO DIFFERENT TYPES of Restriction Enzyme.
typically resulting in sticky ends.
can be cut however many times
more precise
Cloning Use: Preferred for inserting DNA because the two different sticky ends ensure the insert goes in the correct orientation, preventing self-ligation of the vector and the insert.
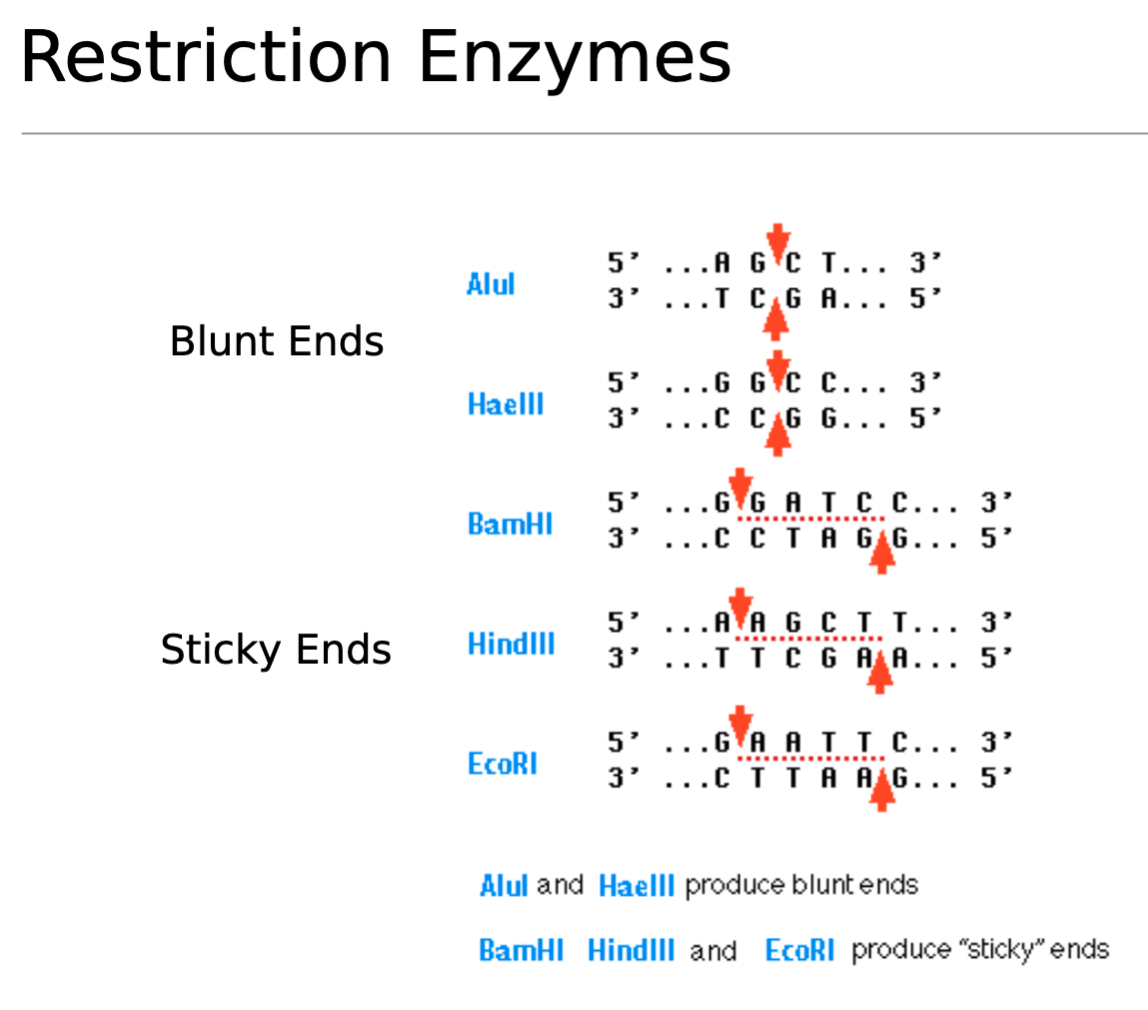
Sticky ends vs Blunt ends

Blunt Ends
Blunt Ends (Flush Ends)
Formation: Created by a straight cut down the middle where both DNA strands are cut at the same position, leaving no overhangs.
Properties: No single-stranded overhangs to guide pairing.
Ligation: Less efficient because ligation relies solely on the DNA ligase enzyme without base-pairing assistance, but they can join any blunt-ended DNA.
Example Enzymes: Alul and Haelll produce blunt ends.
Sticky ends
Sticky Ends (Cohesive Ends)
Formation:
Created by a staggered cut where one DNA strand is longer than the other, leaving a single-stranded overhang (3' or 5').
Properties:
The overhangs are complementary and can form hydrogen bonds with matching sticky ends from another DNA fragment, like Velcro.
Ligation:
Highly efficient due to base pairing, allowing for specific and directional joining.
Example Enzyme:
BamHI, HindIII and EcoRI produce sticky ends.

rDNA
Recombinant DNA
Clones / DNA from another organism.

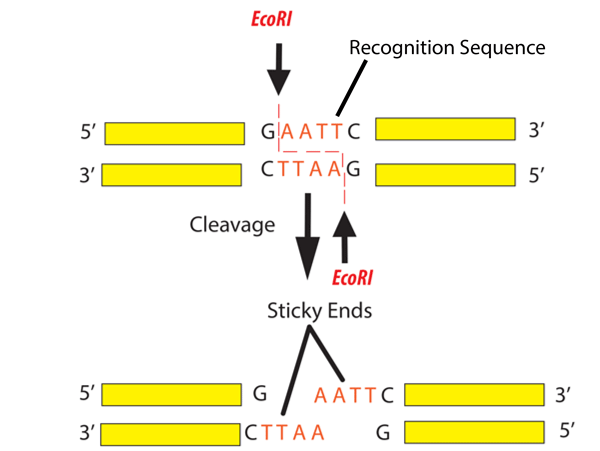
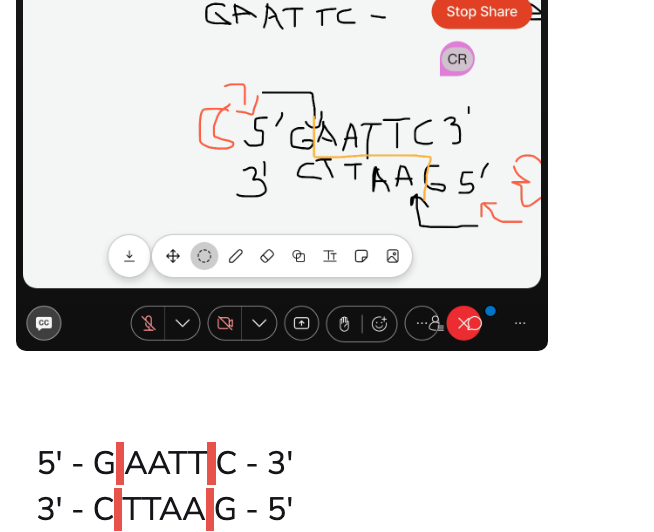
Definition of Palindrome
one way reads the same, the other way reads the same. (racecar)
And that's what your restriction enzyme is looking for.
So where will it cut? It will always cut after the first base..
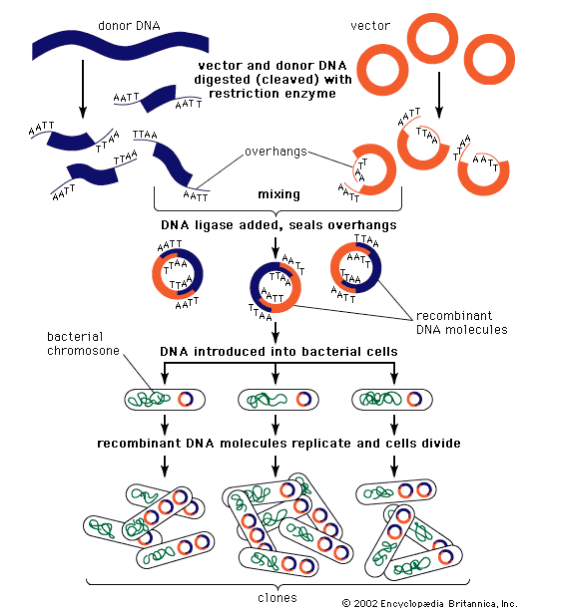
General Procedure in Order
Identify gene of interest from host
Amplify (PCR)
Insert into Vector
Transform secondary host
Identify clones

Step 1. Identify gene of interest from host
•NCBI: National Center for Biotechnology Information
•Search Nucleotide
•Obtain Sequence
•Extract DNA/RNA from host
Step 2. Amplify target sequence
Can be done naturally or in a lab
Natural Cell Replication
PCR (much more efficient) + gel electrophoresis
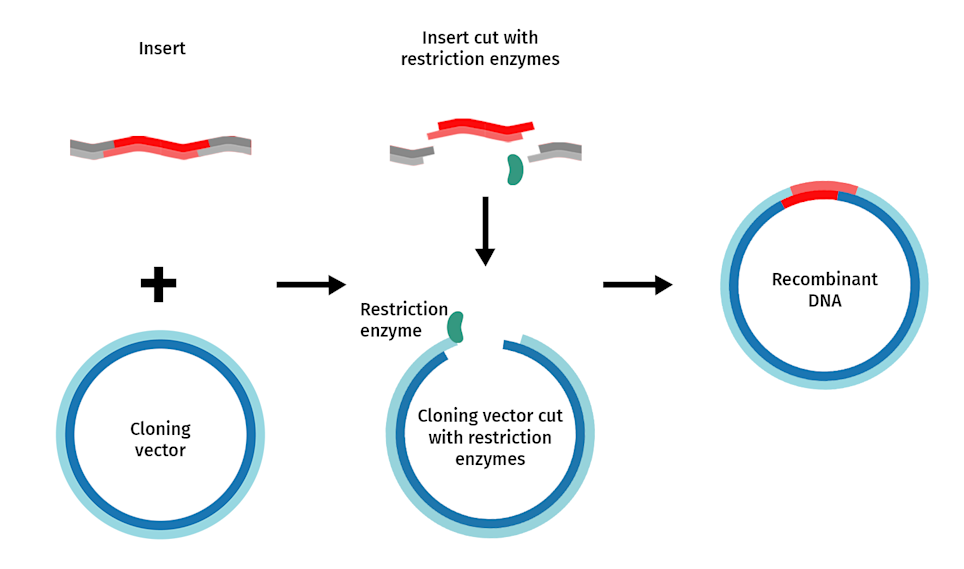
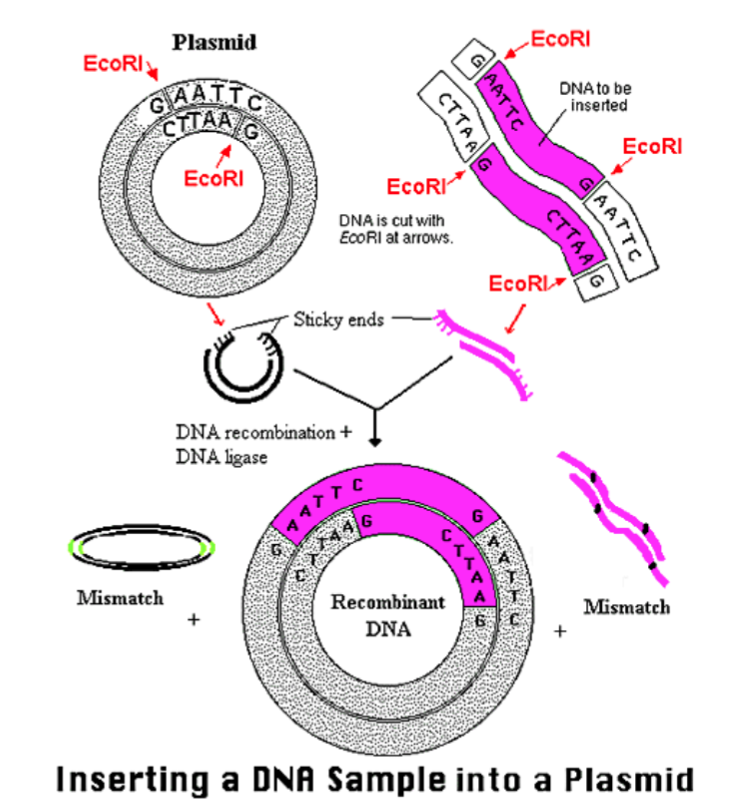
Step 3.) Insert into Vector (plasmid)
The process of incorporating the amplified gene of interest into a plasmid vector, which will later carry the gene into the host cell for expression. This setup allows for proper replication and expression of the inserted gene.
Types of vectors: Plasmids, BAC, YAC. Typicallu have markers
Use depends on size of DNA that you’re inserting
Restriction Enzymes
Ligase; facilitate the joining of DNA fragments, often using techniques like restriction digestion and ligation.
Step 4.) Transform Secondary Host
The process of introducing the vector containing the gene of interest into a secondary host organism, allowing for the expression of the inserted gene. This step enables further propagation or analysis of the genetically modified organism.
Cells must be competent
Must choose appropriate host
Size
Post-translational Modifications
Protein transport
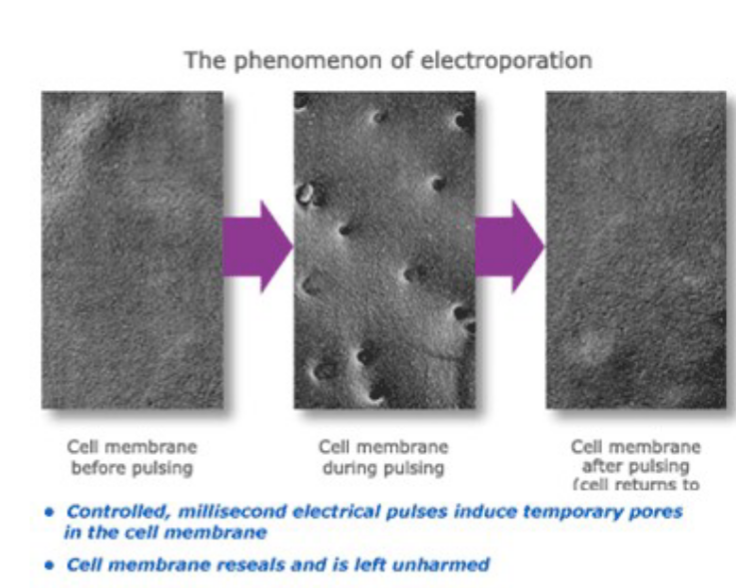
ex.) “Electroporation”
Controlled and quick electrical pulses to induce pores in the cell membrane, allowing vector inside target.
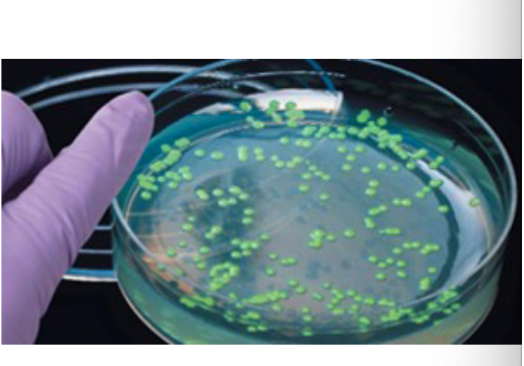
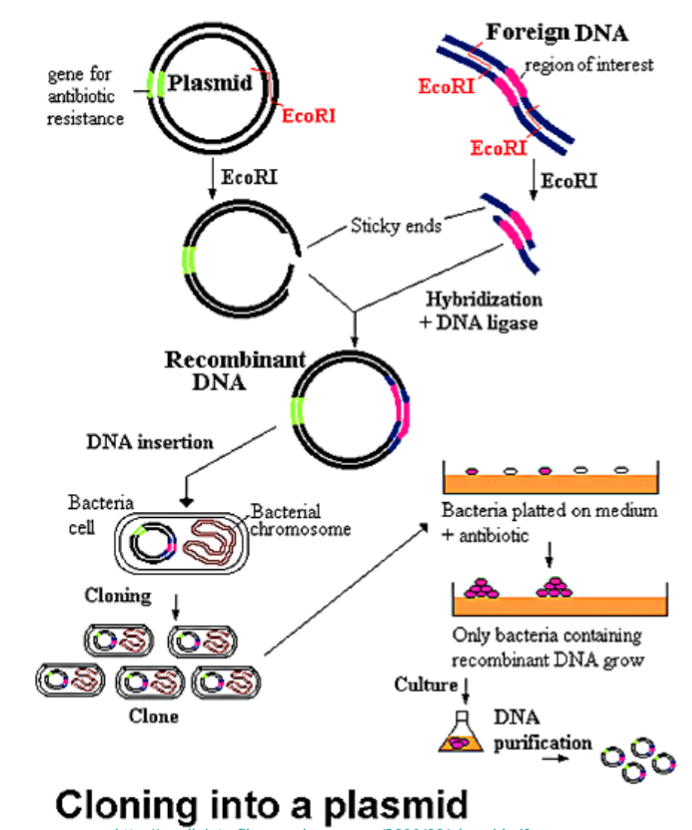
Step 5.) Identify Clones
Using marker, identify clones that successfully obtained recombinant DNA.
Use selective growth mediums ex. antibiotic resistance + bacteria on medium with an antibiotic
Culture the remaining recombinant clones that survive
DNA purification
Left with clones
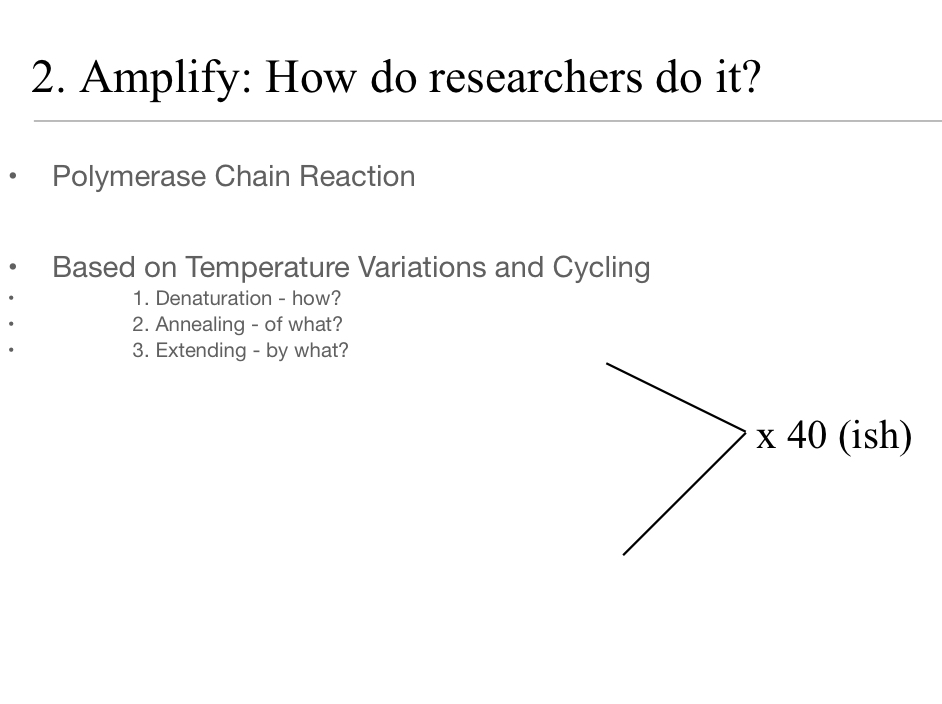
PCR Overview: General Procedure Step 2. Lab-Induced Amplification.
Starts with double stranded DNA.
Form Primers that isolate your target DNA.
Optimize Primers for annealing; T and Tm.
Results in Doubled DNA. (x2 each cycle).
Denaturation, Annealing, Elongation.
Gene- specific primers target area to be amplified
DNA Polymerase adds nucleotides 5’ - 3’.
Cycles x40 times typically

PCR Temperatures
Denaturation: T= 94-95°C
Annealing: T= 5°C below lowest primer Tm (found in optimization guidelines)
Elongation: T= 72°C

PCR Components
DNA
Forward Primer
Reverse Primer
DNA Taq Polymerase
Water
total volume = 25uL
After 2 rounds of PCR, you would expect ____ strand(s) of double stranded DNA?
4 strands of double stranded DNA.
2^n
After 2 rounds of PCR, starting with one double-stranded DNA molecule, you will have 4 double-stranded DNA molecules, as PCR exponentially doubles the target DNA with each cycle, following the formula
2n2 to the n-th power
2𝑛
(where n is cycles), making it 2^2 = 4.

In PCR and cellular DNA replication (amplification), new nucleotides are added to the ______ end?
3’ Hydroxyl End.

True or False? The forward primer is always built off of the 5’ - 3’ strand, and the reverse primer is always built off of the 3’ - 5’ strand.
False.
Both statements are incorrect.
The forward primer is designed to be complementary to the 3’ - 5’ template (antisense) strand and initiates DNA synthesis in the 5’ - 3’ direction.
The reverse primer is designed to be complementary to the 5’ - 3’ coding (sense) strand (specifically, its complementary region on the 3' end when read from the other side of the target) and also initiates DNA synthesis in the 5’ - 3’ direction.
Both forward and reverse primers themselves are always synthesized and written in the 5’ - 3’ orientation.

PCR;
Polymerase Chain Reaction can be used to amplify a specific region of DNA with the use of what?
a.) a forward primer
b.) RNA polymerase
c.) a reverse primer
d.) A and C only
Answer; d.) A and C only.
A forward primer and reverse primer can be used to amplify a specific target sequence of DNA in PCR.

After correctly adding each component to a PCR reaction, a new Biotech researcher programmed the thermocycler for the following temperatures;
Denaturation; 74°C
Annealing; 55°C
Elongation; 72°C
After running a gel of this product, you would expect to see _____?
NO banding pattern.
The programmed denaturation temperature of 74∘C74∘C is significantly lower than the required 94−95∘C94−95∘C
At this sub-optimal temperature, the double-stranded DNA template would not fully denature into single strands.
Without proper denaturation, the primers cannot anneal to their specific target sequences, and consequently, the DNA Taq Polymerase cannot synthesize new DNA strands during the elongation phase.
Therefore, no PCR product would be generated, resulting in no banding pattern visible on a gel.

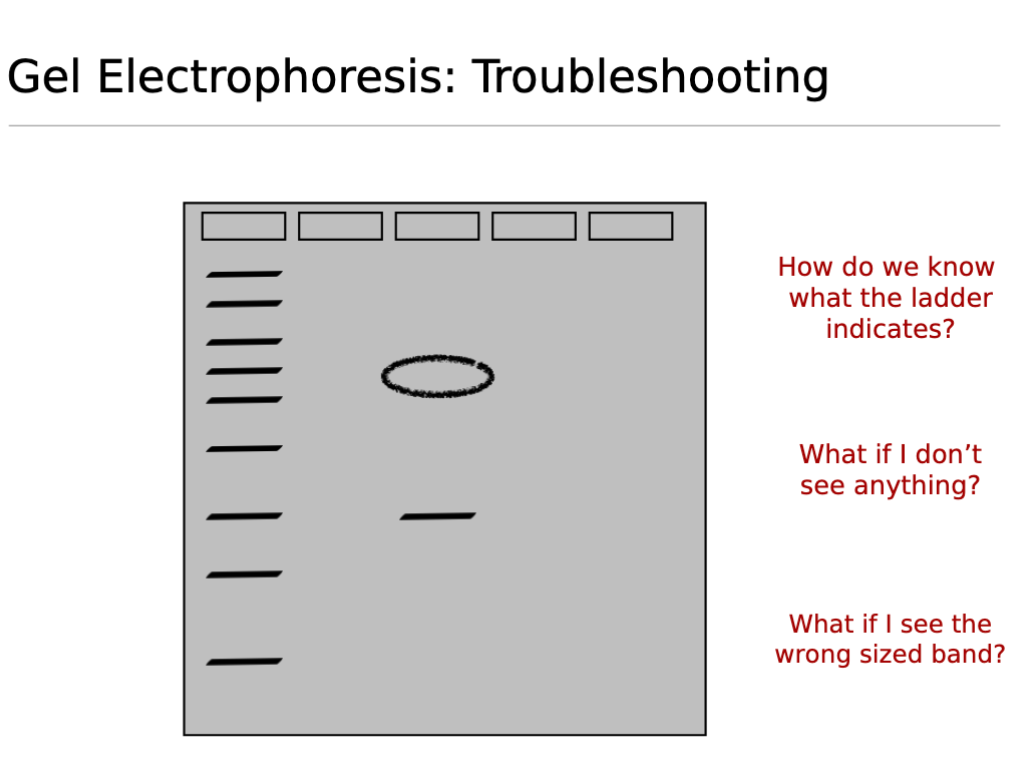
Gel Electrophoresis Troubleshooting;
How do we know what the ladder indicates?
The ladder (also known as a DNA standard or marker) consists of DNA fragments of known molecular sizes (e.g.,bp base pairs or kb kilobases).
It serves as a reference to estimate the size of unknown DNA fragments in your samples by comparing their migration distance to that of the ladder band
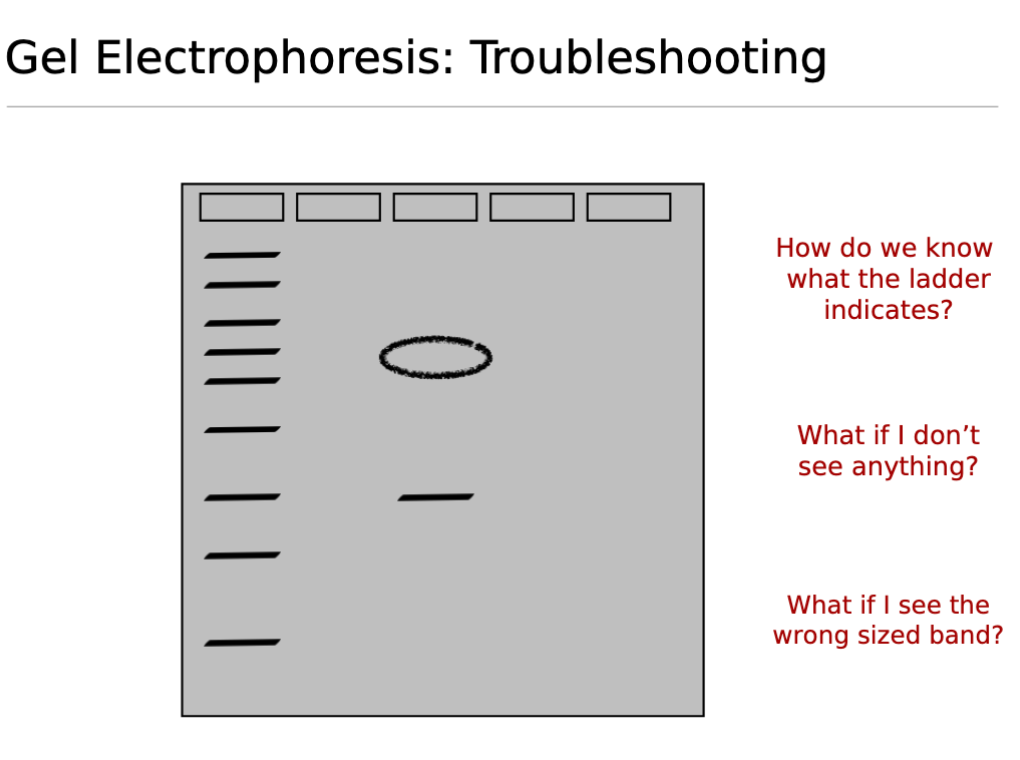
Gel Electrophoresis Troubleshooting;
What does it mean if you don’t see anything/no bands?
What does it mean if you don’t see anything/no bands?
No DNA loaded: The sample was not loaded into the well or was loaded incorrectly.
Reaction did not work: PCR or restriction digest failed due to issues like incorrect temperatures, degraded enzymes, missing reagents, or inhibitors
DNA Degradation: The DNA sample may have degraded before or during the experiment.
- Incorrect Gel Concentration: The gel percentage might be too high (DNA cannot migrate) or too low (small DNA fragments run off the gel).Improper Running Conditions: Too low temperature, insufficient running time, or incorrect buffer composition.
Detection Issues: Problems with DNA staining (e.g., insufficient stain, over-destaining) or the UV illuminator not functioning.
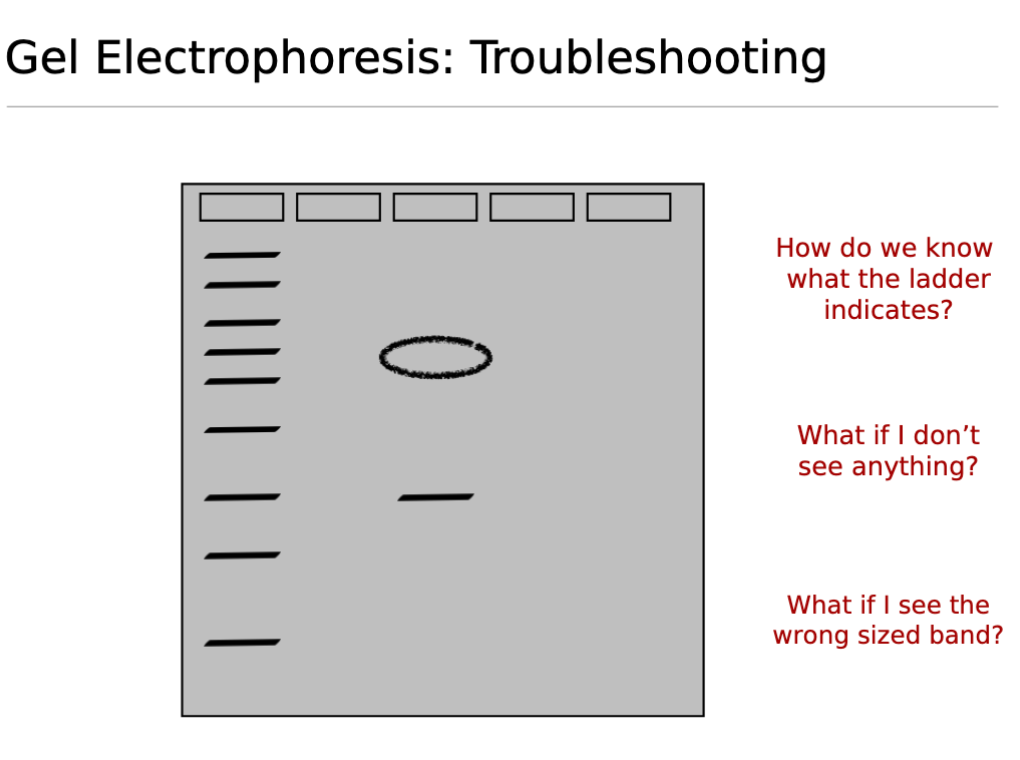
Gel Electrophoresis Troubleshooting;
What if you see the wrong sized band?
What if you see the wrong sized band?
Contamination: Non-target DNA was present and amplified.
Non-specific Annealing/Primer Issues (PCR): Primers bound to unintended sites, amplifying an incorrect fragment, or primer dimers formed (very small bands). Non-optimized primers.
Mutations/Alterations: The target DNA might have an unexpected insertion, deletion, or other mutation altering its size.
Incomplete Digestion (Restriction Digest): If restriction enzymes did not cut at all expected sites, resulting in larger, uncle
What are each of the 3 gel lanes used to detect in PCR gel electrophoresis?

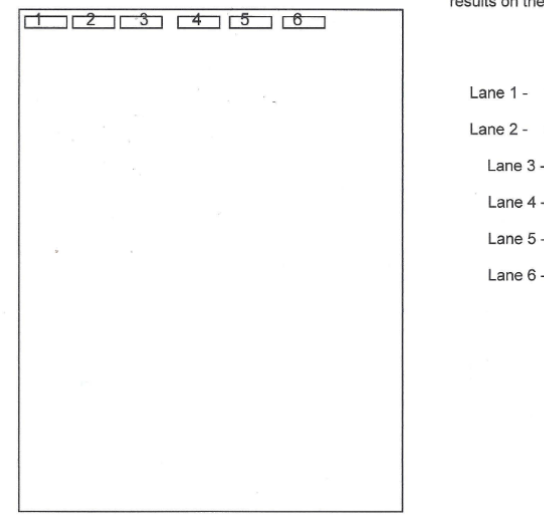
What are each of the 6 gel lanes used to detect in molecular cloning gel electrophoresis?
In molecular cloning gel electrophoresis, a 6-lane gel is typically used to monitor different stages of the cloning process:
DNA Ladder/Marker: This lane contains DNA fragments of known sizes (you make it up). It is used as a reference to determine the approximate size of the unknown DNA fragments (vector, insert, recombinant plasmid) in the other lanes.
Undigested/Uncut Plasmid Vector: This lane shows the plasmid vector before any restriction enzyme digestion. It typically appears as multiple bands representing different topological forms (supercoiled, relaxed, linear) of the plasmid, confirming its integrity and initial size.
Digested Plasmid Vector: After being cut with restriction enzymes, this lane should show the linearized vector as a single, distinct band. This confirms that the restriction enzymes successfully cut the plasmid at the intended sites and allows verification of its linearized size.
Insert DNA: This lane contains the isolated or PCR-amplified gene of interest (the insert). It verifies the size and purity of the DNA fragment intended to be ligated into the vector.
Vector + RE site1 + RE site 2 = Ligation Reaction: This lane contains the mixture after the ligation step (joining the vector and insert). It can show a shift to a larger molecular weight band, indicating the successful formation of the recombinant plasmid (ligated vector + insert). It might also show un-ligated vector or insert, or other ligation products.
Recombinant Plasmid (Ligation of Double-Digested vector with gene insert from Positive Clone): After transformation and selection of positive clones, this lane contains plasmid DNA extracted from a putative recombinant clone, which has then been digested again to excise the cloned insert. The presence of two distinct bands (linearized vector and excised insert) confirms successful cloning of the insert into the vector.

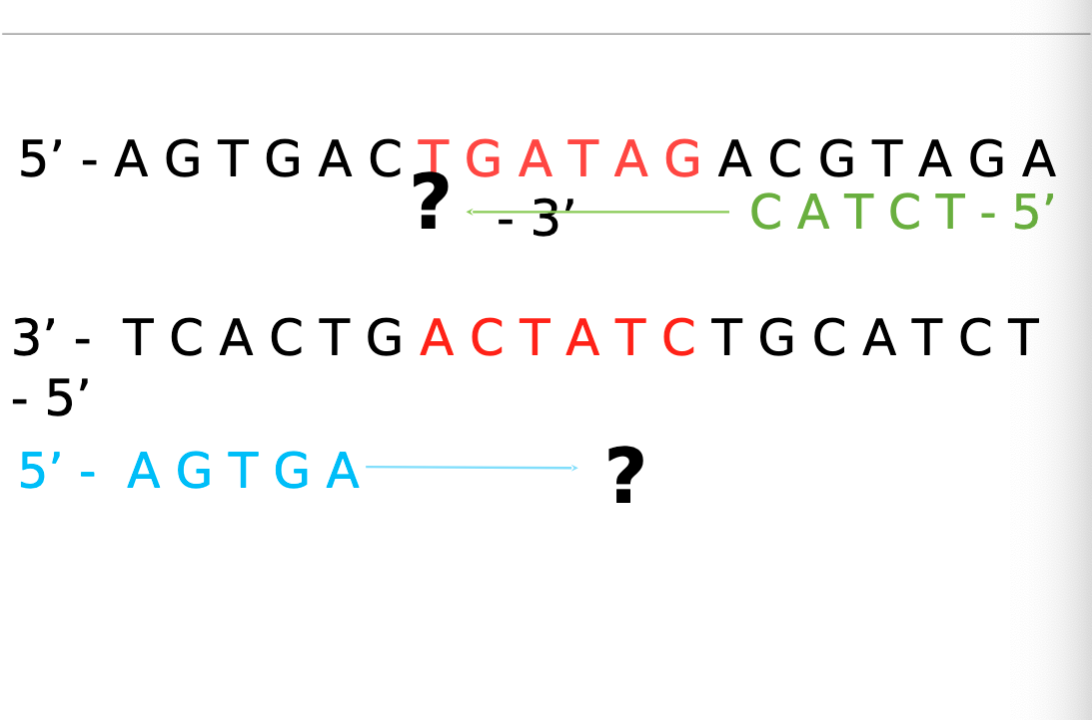
Identifying/Designing Forward and Reverse Primers for PCR
Identify gene of interest/ target sequence (normally 10-20 bps) Starts as double stranded DNA.
Select 14-20 bps upstream and downstream of target sequence
Forward Primer: Blue 5’-3’. attached to left of template/anti-sense strand . Arrow to the right. (3’-5’)
Reverse Primer: Green 3’-5’. attached to right side of non-template/coding strand. Arrow to the left. (5’-3’).

Primer Optimization
Both Primers must be same length- 15-40 bps.
G/C nucleotide content: Each primer must have 40%-60% G/C content.
ex.)
F: 9/20= (G/C / 20 bps total ) = 49%
R: 9/20= (G/C / 20 bps total ) = 49%
Tm = 42℃ - 65℃, and are within 5℃ of eachother.
ex.)
F: 2℃(11/20) + 4℃(9/20) = 58 ℃
R: 2℃(11/20) + 4℃(9/20) = 58 ℃
58℃ and 58 ℃ are within 5 ℃ .
T= 5℃ lower than whatever the lowest primer Tm was.
ex.)
F: 58℃ - 5℃ =53℃ .
(Both F and R Tm just as low)
Calculate Annealing % rate. How often will bps match? Must be under 50%.
ex. )
5’ - 3’
3’ - 5’.
count # of matches vs # of bps.

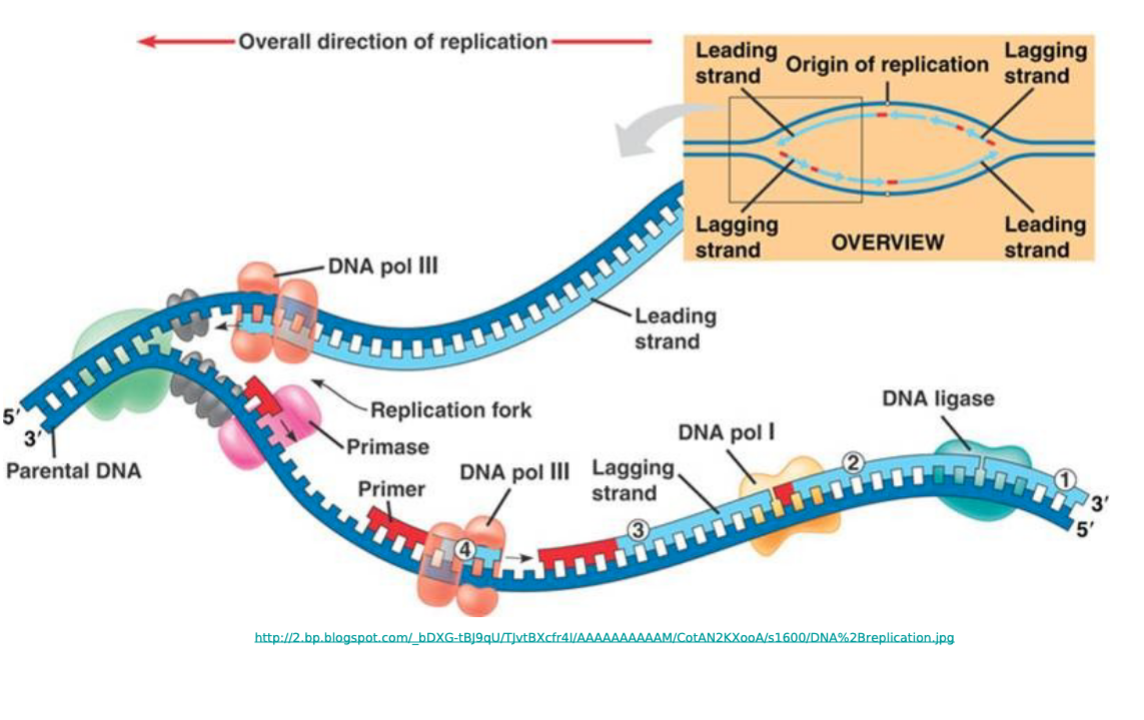
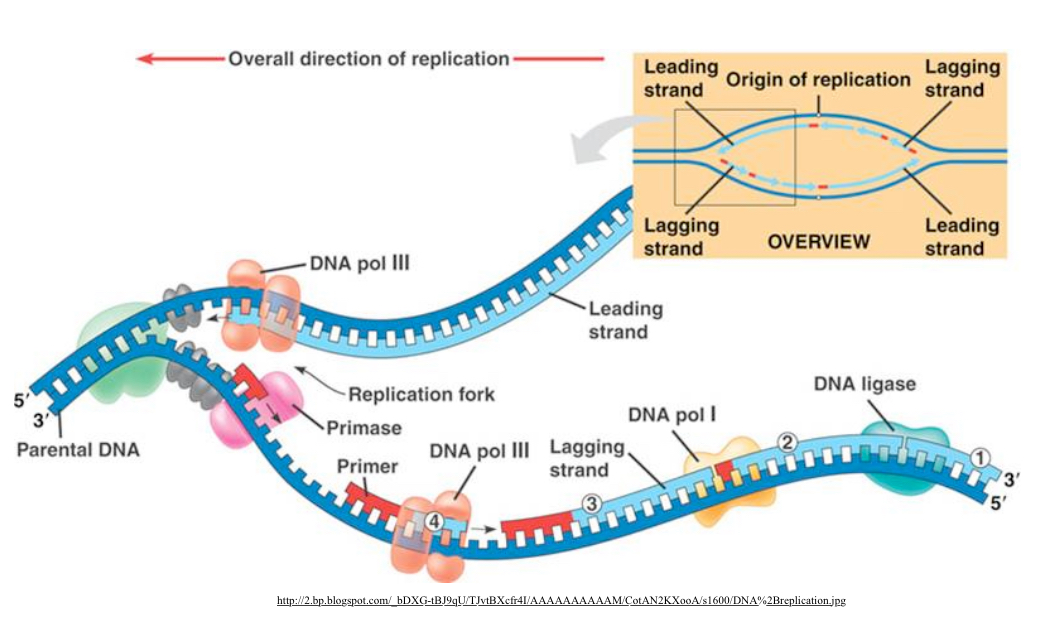
Cellular DNA Replication ;
Parental DNA double stranded.
Double helix of DNA is unwound into 2 separate strands by the Helicase enzyme. One strand gets copied continuously while the other strand is copied backwards
DNA Polymerase III and primase at replication fork
DNA Polymerase III at primer of lagging 3-5 strand. Main replication enzume, synthesizes most new DNA on both the leading and lagging strand. Has 3’-5’ proofreading exonuclease activity.
DNA Polymerase I ; RNA primer removal and fills gap with DNA. Has 3’-5’ exonuclease activity. Slower, specialized clean up for RNA primers and gaps. Replaces primers with DNA.
DNA ligase joins the strands together
Leading and Lagging strand Directions
Leading Strand=Sense Strand=-sense/Non-template Strand: 5’ - 3’ . Synthesized continuously towards replication fork.
Lagging Strand=Anti-Sense=Template Strand: 3’-5’ . Discontinuous synthesis away from fork in fragments. Needs multiple RNA primers. DNA polymerase extends primers. DNA ligase joins the fragments formed.
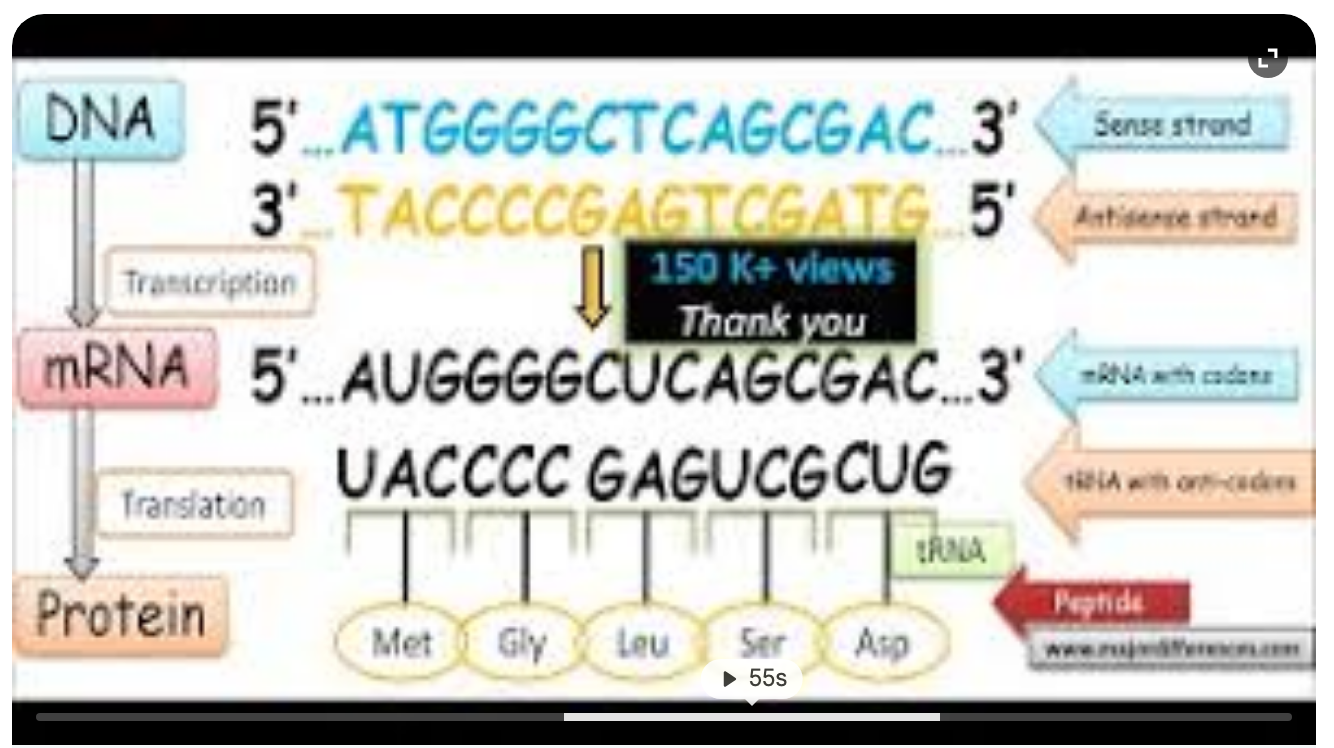
Transcription; mRNA sequence produced is Identical to the DNA’s Sense (leading/Non-Template) Strand - codons
tRNA complementary to 5’ - 3’ mRNA strand is complementary to DNA’s Antisense (lagging/template) strand. - anti codons - translate into protein/amino acids