Lecture 5 - Protein Structure
1/32
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
33 Terms
What are peptides and how are peptide bonds formed?
Peptides and proteins are polymers of amino acids, also called polypeptides or polyamino acids.
A peptide bond is a special type of amide bond that joins two amino acids.
It forms through a condensation reaction, where a molecule of water is released.
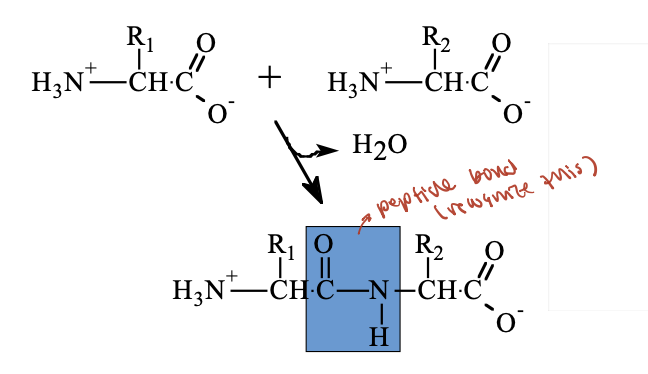
proteins are __ molecules (that can fold) of amino acids joined by amide (peptide) bonds fromed by ‘simple’ removal of H2O from __ and __
linear, carboxyl, amino
What are the ionization and variability properties of a polypeptide?
The repeating backbone is non-ionizable but polar, except at the N-terminus and C-terminus.
Side chains vary depending on the amino acid.
The pKa and pI values of side chains and termini are similar to those of free amino acids.
What happens to α-NH₃⁺ and α-COO⁻ groups when a peptide bond forms?
the α-amino (–NH₃⁺) and α-carboxyl (–COO⁻) groups of amino acids become covalently linked.
As a result, they can no longer ionize in solution.
Only the N-terminus, C-terminus, and ionizable side chains (R groups) can carry charge.
What is a "residue" and how is amino acid sequence written?
An amino acid unit within a peptide or protein is called a residue.
The sequence of a peptide is written from the N-terminus (left) to the C-terminus (right).
Why is folding important for polypeptide chains?
Polypeptide chains are flexible and must fold into a compact 3D structure for proper protein function.
Folding is an essential aspect of protein structure.
What determines how a protein folds?
Protein folding is determined by its amino acid sequence.
This idea is supported by structural data from X-ray crystallography and NMR, along with known folding principles.
primary structure of proteins
linear sequence of amino acid residues (from N-term to C-term)
secondary structure of proteins
regular conformational patterns of contiguous portions of the polypeptide chain, stabilized by hydrogen bonds
hydrogen bond between carbonyl of one aa and amino of another
tertiary structure of proteins
overall fold of the entire polypeptide chain to give a 3D structure
quaternary structure of proteins
the interaction of two or more polypeptides to form a multi-subunit protein
What is the primary structure of a protein?
The primary structure is the sequence (order) of amino acids in a polypeptide chain.
It determines how the protein will fold and function.
This sequence is written from the N-terminus to the C-terminus.
Who first determined a protein’s primary structure, and how is it done today?
Fred Sanger determined the first protein sequence (insulin) in the 1950s (Nobel Prize, 1958).
He later developed a method for DNA sequencing (Nobel Prize, 1980).
Today, most protein sequences are determined by translating DNA sequences using the genetic code.
What are the main steps of Sanger protein sequencing?
Hydrolysis of the polypeptide (digestion) into small fragments using specific cleavage methods (e.g., enzymes or chemicals).
Separation and sequencing of the individual fragments.
Alignment of the fragment sequences to reconstruct the full amino acid sequence of the protein.
What reagents are commonly used to fragment proteins for sequencing?
Protein fragmentation uses sequence-specific cleavage by enzymes or chemicals:
Trypsin – cleaves after Lys or Arg (basic residues)
Chymotrypsin – cleaves after Phe, Tyr, or Trp (aromatic residues)
CNBr (Cyanogen Bromide) – cleaves after Met (methionine) residues
How are peptide fragments sequenced in Sanger protein sequencing?
Sequencing is typically done using Edman degradation.
It uses phenylisothiocyanate (PITC) to react with the N-terminal amino acid of a peptide.
The modified residue is then cleaved off and identified by chromatography.
This process is repeated to determine the sequence one residue at a time.
Can sequence up to >80 (30) residues in favorable conditions.
How does Edman degradation sequence a peptide?
PITC Reaction:
phenylisothiocyanate (PITC) reacts with the N-terminal amino group of the peptide.
Cleavage:
The N-terminal amino acid is cleaved off after the carboxyl group.
Identification:
The cleaved amino acid is identified using chromatography.
Repeat:
The process repeats to identify one amino acid at a time.
How is the complete protein sequence determined after fragmentation?
Multiple cleavage methods (e.g., trypsin, chymotrypsin, CNBr) are used to produce overlapping peptide fragments.
These fragments are compared and aligned by looking for common sequences across digests.
The overlaps help reconstruct the full amino acid sequence.
Today, this process is typically computerized.
What is mass spectrometry used for in protein sequencing?
Mass spectrometry measures the molecular masses of peptide fragments with high accuracy to identify proteins based on their primary sequence.
standard approach: digest protein with protease (like trypsin), then identify peptide based on their mass
What are the 4 main steps in mass spectrometry?
Ion generation
Ion acceleration (via electric/magnetic fields)
Ion separation in the mass analyzer
Ion detection
What is "time of flight" in mass spectrometry?
peptides are ionized and sprayed into a vacuum.
Larger ions move more slowly and reach the detector later.
The time it takes to reach the detector helps determine the mass.
How does mass spectrometry help identify proteins?
Mass spectrometry compares peptide fragments from a protein sample to known sequences from genome databases, enabling rapid and automated protein identification — a key part of proteomics (protein naming/identification)
What is secondary structure in proteins?
It refers to regular folding patterns in the polypeptide backbone stabilized by hydrogen bonds between C=O and N–H groups.
Two main types: α-helix and β-sheet (β-conformation).
These structures help maximize H-bonding and stabilize the protein's shape.
What is the α-helix?
A right-handed helical structure of the polypeptide backbone, stabilized by hydrogen bonds, first described by Linus Pauling.
Why is the peptide bond in an α-helix rigid?
The C–N bond has partial double bond character, making it planar and non-rotatable, which restricts backbone flexibility.
How are R groups arranged in an α-helix?
They are in the trans configuration and point outward from the helix, reducing steric clashes.
How are hydrogen bonds arranged in an α-helix?
Hydrogen bonds form between the C=O of residue i and the N–H of residue i+4, stabilizing the helix.
hydrogen bond every 4th amino acid
What types of residues disrupt α-helices?
Charged, bulky, or small residues (like Glu, Val, Pro, Gly) can disrupt the helix by hindering regular H-bonding or causing steric strain.
What is the α-helix dipole and why is it important?
The helix has a dipole moment (+ at N-terminus, – at C-terminus) that affects:
Helix orientation
Charged residue placement
Membrane interactions
How does electrostatic repulsion disrupt the α-helix?
Like-charged side chains (e.g. Lys–Lys or Glu–Glu) repel each other if placed too closely in the helix, destabilizing the structure.
⚠ However, opposite charges can sometimes stabilize the helix
How do bulky side chains affect α-helices?
Bulky residues like isoleucine cause steric clashes when too close together, disrupting the regular helical structure.
Why do small residues (e.g., Gly, Ser, Ala) disrupt α-helices?
They are flexible and may favor β-sheet formation instead of the rigid α-helix.
Why does Proline disrupt α-helices?
Its amide nitrogen can't form H-bonds (no H).
The ring forces it into a non-planar, rigid conformation.
Often found at the ends of helices, causing sharp bends.