Section 4 - Disorders of Primary Hemostasis (Vasculature & Platelets
1/54
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
55 Terms
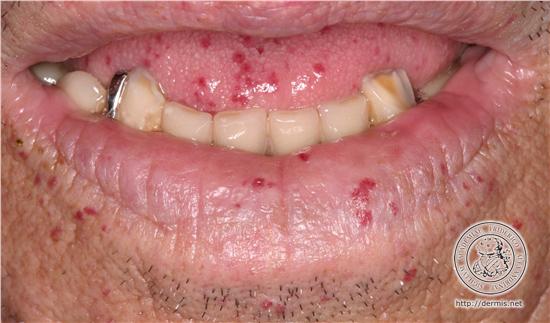
Hereditary Hemorrhagic Telangiectasia (HHT)
Impact on Coagulation
Pathophysiology: Single-layer endothelium with poor structural support → fragile vessels
Bleeding risk: Recurrent bleeding from vessel fragility, not platelet or factor defects
Telangiectasia: Blanching, superficial dilated vessels (vs. petechiae) → vascular origin
Clinical signs: Epistaxis, GI/UG bleeding, oral/mucocutaneous lesions; any organ
Coagulation labs: Normal PT, aPTT, platelet count/function; defect is structural
Hereditary Vascular Disorder
Ehlers-Danlos Syndrome (EDS)
Impact on Coagulation
Pathophysiology: Inherited collagen disorder; variable connective tissue fragility
Bleeding risk: Varies by type; vascular EDS may bleed easily, others do not
Clinical signs: Hypermobile joints, stretchy skin, easy bruising in some subtypes
Coagulation labs: Typically normal; bleeding due to tissue fragility, not platelet/coag defects
Hereditary Vascular Disorder

Allergic and Drug-Induced Purpuras
Impact on Coagulation
Pathophysiology: Autoimmune vascular injury or drug-induced anti-vessel wall antibodies
Bleeding risk: Increased due to immune-mediated vessel damage
Telangiectasia: Absent; lesions appear as non-blanching purpura or petechiae
Clinical signs: Palpable purpura, often in dependent areas; may be drug- or autoimmune-triggered
Coagulation labs: Typically normal; bleeding from vascular inflammation, not factor deficiency
Acquired Vascular Disorder
Henoch–Schönlein Purpura (HSP)
Impact on Coagulation
Pathophysiology: Post–upper respiratory infection; IgA immune complexes deposit in small vessels
Bleeding risk: Immune-mediated vasculitis causes vessel fragility and organ bleeding
Clinical signs: Typically affects children; palpable purpura, joint pain, abdominal pain, hematuria/proteinuria
Progression: Small percentage may develop chronic renal disease
Coagulation labs: Usually normal; bleeding due to vascular inflammation, not factor or platelet defects
Acquired Vascular Disorder
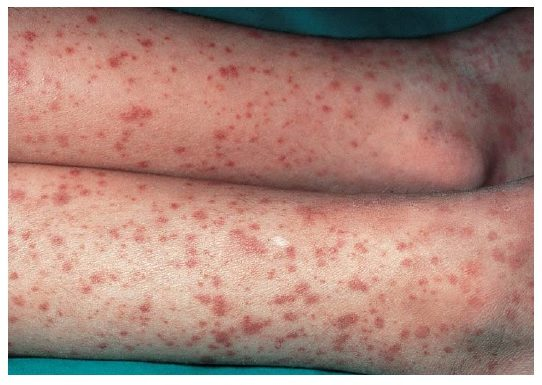
Scurvy
Impact on Coagulation
Pathophysiology: Vitamin C deficiency → impaired collagen synthesis → fragile capillary walls
Bleeding risk: High due to structural vessel weakness, especially in skin and mucosa
Clinical signs: Easy bruising, perifollicular hemorrhages, bleeding gums, poor wound healing
Coagulation labs: Normal; bleeding from defective vascular support, not platelet/coag factor issues
Acquired Vascular Disorder
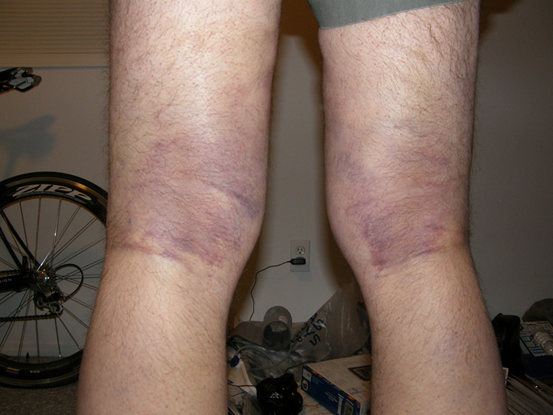
Senile Purpura
Impact on Coagulation
Pathophysiology: Age-related loss of collagen and subcutaneous fat → reduced vessel support
Bleeding risk: Mild to moderate; bruising from minor trauma due to capillary fragility
Clinical signs: Elderly, especially on forearms and hands; dark, irregular purpura
Coagulation labs: Normal; bleeding is mechanical, not hematologic
Acquired Vascular Disorder
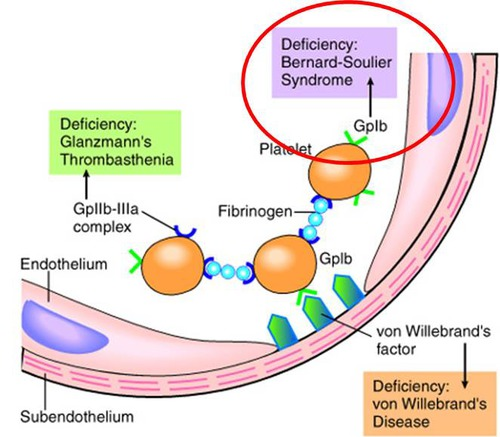
Bernard–Soulier Syndrome
Defect in Platelet Adhesion
Primary defect: Absent or dysfunctional glycoprotein Ib (GPIb) on platelet surface
Mechanism: Platelets cannot bind von Willebrand factor (vWF) → impaired adhesion to subendothelium
Lab findings:
↑ Bleeding time/PFA
↓ Platelet count, large platelets
Normal aggregation with all agents except ristocetin
Bernard–Soulier Syndrome
Laboratory Diagnosis
Platelet count: ↓ Mild thrombocytopenia
Platelet morphology: Giant platelets seen on peripheral smear
Bleeding time / PFA-100: Prolonged (defective adhesion)
Platelet aggregation studies:
Normal with ADP, collagen, epinephrine
Absent response to ristocetin (does not correct with vWF addition)
Flow cytometry: Shows ↓ or absent GPIb expression
von Willebrand Disease (vWD)
General Defect Overview
Inherited bleeding disorder due to deficiency or dysfunction of vWF
vWF is critical for platelet adhesion and carries factor VIII
Affects both primary and secondary hemostasis
Stored in alpha granules of plts & present in plasma
vWD Type I – Classic
Most common type
Quantitative deficiency (↓ all multimers)
vWF structure is normal
Labs: ↓ vWF, ± ↓ FVIII:C, Prolonged APTT possible
vWD Type II – Qualitative Defects
Subtypes: abnormal structure or function of vWF
2A: ↓ HMW multimers, impaired platelet binding
2B: ↑ affinity for GPIb → platelet clumping & ↓ platelet count
2M: ↓ platelet binding (normal multimers)
2N: ↓ FVIII binding (mimics mild hemophilia A)
APTT: often prolonged, FVIII:C low or normal
vWD Type III – Severe
Near absence of all vWF multimers
Severe bleeding, very low FVIII:C
APTT: prolonged, bleeding resembles hemophilia A
Platelet-Type vWD
Gain-of-function defect in platelet GPIb
Platelets bind vWF spontaneously → thrombocytopenia
Multimers are normal, but appear consumed
vWD Type I – Lab Findings
PT: Normal
APTT: ↑ (due to ↓ factor VIII)
PFA: ↑ (no longer routinely used)
Other tests: Needed to distinguish subtypes (e.g., vWF antigen, activity, multimer analysis)
vWF:Ag – Antigen Level
Purpose/Principle: Measures quantity of von Willebrand factor (vWF) protein in plasma.
Diagnostic Utility: Distinguishes quantitative (Type I, III) vs. qualitative (Type II) defects.
Result Interpretation:
↓ in Type I (partial deficiency)
≈ 0 in Type III (near-total absence)
Normal or slightly ↓ in most Type II subtypes
Contextual Notes:
Does not assess vWF function
Measured via ELISA, latex agglutination, chemiluminescence
vWF:RCo – Ristocetin Cofactor Activity
Purpose/Principle: Measures functional activity of vWF by its ability to agglutinate donor platelets in the presence of ristocetin.
Diagnostic Utility: Confirms functional impairment of vWF.
Result Interpretation:
↓ in all types except possibly 2N
Disproportionately lower than Ag in Type II
Corrects with normal plasma (Type I), not in Type II or III
Contextual Notes:
Most important functional test
Bernard-Soulier will have normal RCo with abnormal platelets
vWF Multimer Analysis
Purpose/Principle: Separates vWF multimers by size using agarose gel electrophoresis or immunoelectrophoresis.
Diagnostic Utility: Identifies structural abnormalities in vWF multimers.
Result Interpretation:
↓ in all multimers: Type I
↓ or absent HMW: Types 2A, 2B
Normal multimers: Type 2N, 2M
Absent or nearly absent multimers: Type III
Contextual Notes:
Useful for subtyping Type II disorders
Not routinely done in all labs
Platelet Aggregation Patterns: vWD vs Bernard-Soulier vs Glanzmann
von Willebrand Disease
Normal aggregation w/ all agonists except ristocetin
Ristocetin abnormal (corrects w/ normal plasma)
Bernard-Soulier Syndrome
Normal aggregation w/ all agonists except ristocetin
No correction with normal plasma (due to GPIb defect)
Glanzmann Thrombasthenia
Absent aggregation with all agonists except ristocetin
Ristocetin: normal (does not require GPIIb/IIIa)
Key Mechanisms:
vWD: ↓ vWF (defective ligand)
Bernard-Soulier: ↓ GPIb (defective receptor)
Glanzmann: ↓ GPIIb/IIIa (defective fibrinogen receptor for aggregation)
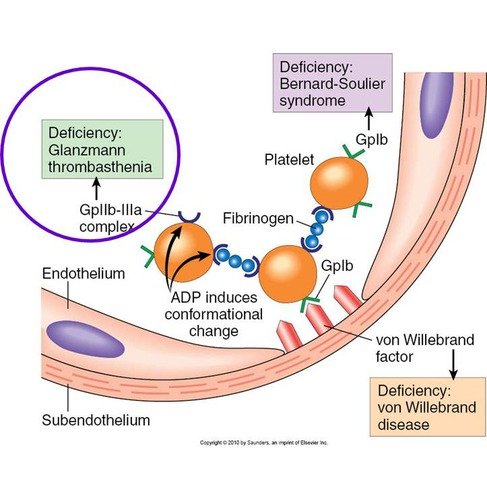
Glanzmann’s Thrombasthenia – Defect
Defect:
Platelets lack or have dysfunctional GPIIb/IIIa complex (integrin αIIbβ3)
Prevents fibrinogen bridging between platelets
Results in impaired platelet aggregation, despite normal adhesion
Clinical Type:
Most common inherited aggregation disorder
Genetics:
Autosomal recessive
Glanzmann’s Thrombasthenia – Laboratory Diagnosis
Platelet Function Analyzer (PFA):
↑ Prolonged — due to defective aggregation.
Prothrombin Time (PT):
Normal — coagulation cascade unaffected.
Activated Partial Thromboplastin Time (APTT):
Normal — intrinsic pathway intact.
Platelet Count:
Normal — platelet number unaffected.
Platelet Aggregation Studies:
Abnormal with all agonists except ristocetin
Normal aggregation with ristocetin — ristocetin tests adhesion (via vWF/GPIb), not aggregation.
von Willebrand Disease – DDAVP Therapy
Mechanism:
DDAVP (1-deamino-8-d-arginine vasopressin) stimulates endothelial cells to release stored von Willebrand factor (vWF) from Weibel–Palade bodies, increasing circulating vWF levels.
Best Use:
Effective for Type 1 vWD (mild to moderate forms)
Not recommended for Type 2B (risk of thrombocytopenia) or Type 3 (vWF absent)
Note:
Not a replacement for vWF; boosts endogenous stores.
Bleeding in Plasma Cell Dyscrasias (MM/Waldenstrom’s)
Mechanism:
Excess IgG or IgM coats:
Platelet membranes → impairs aggregation
Collagen fibers → interferes with platelet adhesion
Result:
→ Impaired primary hemostasis
→ Prolonged bleeding time/PFA despite normal platelet count and coag factors
Contextual Note:
This mimics platelet adhesion disorders (like vWD or Bernard–Soulier) but is due to paraprotein interference, not intrinsic platelet defects.
Effect of Aspirin and Ibuprofen on Platelet Function
Aspirin irreversibly inhibits cyclooxygenase (COX), blocking thromboxane A₂ (TXA₂) synthesis → impaired platelet aggregation
• Ibuprofen reversibly inhibits COX, causing transient platelet dysfunction (less potent than aspirin)
• Aspirin Resistance:
• Up to 22% of patients have reduced aspirin response
• Leads to higher risk of MI and stroke
• Detect with platelet aggregation studies—arachidonic acid curve is most sensitive
• Aggregation studies can also track aspirin use/effectiveness
Platelet Antibodies
• IgG antibodies target platelet membrane receptors (e.g., GPIIb/IIIa, GPIb/IX)
• Lead to platelet destruction by splenic macrophages → ↓ platelet count
• Some antibodies block receptor function → impaired aggregation or adhesion
• Result: thrombocytopenia and/or platelet dysfunction → mucocutaneous bleeding
Hermansky-Pudlak Syndrome
Dense granule deficiency
• Dilated canalicular system on platelet surface
• Electron microscopy shows "Swiss cheese platelets"
• Defective release reaction → abnormal aggregation
Gray Platelet Syndrome
Marked ↓ in platelet alpha granules
• Platelets appear hypogranular or agranular
• Impairs platelet aggregation and clot formation
Wiskott-Aldrich Syndrome
• Microplatelets
• ↓ alpha and dense granules
• Platelet sequestration → ↓ platelet count
• Immunodeficiency: recurrent infections, ↓ serum IgM
Chediak-Higashi Syndrome
Platelets lack normal dense granules
• Giant, abnormally formed lysosomal granules in WBCs
• Impairs platelet function and immune cell activity
Storage Pool Diseases (SPD)
• Deficiency of platelet dense or alpha granules
• Impaired release reaction
• Results in abnormal platelet aggregation
Thrombocytopenia
Low platelet count
• Caused by:
– Decreased production → IPF (Immature Platelet Fraction) low
– Increased destruction → IPF high (↑ immature “retic” platelets)
Thrombocytosis
Reactive: due to blood loss, surgery, childbirth, necrosis, inflammation, or exercise
• Platelets ↑ but function normally
• Myeloproliferative: seen in PV, CML, primary myelofibrosis, ET
Essential Thrombocythemia (ET)
• PLT >600K, often >1 million
• Bone marrow: ↑ megakaryocytes
• Peripheral smear: giant, bizarre, and small platelets; large aggregates
• Spontaneous aggregation
• Platelet dysfunction → ↑ risk of thrombosis and bleeding
Four causes of thrombocytopenia
• Decreased production (e.g. Fanconi syndrome, May-Hegglin, marrow failure)
• Increased destruction (e.g. ITP, drug-induced antibodies)
• Platelet sequestration (e.g. Wiskott-Aldrich)
• Ineffective thrombopoiesis (e.g. May-Hegglin: large, bizarre plts, Dohle-like bodies)
3 Causes of congenital platelet hypoplasia
Fanconi syndrome – congenital aplasia
• Wiskott-Aldrich – platelet sequestration, small plts, ↑ apoptosis signaling
• May-Hegglin – ineffective thrombopoiesis, giant plts, Dohle-like bodies
List 9 Causes of acquired platelet hypoplasia
Irradiation
Drugs
Ethanol
Early aplastic anemia
Pernicious anemia and folate deficiency (DNA synthesis defects impair megakaryocyte function)
Viruses
Bacterial infections
Malignancies
Myelodysplastic syndromes (may also cause increased platelet destruction)
Idiopathic Thrombocytopenic Purpura (ITP)
Autoimmune platelet destruction via anti-platelet antibodies
• Most common mechanism: IgG-mediated opsonization → splenic phagocytosis
• Isolated thrombocytopenia (<100,000) without another underlying cause
• Can be acute (self-limited) or chronic (relapsing/persistent)
Laboratory Findings in ITP
• PLT count often < 20,000
• Peripheral blood: large platelets with variable size/shape
• Bone marrow: megakaryocyte hyperplasia
• IPF: low to very low
• Bleeding time or PFA: prolonged due to low PLT
• Deficient clot retraction
Acute vs Chronic ITP
Acute ITP
• Most common in children
• Often follows a viral infection (2–21 days prior)
• May occur post-immunization
• Usually self-limited
• 80% spontaneous remission
Chronic ITP
• Most common in adults (20–50 yrs)
• Fluctuating disease course
• Bleeding episodes may last days or weeks
• Spontaneous remissions are rare
• May be early manifestation of AIDS
Drug-Induced Immune Thrombocytopenia
Caused by formation of drug-related antibodies that affect platelets
True autoantibody may form that targets platelets even without drug presence
Hapten mechanism: drug binds platelet → antibody forms to drug-platelet complex
Drug-antibody complex mechanism: Ab-drug complex attaches to platelet
Common drugs: Heparin, Quinine, Quinidine
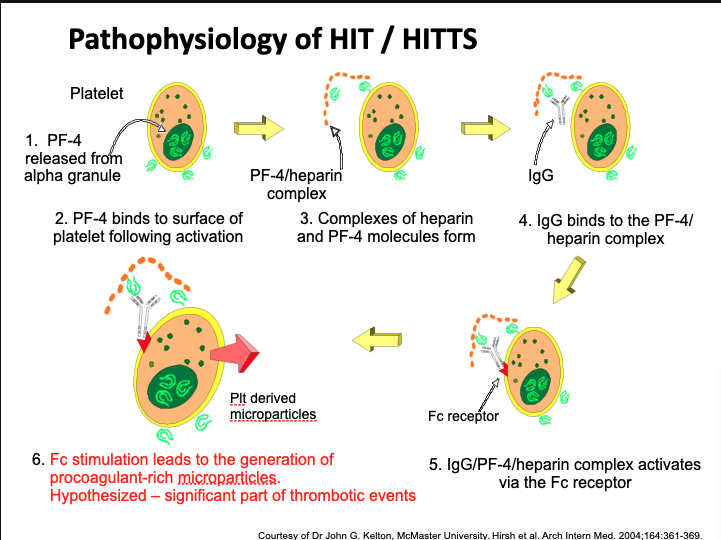
Heparin-Induced Thrombocytopenia (HIT) Mechanism
PF4 is released from platelet alpha granules upon activation
Heparin binds to PF4 → forms PF4/heparin complex
IgG antibody binds to this complex
Fc portion of IgG binds Fc receptor on platelet
This activates platelets → aggregation, granule release
Results in thrombocytopenia and procoagulant microparticles
These microparticles are thought to drive thrombosis
Removal of heparin leads to improvement
Heparin-Associated Thrombocytopenia (HAT)
Direct non-immune activation of platelets by heparin
Develops within 1–3 days of treatment
PLT rarely falls below 100 × 10³/mm³
Benign and transient
No thrombosis risk
Not antibody mediated
Heparin-Induced Thrombocytopenia (HIT)
Immune-mediated drop in PLT count (~30–50%)
Develops 5–10 days after heparin initiation
Antibody to PF4/heparin complex forms
PLT activation via Fc receptor causes consumption and thrombosis
Remove heparin → PLT count rebounds
May lead to severe thrombocytopenia and hypercoagulable state
HIT Type II (HITTS)
Subset of HIT with thrombotic complications
PLT can fall as low as 20 × 10³/mm³
PF4/heparin complex + IgG → strong platelet activation
Produces procoagulant microparticles
May occlude microvasculature → thrombosis
Pathophysiology not fully understood
HIT Ab may bind heparan on endothelium → TF expression
Patients may develop only thrombocytopenia or also thrombosis
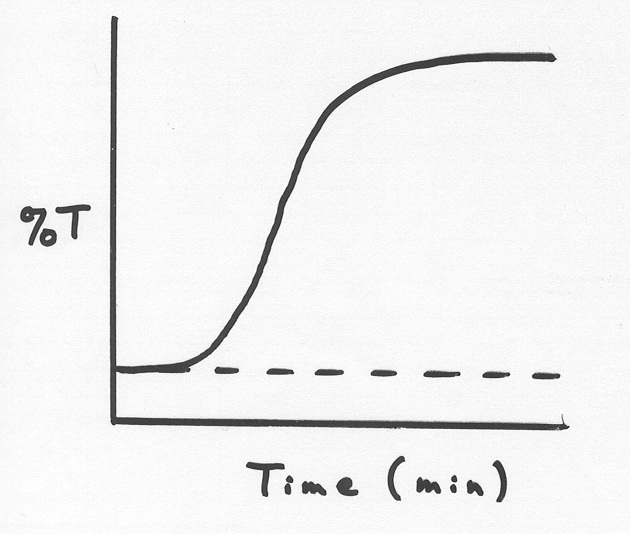
Heparin Induced Platelet Aggregation (HIPA) Test
Detects heparin-induced antibodies in patient plasma
Patient must be off heparin for ≥ 8 hours
Uses platelet aggregometer
Mix: normal donor platelets + patient platelet-poor plasma + heparin dilutions
If Ab present, platelets aggregate (measure %T – transmission)
Negative control: donor platelets + patient PPP + saline
Interpretation:
< 20%T → unable to demonstrate (not reported as “negative”)
20–40%T → weak positive
40%T → positive
Neonatal alloimmune thrombocytopenia (NAIT)
Caused by maternal IgG antibodies against fetal platelet antigens (usually HPA-1a)
Mother lacks antigen; fetus inherits it from father
Fetal platelets enter maternal circulation → antibody formation
Maternal antibodies cross placenta and destroy fetal platelets
Newborn appears healthy at birth → develops petechiae/purpura
Mechanism is similar to HDFN
Post-transfusion purpura (PTP)
Alloimmune thrombocytopenia developing 3–12 days post transfusion
Caused by antibodies (usually anti-HPA-1a) against platelet antigens not present in the recipient
Initial sensitization from pregnancy or prior transfusion
Re-exposure → anamnestic response → rapid thrombocytopenia
Can also impact future pregnancies if antibody crosses placenta
Confirm via antibody ID (e.g., ELISA) and antigen phenotype (e.g., Flow)
ITP in Pregnancy
Autoantibodies target maternal platelets → thrombocytopenia
IgG autoantibodies cross placenta → fetal platelet destruction
Risk of neonatal thrombocytopenia and intracranial hemorrhage
Pregnant patient may require treatment (e.g., IVIG, steroids) to maintain safe PLT count
Platelet transfusion typically ineffective unless combined with immune suppression
Context: Differentiate ITP from incidental or non-immune causes (e.g., HELLP, TTP, HUS) — ITP is immune-mediated and can affect both mom and fetus.
HELLP Syndrome Overview
Variant of preeclampsia w/ Hemolysis, Elevated Liver enzymes, Low Platelet count
Labs:
↓ Hgb/Hct, ↓ haptoglobin, ↑ LDH, ↑ bilirubin (hemolysis)
↑ AST, ↑ ALT (hepatic injury)
PLT < 200,000/mm³ (thrombocytopenia)
Schistocytes on smear
Onset: 27–36 wks (can be postpartum)
Tx: Delivery, transfusion, corticosteroids (for lung maturity)
HELLP Pathophysiolog
Endothelial damage + vasospasm → platelet activation
Thromboxane A₂ → vasoconstriction
Liver microvascular injury → ↑ AST/ALT
Risk: DIC due to systemic clotting
HELLP Syndrome Symptoms
Hypertension, edema
RUQ/epigastric pain, malaise, nausea
Proteinuria, oliguria (↓ urination)
Visual/cerebral disturbances
Symptoms overlap w/ preeclampsia but often more severe
Thrombotic Thrombocytopenic Purpura (TTP)
Pentad: thrombocytopenia, microangiopathic hemolytic anemia (MAHA), neurological changes, renal dysfunction, fever
Labs: ↓ platelets, schistocytes, ↑ LDH, ↑ bilirubin, ↓ haptoglobin
Platelet function normal on aggregometry
Hemolytic Uremic Syndrome (HUS)
Triad: MAHA, thrombocytopenia, acute renal failure
Often post-E. coli O157:H7 infection (children 6 mo–4 yrs)
Labs: schistocytes, hematuria, proteinuria, casts
Minimal/no neurologic symptoms (vs. TTP)
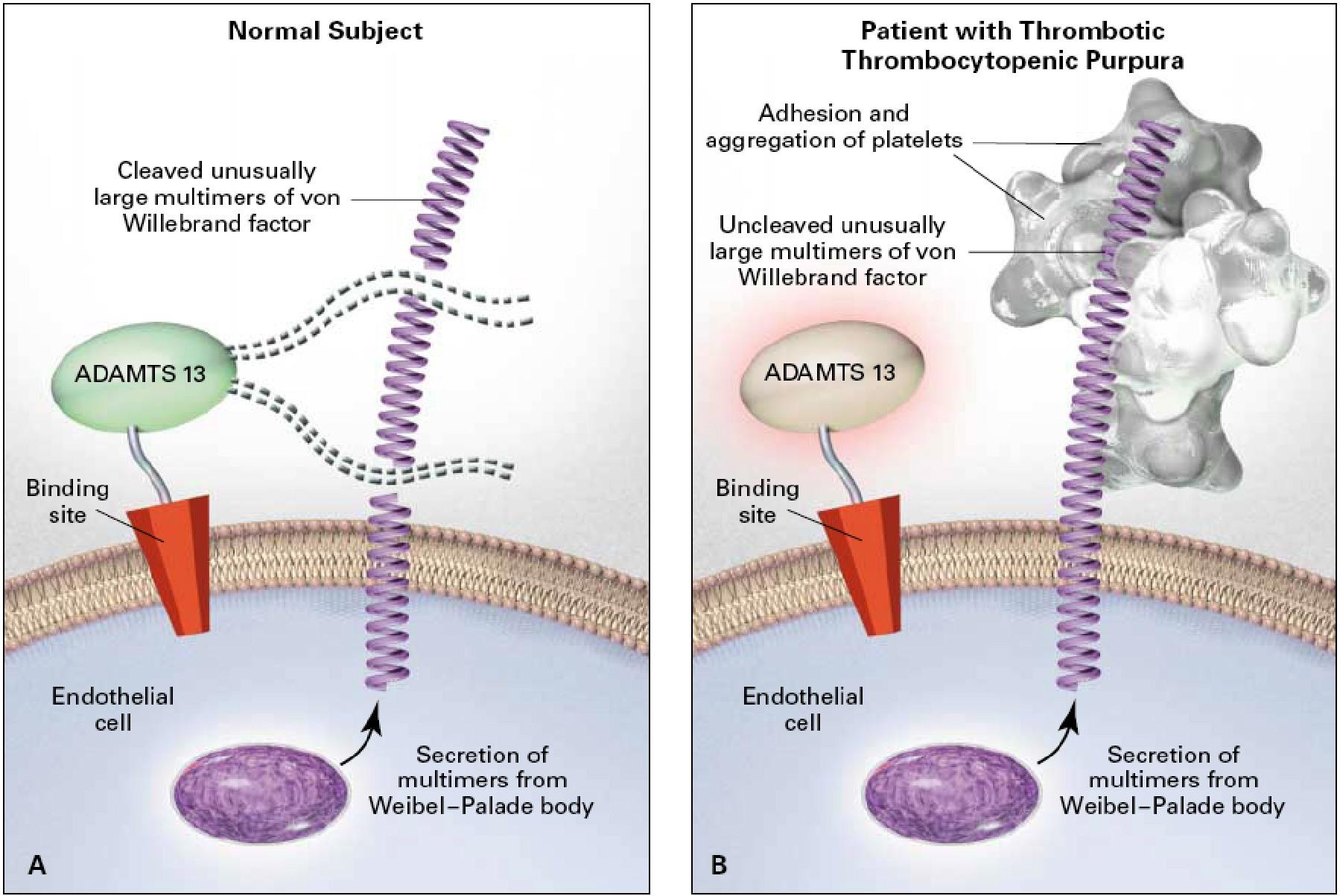
TTP Mechanism
↓/inhibited ADAMTS13 → uncleaved ultra-large vWF multimers
→ platelet microthrombi in small vessels
Acquired: anti-ADAMTS13 autoantibody
Congenital: inherited ADAMTS13 deficiency
TTP vs. HUS
Both: MAHA + thrombocytopenia
TTP: prominent neuro symptoms, mild renal impairment, adults
HUS: severe renal failure, minimal/no neuro signs, children (esp. post-E. coli)
The hallmark finding of Essential Thrombocythemia (ET):
Sustained thrombocytosis
Platelet count >600,000/µL, often >1 million/µL
No reactive cause (e.g., inflammation, iron deficiency)
Bone marrow shows increased megakaryocytes, often large and atypical