Pantothenic Acid, Biotin, Vit B6, Folate, and Vit B12
1/85
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
86 Terms
Pantothenate Acid
Ionized form of pantothenic acid
Pantothenic acid
precursor of Coenzyme A (CoA)
Biosynthesis of CoA uses pantothenate, ATP, and cysteine as substrates
There are 5 enzymatic steps in CoA biosynthesis
Pantothenate —> pantothenate kinase ( along with Mg2+ and ATP to ADP)—> 4’ phosphopantothenate
This is also the rate-limiting steps
Pantothenic Acid Sources
virtually all foods
excellent sources: animal organs (liver and kidney), fish, shellfish, milk products, eggs, avocados, legumes, mushrooms, and sweet potatoes
Produced by bacteria in the colon
Supplements
Calcium or sodium pantothenate, or panthenol
Multivitamin: 10 mg
Single-nutrients: 5-500 mg
Pantothenic Acid Stability
Stable when dry and in the solution with a neutral pH
Destroyed
heating and freezing
acidic and alkaline solutions
refining of grains, freezing, and canning lowers pantothenic acid content up to 75%
How is Pantothenic Acid found
present in food in free and bound forms
85% of PA is found as a component of CoA or 4’ phosphopantetheine
Digestion of CoA
CoA → hydrolyzed by pyrophatase → 4’-phosphopantethine → phosphatase → pantetheine → pantotheinase → pantothenic acid
Pantothenic Acid Absorption
is absorbed primarily in the jejunum
~50% of PA is absorbed (range: 40-61%)
High [ ] = Passive diffusion
Low [ ] = absorbed by Na-dependent shared multivitamin transporter (SMVT)
Panthenol (supplements) - diffusion and converted to PA
~10% absorbed with supplement if ingesting 10 x AI (50 mg or more)
Pantothenic Acid Transport, Uptake, and Storage
Transport
Transported freely in blood from the small intestine enterocyte to liver then the rest of the body
Primarily within RBCs
[PA] = 30 - 60 micrograms/dL
Uptake
RBC and brain: passive diffusion
Other tissues: SMVT (in intestine)
Storage
Present within cells as PA and 4’- phosphospantothenic acid
Most PA is used to synthesize CoA, which is found in all tissues
high [ ] in liver, adrenal gland, kidneys, brain, heart
Majority of CoA is located in the mitochondria
Pantothenic Acid: Functions and Mechanisms of Actions (CHO)
Acetic acid → Acetyl CoA, Malonic acid → Malonyl CoA, Propionic acid → Propionyl CoA, Methylmalonic acid → Methylmalonyl CoA, Succinic acid → Succinyl CoA
CoA is involved in nutrient metabolism: CHO, fat and protein
CHO
Oxidative decarboxylation of pyruvate to acetyl CoA using pyruvate dehydrogenase complex along with NAD+ to NADH
glycolysis
Oxidative decarboxylation of A-keto-glutarate to succinyl CoA using using NAD+ to NADH, FAD, thiamin pyrophosphate, and Lipoic acid
Kreb’s cycle / TCA cycle
Pantothenic Acid: Functions and Mechanisms of Actions (Lipid Metabolism)
Synthesis
Cholesterol, ketone bodies, fatty acids, phospholipids, steroid hormones, and sphingolipids
Cholesterol and ketone bodies
2 acetyl-CoA → Acetyl-CoA acetyltransferase → acetoacetyl CoA → HMG-CoA synthase 1 → 3-hydroxy-3-methylglutaryl-CoA → (Rate Limiting Step, needs NADPH as a coenzyme) HMG-CoA reductase → mevalonate → cholesterol
statins inhibit the RLS to decrease production of cholesterol
Keto acid is used to assess ketosis because it has a longer half life
glutaryl Co A is important for making ketine bodies
Pantothenic Acid: Functions and Mechanisms of Actions (FA synthesis)
Condensation of acetyl CoA with activated CO2 to form malonyl-CoA
needed for the repetitive addition of 2 carbons on fatty acid synthesis
Malonyl-CoA binds with ACP and the condensing enzyme combines with acetyl CoA
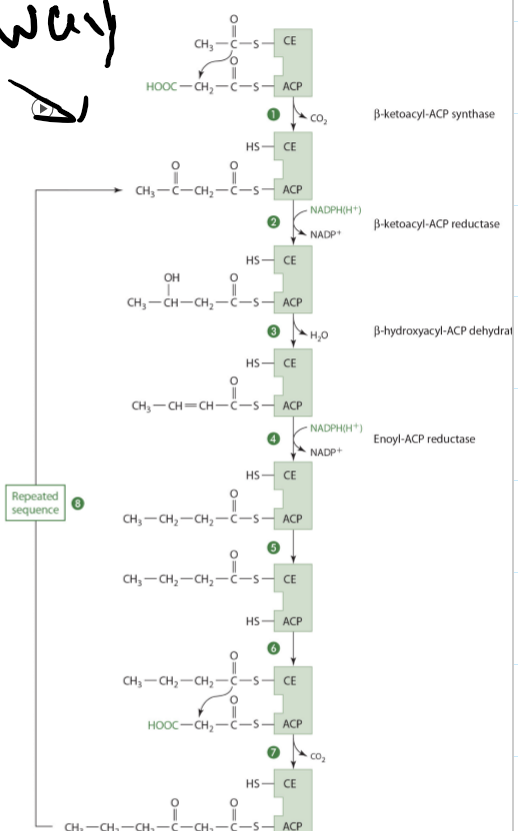
Pantothenic Acid: Functions and Mechanisms of Actions (Acylation and Acetylation or proteins, sugars, and drugs)
Acylation and Acetylations
Post transitional modification of proteins
Acylation: affects protein function, activity, and location
Acetylation: affects enzyme activity and function
when a acetyl is added to a substrate
Acetylation
Extensive with proteins - Liver
prolongs the ½ life of proteins
Usually occurs on lysine residues
acetylated aminosugars can provide recognition sites on cell surfaces or direct proteins for membrane functions
choline is a acetylated to the neurotransmitter acetylcholine
Pantothenic Acid: Functions and Mechanisms of Actions (Folate)
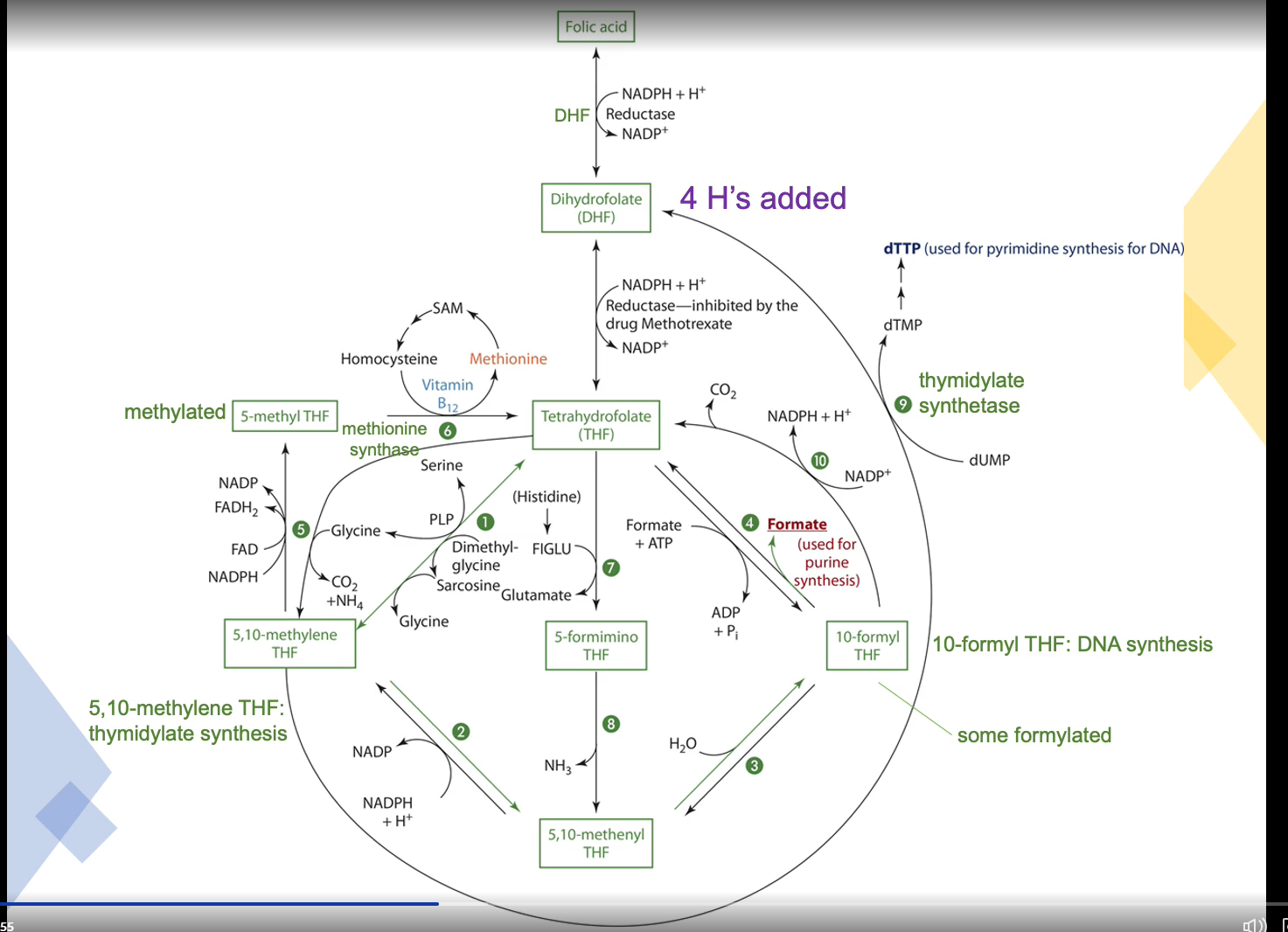
10-formyl tetrahydrofolate dehydrogenase (#10)
Requires 4’-phosphopantethiene for activity
NADP is the oxidizing agent and CoA is needed
Patonthenic Acid Excretion and Adequate Intake (AI)
excreted as pantothenate or pantothenic acid
AI
Adults: 5 mg/day
Pregnancy 6 mg/day
Lactation 7 mg/day
Patothenic Acid Deficinecy
Unlikely'; occur multiple deficiencies
Burning Foot Syndrome (rare)
numbness of toes and burning sensation in feet and nerve inflammation
Exacerbated by warmth and diminished with cold
Vomiting, fatigue, muscle weakness, restlessness, irritability
Treated with calcium or sodium, pantothenic
At risk of deficiency
alcoholism (low intake/increased excretion), diabetes (increased excretion) inflammatory bowel disease ( decreased absorption)
Pantothenic Acid Toxicity
No UL
No toxic level
10 g daily for up to 6 weeks - no side effects
> 15 to 20 g associated with mild intestinal distress, including diarrhea
Assessment of nutriture
blood concentration <100 mg/dL may reflect low dietary intake
do not correlate well with changes in OA intake and status
Urinary pantothenate excretion better indicator of status
<1mg/day indications poor PA staus
Biotin Sources
Commonly found free or bound to proteins in liver, soybeans, egg yolk, legumes and nuts
there is a glycoprotein in raw egg called avidin that binds biotin. It’s an irreversible bond, unless it is cooked
Biotin Digestion
Biotin is bound to protein in food, must be removed from the protein bond
pepsin in your stomach and proteases either on the brush border or from your pancreas will break the bonds, and you end up with free biotin or biocytin (biotin bound to lysine).
Biocytin can be further digested to free biotin in the presence of the enzyme biotinidase to biotin and lysine
there is a genetic mutation that leads to biotinidase deficiency
symptoms would be lethargy, hypotonia, seizures, ataxia, dermatitis, and alopecia
Biotin Absorption
absorbed primarily as free in the proximal small intestine
duodenum is the preferred area of absorption
In physiological intakes biotin will cross the brush border of the small intestine or the colonic cell membrane with a carrier
carrier is a sodium dependent transporter (SMVT) the same transporter for pantothenic acid
High levels of biotin suppresses the transcription of the transporter gene
Undigested biocytin may be absorbed by peptide carriers
alcohol decrease absorption
biotin synthesized by colonic bacteria is absorbed in proximal and transverse colon (SMVT)
transport across the basolateral membrane is carrier-mediated
100% of oral free biotin is absorbed
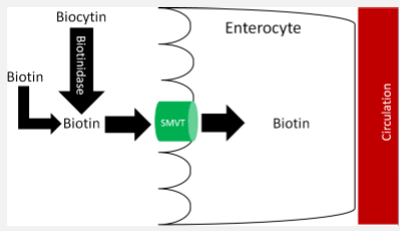
Biotin Transport, Uptake, and Storage
Transport
In plasma 80% is free biotin, 20% is bound to proteins (albumin, globulins, and biotinidase)
Blood (biotin): 200 759 pg/mL
Uptake
Liver and probably other tissues: SMVT and monocarboxylate transporter (MCT) I
Storage
Small quantities stored in muscle, liver, and brain
Biotin AI (Adequate Intake) and UL
Adults: 30 micrograms/day
Pregnancy: 30
Lactation: 35
UL: none
up to 200 mg without side effects
At risk for biotin deficiency
Those consuming excess raw egg whites
Those with GI disorders
People who consume excess alcohol
Pregnant and lactating women
Those on anticonvulsant drug
Biotin Assesment of Nutriture
Decreased urinary biotin excretion (< 6 micrograms/day) is a sensitive and early indicator of biotion deficiency
a diet devoid of biotin can decrease biotin in plasma and urine in ~2-4 weeks
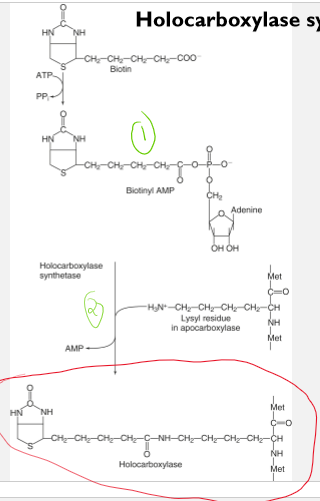
Biotin as a coenzyme in a holocarboxylase synthetase reaction
biotin is a covalently attached as a coenzyme to form several holoenzymes
holoenzymes function as carboxylases
the carboxylases facilitate the incorporations of a carboxyl group into a substrate
Each holocarboxylase is formed in a reaction catalyzed in two sequential steps by holocarboxylase synthetase
formation of biotinyl-AMP from biotin and ATP
Formation of an amide bond between the carboxyl group of biotin and the e-amino group on a specific lysine residue in each apocarboxylase with the release of AMP
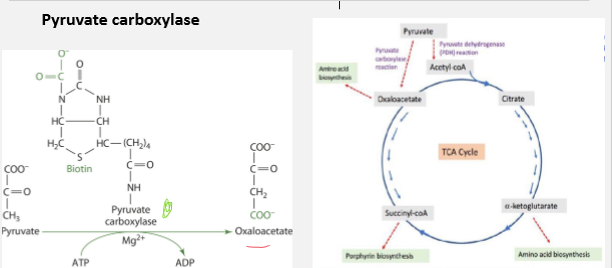
Biotin and Pyruvate Carboxylase
Role: Converts pyruvate t oxaloacetate
Significance: Replenishes oxaloacetate for TCA cycle. Necessary for glucogenesis. Important for its regulatory function
Decrease in levels of ATP promotes TCA cycle
An increase in levels promotes glycogenesis
Biotin and Acetyl-Coa Carboxylase
Role: Forms malonyl-CoA from acetate
Signifigance: Commits acetate units to fatty acid synthesis to start the synthesis
Rxn needs ATP and biotin
Acetyl-Coa Carboxylase is considered and regulatory and rate limiting enzyme
Malonyl CoA will hook up with a acyl carrier protein and acetyl CoA will hook up with a condensing enzyme to create the complex that is important for fatty acid synthesis
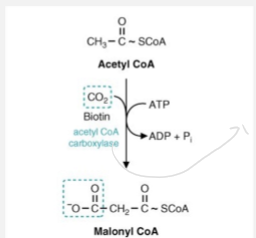
Biotin and Propionyl-CoA Carboxylase
Role: Converts propionyl-CoA to methylmalonyl-CoA
Significance: Provides mechanism for metabolism of some amino acids and odd-chain fatty acids to become Propionyl CoA
Important for converting propionyl CoA into Methylmalonyl CoA
the catabolism of Threonine, Methionine, Isoleucine, and Valine lead to the product of propionyl CoA
Beta oxidation of fatty acids involves pulling off 2 C at a time. So when when there is an even # of C the end product is Acetyl CoA
In order to go to Methylmalonyl CoA requires an enzyme, biotin, magnesium, and ATP as a cofactor
Methylmalonyl is then converted to Succinyl CoA to be used in the Kreb’s cycle by mutase and B12
Biotin and B-methylcrotonyl-CoA Carboxylase
Role: Converts B-methylcrotonyl-CoA to methylglutaconyl-CoA
Significance: Allows catabolism of leucine and certain isoprenoid compounds
during catabolism of leucine B-methylcrotonyl-CoA carboxylase
B-Methylcrotonyl-CoA is carboxylated in the presence of ATP, Mg as a cofactor, and enzyme that is biotin dependent
There can be a deficiency and the beta methylpropyl choice carboxylase or there could be a decline in activity.
Therefore this beta methyl crotonyl COA is shunted to other pathways and there are compounds that are made that you could measure in urine that would tell you that this carboxylation reaction is not occurring.
Vitamin B6
exist as 6 vitamers Pyridoxine (PN; Alcohol form), Pyridoxal (PL; aldehyde form), Pyridoxamine (PM; amine form), Pyridoxine Phosphate (PNP), Pyridoxal Phosphate (PLP) Pyridoxamine Phosphate (PMP)
PMP is the most active enzyme
Vitamin B6 Sources
Plant Foods
May be present as a glucoside
Vegetables (potatoes), some fruits (bananas), and nuts as well fortified cereals
Animal products: PL, PLP, PM, PMP
Beef, fish, pork, and chicken
Fortified foods and supplements: pyridoxine hydrochloride
Multi: 2 mg
single 2-300 mg
Fairly stable with cooking
Lost in prolonged heat (sterilizing and canning)
lost in refining/milling of grains (not added back)
lost in storage
Vitamin B6 Digestion
PLP, PNP, PMP must be dephosphorylated to PL, PN, PM
Alkaline phosphatase, a zinc dependent enzyme at brush border and other intestinal phosphatases remove phosphates from PLP, PNP, PMP to make the free forms
Vitamin B6 Absorption
PL, PN, and PM (the free forms) are absorbed in jejunum by passive diffusion
can be absorbed anywhere in GI tract, just prefers the jejunum
glucosidases can dephosphorylate the vitamers to the free forms
75% (61-97%range) absorbed
little metabolism in intestinal cells
Sometimes pyridoxine can be converted to pyridoxine phosphate or to pyridoxal phosphate
PN, PL, and PM are released into portal blood and then transported to the liver
The liver mainly absorbs and metabolizes B6
Vitamin B6 Transport
PLP and PL are major forms in systemic blood (75-90%)
Most PLP (and other vitamers) transported bound to albumin
Liver takes up newly absorbed B6 by passive diffusion
Oxidase is FMN dependent
Steps
PL and PLP are released from the liver to other tissues
PLP is hydrolyzed to PL in blood for cellular uptake
PL is then phosphorylated by an intracellular kinase
free forms are taken by tissues
PLP then binds a protein to prevent its degeneration
functions as a coenzyme
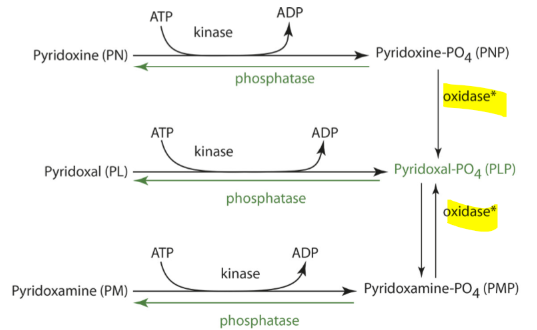
Vitamin B6 Storage
5-10% in liver
75-80% in muscles
As PLP bound tp glycogen phosphorylase
phosphorylation of B6 traps it in the cell
Binding to protein prevents hydrolysis by phosphatases
important for adequate amount of B6 to be available
Other tissues with B6: brain, kidney, spleen
typically, phosphorylated and bound to enzymes or PLP binding proteins in the cytosol and mitochondria
Vitamin B6 and Non Coenzyme role (Non PLP role)
Gene expression
Modulates steroid hormone binding and transcription factor binding to regulatory DNA regions
Vitamin B6 and Coenzymes (primarily as PLP)
Transamination
Deamination
Decarboxylation
Transulfhydration
Heme synthesis
Tryptophan → niacin
Glycogen degradation
Transelenation
Folate
PLP as a coenzyme and reactions will attached by a shift based linkage to the amino group on the enzyme’s active site.
Is usually lysine
A shift base is a compound where there’s the presence of a double bond linking C and N
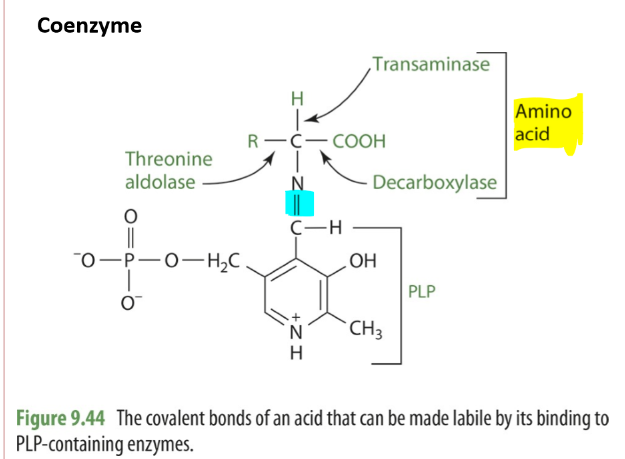
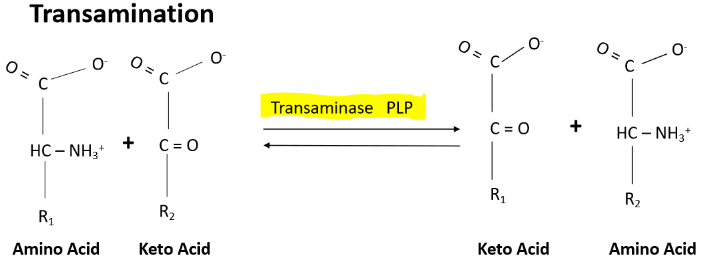
B6 and Transamination
One of the main roles of vitamin B6 is when you have a transfer of an amino group from one amino acid to an alpha ketoacid.
This makes a new alpha ketoacid and a new amino acid
The enzyme that is necessary for transamination is called transaminase or Aminotransferase
PLP is the coenzyme that's important in the involvement of the transferring of the amino group
EX.
Glutamate (keto acid) + pyruvate → a-ketogluterate (keto acid) and alanine (amino acid)
Enzyme-PLP Schiff base + amino acid → PMP-Enzyme + a-keto acid product → a keto acid substrate → PLP enzyme
B6 and Demanimation
Is the removal of the amino group
Amino acid in the presence of dehydrogenase and coenzyme of PLP turn into a Keto acid + NH3
a desulfhydrase is used instead if the amino acid contains sulfur
NH3 is either converted to urea or added to glutamate to make glutamate or glutamin
this is how toxic NH3 is removed
B6 and Decarbxylation
The elimination of the CO2, the carboxy from a compound usually results in a drop in energy free energy.
Dopamine, epinephrine, norepinephrine
Tyrosine → Tyrosinase and Biopterin/Vit C → Dihydroxyphenylalanine (DOPA) → Decarboxylase and PLP (Rate Limiting Step)→ Dopamine → Dopamine hydroxylase and Vit C → Norepinephrine → Transmethylase and S-adenosylmethionine → Epinephrine
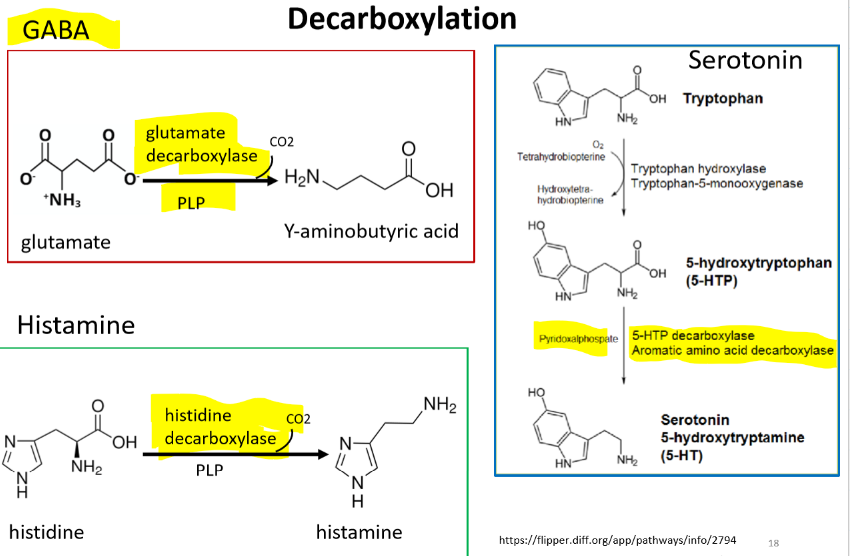
GABA - is a major inhibitory neurotransmitter, and his primary role is to reduce neuron excitability.
Glutamate → glutamate decarboxylase and PLP → Y-aminobutyric acid
Serotonin
Tryptophan → 5-hydroxytryptophan → 5-HTP decarboxylase/aromatic amino acid decarboxylase/PLP → serotonin/5-hydroxytryptamine (5-HT)
Histamine - is a major inhibitory neurotransmitter, and his primary role is to reduce neuron excitability. Ex. important for gastric acid secretion and it regulates vasodilation secretion and bronchoconstriction
Histidine → Histidine decarboxylase/PLP → histamine
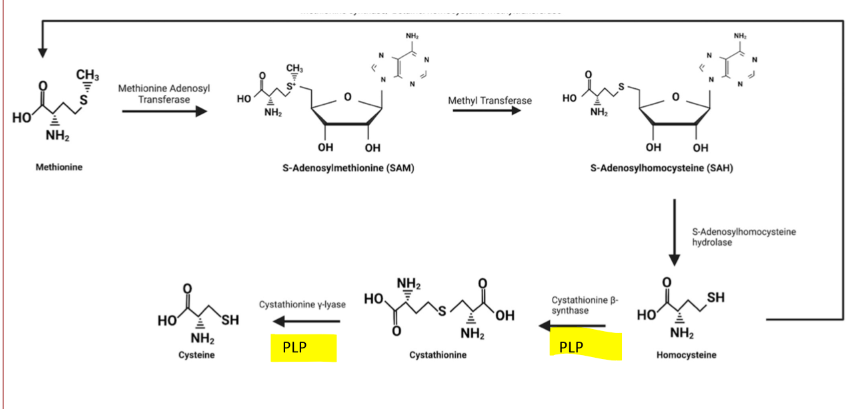
B6 and Transulfhydration
PLP is required for the enzymes Cystathione B-synthase and Cystathionine y-lase
is needed for transulfhydration pathway of methionine to Cysteine
methionine is an essential amino acid and when thiamine levels are low cysteine levels become important
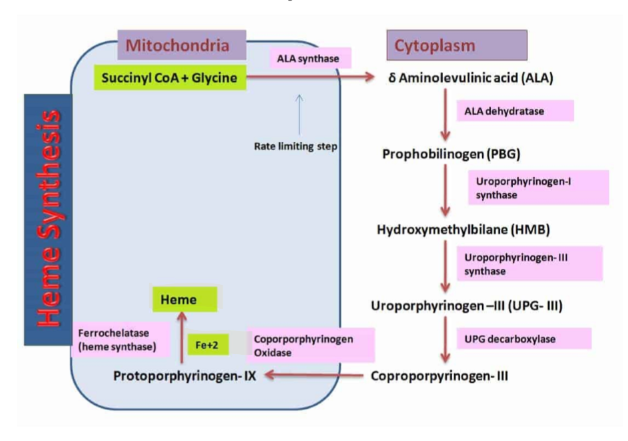
B6 and Heme Synthesis
Glycine + Succinyl Coa → ALA synthase and PLP (Rate Limiting Step) → aminolevulinic acid + CO2 +CoASH
occurs in mitochondria
zinc and iron are involved
If there is not an adequate supply of vitamin B6 then there will be impairment in making Heme
therefore B6 is associated with some anemias
Vitamin B6 and Tryotophan to Niacin Pathway
Kynureninase needs B6 (PLP) as a coenzyme)
Myosin, riboflavin, ATP, and niacin are involved in this process
If there is not an adequate supply of PLP there will be a build up of 3-OH Kynurenine
B6 and Glycogen Degradation
PLP is a coenzyme for Glycogen Phosphorylase
If there were inadequate amounts of PLP there would be build up of glycogen
In this process, the phosphate of PLP is thought to stabilize the compound and permit covalent bonding of the phosphate to form glucose 1 phosphate.
B6 and Transelenation
seleneomethionine → Selenocysteine → PLP/B-lyase → Selenide
Selenide is important for the making of various selenoproteins.
B6 and Folate
Selene hydroxymethyltransferase requires PLP as a coenzyme to make Tetrahydrofolate (THF) from 5,10-methlyene THF
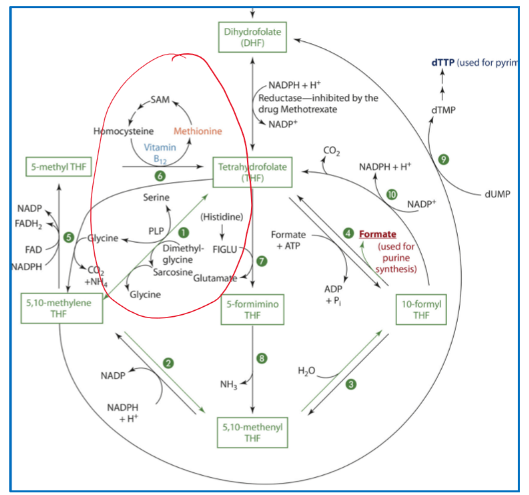
B6 Metabolism and Excretion
Vitamin B6 is excreted primarily in urine and very little is excreted in feces
4-pyridoxic acid is major urinary metabolite and is derived from the oxidation of PL
The oxidated PL comes from a dehydrogenase that is NAD dependent or an oxidase that is FAD dependent
Vitamin B6 Assessment of Nutriture
Plasma PLP concentration best indicator of tissue store (best way to asses status)
< 5 micrograms/L indicates a vitamin deficiency
5-7.5 indicate marginal status
>7.5 indicate adequacy
Urinary B6 and 4-pyridoxic acid
4-pyridoxic acid: short term indicator of b6 status
Erythrocyte transaminase activity
Vitamin B6: RDA
Adults aged 19-50: 1.3 mg/day
Preganacy: 1.9 mg/day
Lactation: 2.0 mg/day
RDA increases with age
males aged >51: 1.7 mg/day
females aged > 1.5 mg/day
Vitamin B6 Deficiency and Treatment
Rare in the US
Deficiency may occur in2-3 weeks, but it may take up to 2.5 months
Symptoms
weakness, fatigue, cheilosis, glossitis, angular stomatitis
neurological problems: depression, confusion, peripheral neuropathy, seizures
Deficiency causes
Hypochromic microcytic anemia
Impaired Niacin synthesis from tryptophan INhibits Homocysteine metabolism
Treatment
oral supplements 2.5-25 mg (up to 100 mg) daily for a few weeks
At Risk For B6 Deficiency
Elderly
poor intake, accelerated hydrolysis of PLP, and oxidation of paradoxal
Excessive alcohol consumption
can impair the conversion of paradoxes and pyridoxamine to PLP and the accumulation Of acetaldehyde, which is formed from alcohol metabolism, remember alcohol and the presence of alcohol dehydrogenase and NAD makes acetaldehyde and acetaldehyde. Thus enhances coenzyme degradation
Systemic inflammation
may increase catabolism, and there also may be an increased need for people during inflammation and then malabsorptive conditions.
Malabsorption condition (inhibits)
Medication/Drugs
INH (isoniazid)
Penicillnamine
Corticosteroids - prednisone
Anticonvulsants
Oral contraceptives
Vitamin B6 Dependency Syndrome
pyridoxine-dependent seizure (PDS)
autosomal recessive neurometabolic disorder
Vitamin B6 toxicity
>200 mg/day: sensory and perpheral neuropathy
unsteady gait, paresthesia (tingling/numbness) in extemities, impaired tendon reflexes
>2 g/day
May cause degeneration of neurons in spinal cord and impaired coordination
UL = 100 mg/day
to reduce the risk of developing neuropathy
Vitamin B12 (Cobalamin)
Macrocylic (corrin) ring made of four reduced pyrrole rings linked together
Contains cobalt in ring center attached to the nucleotide 5,6 dimethylbenzimidazole
Vitamin B12 Sources
Primarily from animal products
meat and meats products
fish and shellfish
dairy contains less, nut may be more bioavailable
Fortified plant based foods (cereals, soymilk, nutritional yeast)
Fairly stable and resistant to light, heat, oxidation
Cynocobalamin and hydroxyocobalamin are th main forms in supplements
readily cinverted to methylcobalamin and adenosylcobalamin
Vitamin B12 Dijestion
The body absorbs vitamin B12 from food in a two-step process. First, hydrochloric acid and pepsin in the stomach separates vitamin B12 from the protein that it's attached to. Second, the freed vitamin B12 then combines with a protein made by the stomach, called intrinsic factor, in the duodenum, and the body absorbs them together.
Vitamin B12 Absorption
Carrier mediated intestinal absorption is saturated with 1.5-2 micrograms per meal
With pharmacological doses, 1-3% absorbed vias passive diffusion through the small intestine (does not need intrinsic factor complex)
Overall, 50% (11-65% range_ absorbed with usual uptake
Absorption efficiency decreases as intake increases
protective mechanism so too much is not absorbed
Vitamin B12 Deficiency
Can take up to 5 years for deficiency to occur
Enterohepatic Circulation
The vitamin is excreted in the bile: however, it can bind to intrinsic factor (IF) in the duodenum and be absorbed in the ileum
Related to malabsorption syndrome
Vitamin B12 Transport
Appears in the blood 3-4 h after absorption and peaks in 8-12 h
60%-80% methylcobalamin and 20% adenosylcobalamin (2 enzyme forms)
Circulates in blood bound to transcobalamin (TCII) and haptocorrin
TCII transports newly absorbed B12 and accounts for 20% of cobalamin in the blood
Half-Life: <2hrs
Haptocorrin is synthesized in WBCs and transports and accounts 80% of cobalamin in the blood (does not transport newly absorbed B12 but stored B12 )
This complex is not taken up by tissues
Half life: 10 days
Circulating storage and delivering cobalamin from peripheral tissues back to the liver
Insertion of arginine of proline diminishes the ability of TCII to bind and transport B12 to tissues
Vitamin B12 Uptake
Taken up by tissues via tanscobalamin recepeter mediated endocytosis
Transport proteins that are in the cell and they're called chaperones and those proteins are thought to carry the vitamin within the cell to the various organelles and various compartments within the cell.
Metabolized to either coenzyme forms methylcobalamin (cofactor in cytoplasm) or adenosylcobalamin (coenzyme in mitochondria)
Vitamin B12 Storage
2-3 mg stored in the body
50% in liver
30% in muscle
smaller amounts in pituatary gland, bone, kidneys, heart, brain, spleen
Mainly as adenosylcobalamin with small amounts if hydroxocobalamin ad methylcobalamin
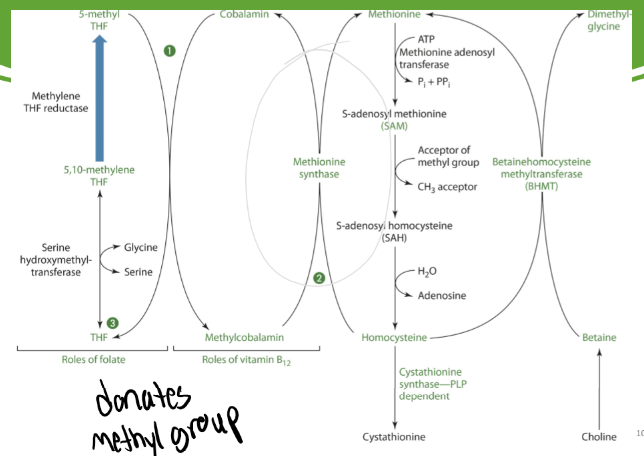
B12 to make Methionine
Methionine synthase needs B12 as a coenzyme to make methionine from homocysteine and Cobalamin from Methylcobalamin
Methionine synthase takes off the methyl group that methylcobalamin gained from 5-methyl THF and gives it to homocysteine to make methionine
Cobalamin is easily oxidized meaning methionine synthase easily becomes inactive
There is a NADPH-dependent enzyme that reduces methionine synthase to its active form
Folate is needed to turn THF to 5,10-methylene THF to % methyl THF
5-methyl THF is needed to turn cobalamin into methylcobalamin
B12 and the Oxidation of L- methylmalonyl-CoA
Startes with a odd #chain fatty acid
Propionyl → L-methylmalonyl-CoA → Methylmalonic acid or Succinyl CoA
Methylmalonic CoA is made without B12
Is measured to see if B12 levels are low
Succinyl CoA is made with Methylmalonyl CoA mutase which consists units that need 5’deoxyadenosyl cobalamin form of vitamin B12
Then enters the Kreb’s cycle
Vitamin B12 Metabolism and Excretion
Undergoes little to no degradation prior to excretion
0.1% is excreted in bile, but (75%) reabsorbed in the ileum
Only small amounts (~0.25 micrograms/day) lost in urine
Trace losses through the skin may also occur
Vitamin B12 RDA
Adults: 2.4 micrograms/day
Pregnancy: 2.6
Lactation: 2.8
Aged>51 fortified or B12 supplements (25-100)
due to decrease in consumption of animal products and the ingesting of PPI or antiacids that affects absorption
Vitamin B12 Deficiency
Megaloblastic Macrocytic Anemia (MA)
It can be B12 and/or folate deficiency, levels would have to be tested to determine
Appears in stages
decrease in B12 serum levels
decrease in B12 Cellular levels
decrease in B12 dependent enzymes
decrease in DNA synthesis
increase in plasma (homocysteine)
increase plasma methylmalonic acid
B12 serum levels and plasma methylmalonic acid levels should be measured together
Morphological and functional changes occur in RBCs
Neurological impairments may also occur
Pernicious anemia (is only related to B12)
autoimmune destruction of gastric parietal and mucosal cells
decreased HCl and IF production

B12 AT Risk for Deficiency
Inadequate intake (vegan; especially infants/children)
Altered gastric pH
Destruction of gastric parietal cells
Atrophic gastritis
caused by advancing age or by H.pylori infection of stomach
loss and inflammation of gastric parietal cells
decreased HCl and IF production
Altered intestinal pH
pancreatic impaired exocrine function and Zollinger-Ellison syndrome
more acidic pH of small intestine impairs release of B12 from R protein
Defects in cubam receptors
Impaired intestinal integrity or function
Malabsorptive syndromes resections of portions stomach and small intestine
Competition
parasitic infections (tapeworms)
Bacterial overgrowth associated with GERD and ulcer medications
Vitamin B12 Deficiency Treatment
Inadequate intake with neurological symptoms
Uptake to 1 mg B12 for 5 weeks followed by lower doses for 1-2 months
Pernicious anemia and deficiency secondary to malabsorption
Monthly IM injections of 500 -1,000 micrograms ot
Oral ingestion 1,000-2,000
or nasal spray 500
Improvements may be evident within 10 days
Serum [metholamlonic] decreases in the first week
Hematological response: may take up to 2 months
Vitamin B12 Toxicity
None observed
No UL
excessive amounts can cause GI distress
Vitamin B12 Assessment of Nutriture
Total serum B12 (B12 TCII + B12 haptocorrin)
most common way to measure B12 status
you would see low B12 serum and high serum methylmalonic acid and an increase in methyl malonic acid in urine
Increased serum or urinary methylmalonic acid indicates deficiency
Increased serum homocysteine> 20 micromol/L if folate and B6 are adequate
Breath test: subnormal production of labeled CO2 with ingestion of labeled propionate
Schilling test
orally administer radioactive B12 and measure urinary excretion
Below normal urinary excretion indicates impaired absorption
2 factor when measuring B12
when measuring B12 and methylmalonic acid folate should be measured too
calcium status is important too bc its responsible for the bonding of intrinsic factor to receptors
Folate
Folic Acid: Oxidzed form found in fortified foods and supplements
Folate: Reduced form found in naturally in foods and tissues
Folate Metabolism
Folate Sources
Mushrooms and green leafy vegetables, brussel sprouts, broccoli, asparagus
peanuts, beans (especially pento), lentils, stawberries, oranges, cantaloupe
liver
Fortified foods: bread, cereal, grain products (140 micrograms/100g)
Produced by bacteria in colon
Raw foods better than cooked
Destroyed by heat, oxidation, UV light
50-80% lost with certain food processing/preparation
Bioavailability of folate from foods
Intestinal pH
Genetic variability in enzymatic activity
Presence of inhibitors
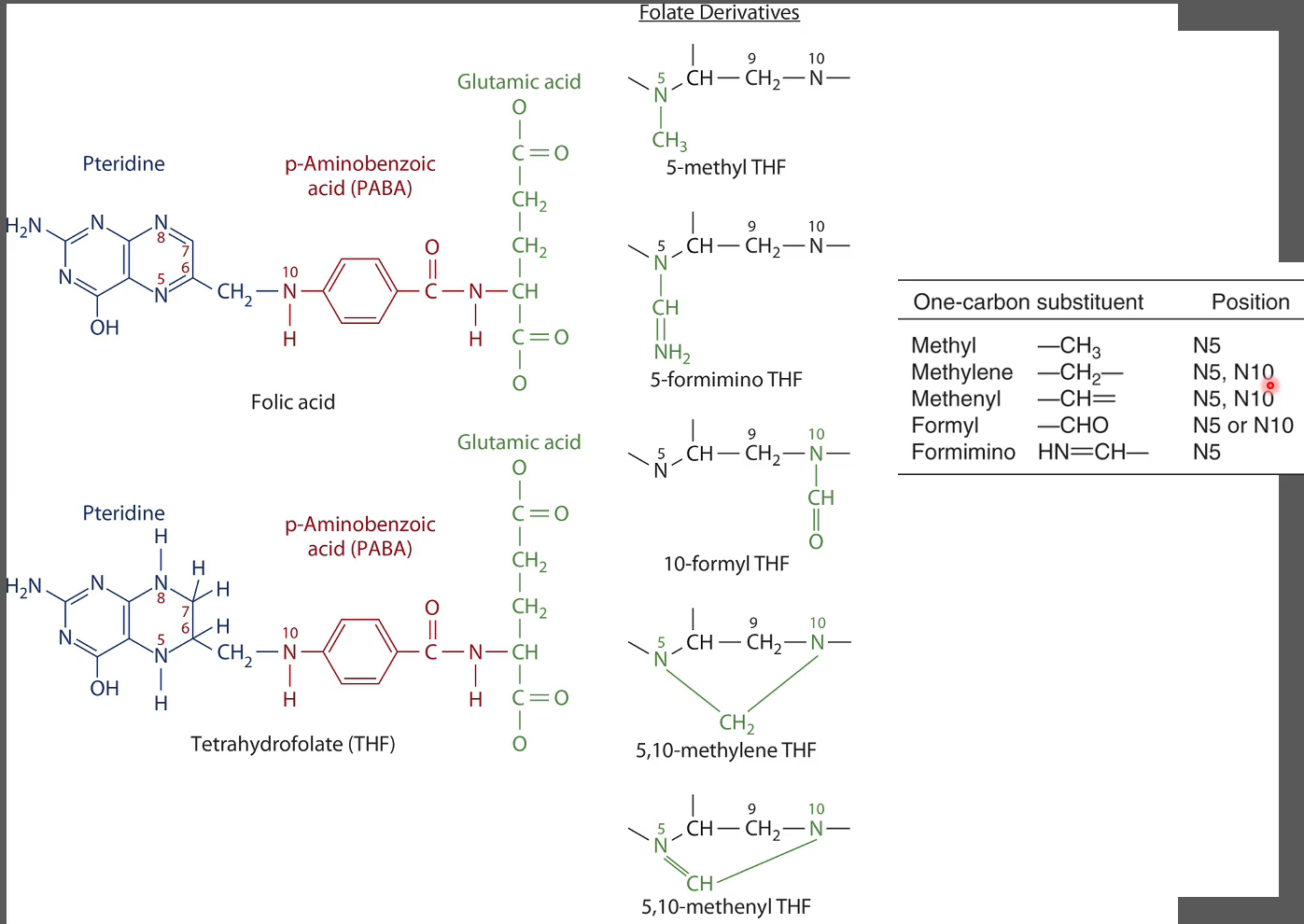
Folate Forms
Naturally Occuring in foods:
5-methyl tetrahydrofolate (THF)
5-formyl THF
10-formyl THF
Over 75% attached to multiplr glutamic acid residues
Overall absorption is about 50% (10%-90% range)
Supplements
Folic acid (only one glutamic acid residue)
monoglutamate
5-formyl THF
%-methyl THF
Multi: 400 micrograms
Single ingredient: 1,000 micrograms
100% absorbed (especially on empty stomach
Consumed with food sources absorption is about 85%
Folate RDA
Adults: 400 micrograms DRE/day
Pregnancy: 600
Lactation: 500
Bioavailability
1 mcg DFE = 1 mcg food folate
1 mcg DFE = 0.6 mcg folic acid from fortified foods or dietary supplements consumed with foods
1mcg DFE = 0.5 mcg folic acid from dietary supplements taken on an empty stomach
Folate Absorption
occurs in the duodenum and upper jejunum
bc its transporter (PCFT) prefers a more acidic environment
Folate or Folic acid → Monoglutamate → PCFT → Enterocyte
In the enterocyte, the monoglutamate is methylated at the glutamic acid residue end
MRP3 or 5 transports the molecule to the bloodstream
Pharmacological doses of folic acid are absorbed through passive diffusion
In the colon uses the transporter RFC
Folate Digestion
Polyglutamate →glutamate carboxypeptidase and zinc → monoglutamate
Enzyme is affected by
zinc deficiency
Acidic pH
Alcohol ingestion
inhibitors in legumes, cabbage, oranges
Folic acid in fortified foods and supplements already in monoglutamate form
Folate Transport
Blood
Folate is bound as free monoglutamate (~1/3) or bound to proteins (~2/3) such as albumin and macroglobulins and folate binding protein
found primarily as 5-methyl THF and smaller amounts of THF, 10formyl THF, and other THF derivatives
Higher [folate] in cerebral spinal fluid and RBCs than plasma itself
Folate in RBCs attained during erythopoiesis: index of long-term folate status (2-3 months)
Plasma levels represent folate intake
Folate Uptake
uptake of folate into tissues: carrier mediated
PCFT: liver, pancreas, kidney, and spleen
RFC: folate, especially 5-methyl THF for systemic circulation
Organic anion transporting poly pepetides (OATP) B1 and B3: liver
Folate receptors mediate uptake via endocytosis in tissues including the brain
mutation prevent folate from crossing the BBB
low cerebral spinal fluid [folate] and neurological problems
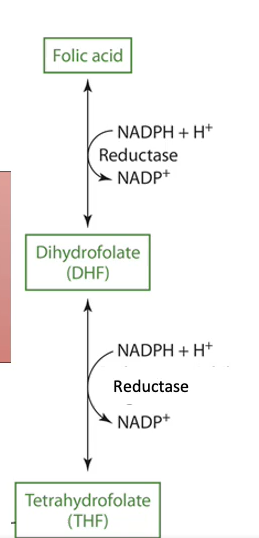
Folate Hepatic Metabolism
THF in hepatocytes
THF (33%)
5-methyl THF (~33%)
5- and 10-formyl THF (~33%)
Folic acid → DHF → THF (cytosol)
NADPH dependent DHF reductase
THF and THF derivatives: glutamate residues added (one at a time) by folylpolyglutamate synthase (ATP dependent)
Traps THF in hepatocytes → prevents degradation and enables storage
y-glutamyl hydrolases remove glutamate residues before release into the blood or secreted into bile
50% secreted into bile by MRP2 and breast cancer resistant protein (BCRP) carriers
Folate Storage
Body stores: 7-30 mg
Half of body’s folate is found in the liver
Stored wth intracellular folate-binding proteins
Main storage forms:
Polyglutamate forms of THF and 5-methyl THF
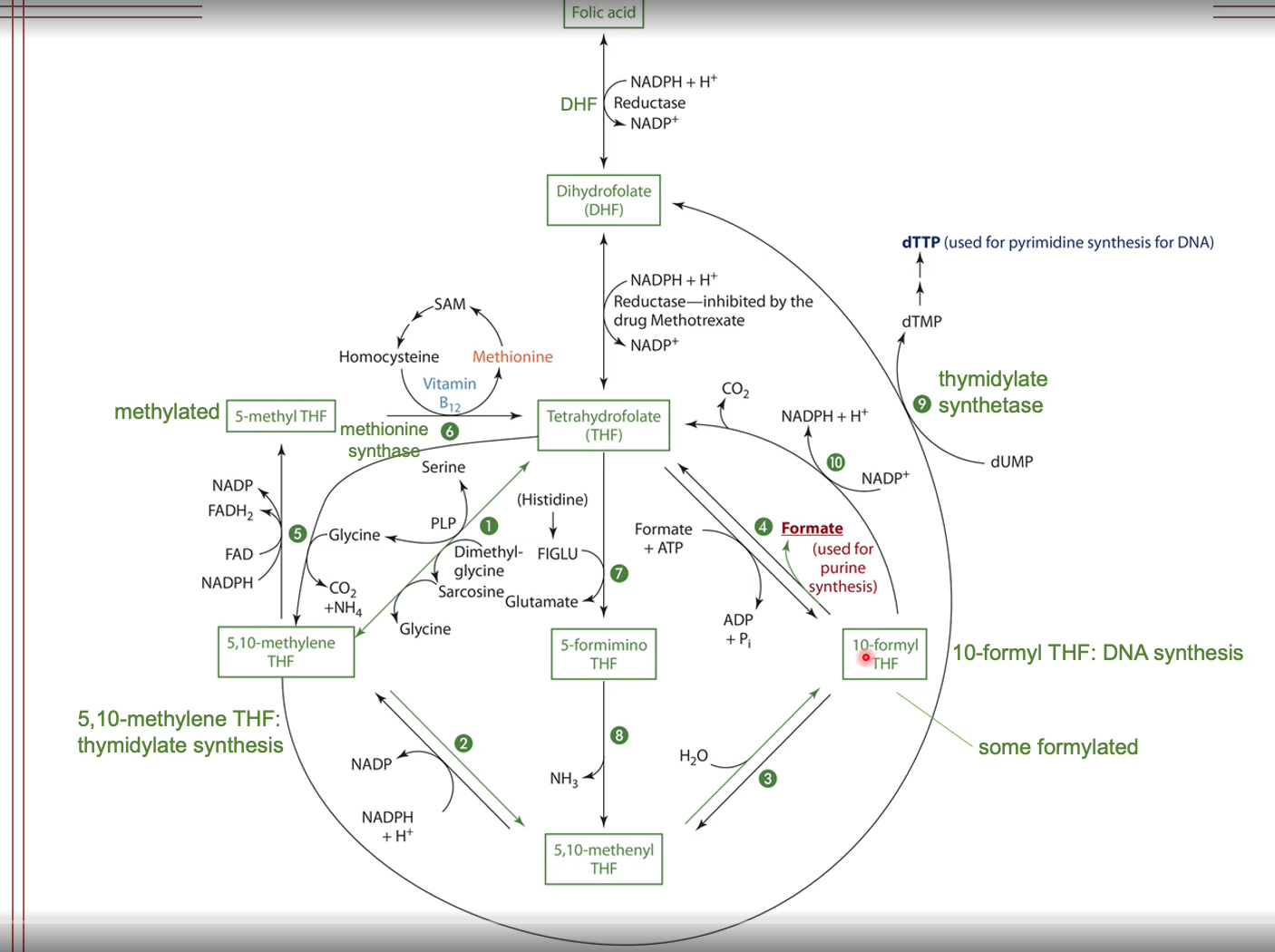
Folate Functions
One-carbon metabolism
Accepts and donates one carbon units
Amino acid metabolism
DNA synthesis metabolism
Cells with short lifespans are particularly affected by folate inadequacy
Methyl group (CH3) carrier
Impacts gene expression
Tetrahydrofolate functions as a coenzyme in the mitochondria and the cytosol
THF is generated from amino metabolism
THF derivates serve as donors of one carbon units in synthetic reactions such as amino acid, purine, and pyrimidine synthesis
Poor folate statues is associated with decreased DNA methylation
Folate and Disease
Low folate intake associated with increased plasma [homocysteine]
Increase in [homocysteine] associated with premature heart disease and stroke
cognitive dysfunction
Folate deficiency associated with increased initiation of colon ( and other) cancer
can also inhibit gene translation
decrease serum and RBC [folate] associated with depression
Neural tube defects
Spina bifida
anencephaly
Folate Metabolism and Excretion
Excreted in urine intact and as metabolites
In kidneys, folate binding proteins in renal brush border membrane aids tubular reabsorption
Folate is secreted by liver into bile
most is reabsorbed with enterohepatic recirculation
Folate losses in feces are minimal
Bacteria in intestine can synthesize folate, and that can be excreted in feces
Folate Deficiency
Megaloblastic macrocytic anemia (MA)
few, abnormally nucleated (immature) and large RBCs
fatigues, weakness, headaches, irritability, difficulty concentrating, shortness of breath, heart palpitations
can rise from deficiency of folate or B12
disrupt DNA synthesis (replication) and cell division
Purine and pyrimidine synthesis compromised → macrocytic and megaloblastic cells
Treatment: 1-5 mg (oral ingestion) folate daily
5 methyl THF more effective than folic acid
Diagnosis and progression
1-2 months: decrease plasma [folate] and increase [homocysteine
3-4 months: decrease in RBC [folate]
4-5 months: rapidly dividing cells become megaloblastic
increase MCV, hyper segmentation of WBC and decrease blood cell count
blood’s oxygen-carrying capacity
At Risk for Folate Deficiency
Excessive alcohol ingestion
Malabsorption disorders
Gastric bypass
Medications
diuretic
anticonvulsants
methotrexate
cholestyramine
sulfasalazine
Folate Toxicity
UL = 1,000 mcg synthetic (non-natural) folic acid
Folate can mask B12 deficiency
Folic acid supplements can alleviate the MA caused by B12 deficiency, but the neurological damage progresses undetected and is irreversible
15 mg folate daily
insomnia, malaise, irritability, GI distress
Some studies show increased risk of cancer with 1,00 mcg folic acid daily
Folate Assessment of Nutriture
Plasma, serum, or RBC concentration
Serum or plasma [folate] reflect recent dietary intake
True deficiency diagnoses requires repeated measures of serum/plasma folate
RBC [folate] better indicator of folate status
Formiminoglutamate excretion (FIGLU) excretion
A functional marker of folate and vitamin B12 deficiencies is elevated plasma homocysteine concentration
![<ul><li><p>Plasma, serum, or RBC concentration</p></li><li><p>Serum or plasma [folate] reflect recent dietary intake</p><ul><li><p>True deficiency diagnoses requires repeated measures of serum/plasma folate</p></li></ul></li><li><p>RBC [folate] better indicator of folate status</p></li><li><p>Formiminoglutamate excretion (FIGLU) excretion</p></li><li><p>A functional marker of folate and vitamin B12 deficiencies is elevated plasma homocysteine concentration</p></li></ul>](https://knowt-user-attachments.s3.amazonaws.com/a16329dd-6aa7-472e-a2e7-523b66565cc2.jpeg)