Proteome Data Analysis 1
1/5
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
6 Terms
Protein Identification Techniques
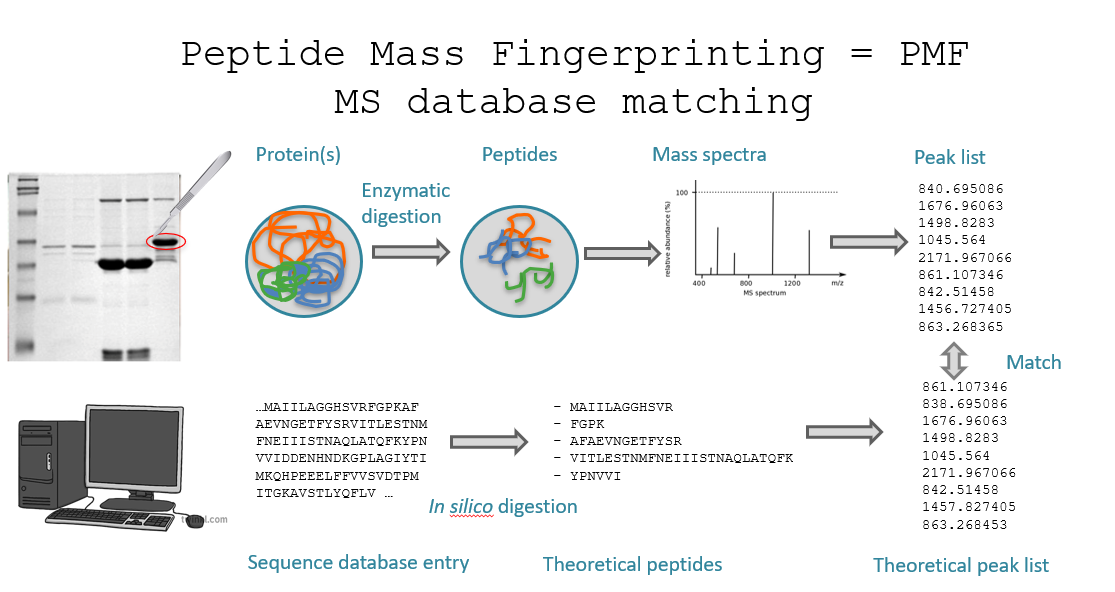
Peptide mass fingerprinting
Tandem MS (MS/MS)
Peptide mass fingerprinting
Peptide Mass Fingerprinting (PMF) is a protein identification technique where a protein is enzymatically digested into peptides, the masses of these peptides are measured, and the resulting “fingerprint” is compared to theoretical peptide masses from a protein database.
The idea is that each protein has a unique pattern of peptide masses after digestion, like a fingerprint, that can be matched computationally.
Workflow
Protein separation: using 2D gel electrophoresis or SDS-PAGE. Each spot or band ideally contains a single purified protein.
Enzymatic digestion: the isolated protein is digested with an enzyme, usually trypsin. Trypsin cleaves at the C-terminal of lysine and arginine, unless followed by proline. This results in a set of peptides of known cleavage rules.
Mass spectrometry (MS): the resulting peptides are introduced into a mass spectrometer. This produces a mass spectrum: a list of mass/charge (m/z) values. This set of masses is the peptide mass fingerprint.
Database search: a computational algorithm (e.g. MOWSE, Mascot) compares the measured peptide masses to theoretical masses from in silico digestion of proteins in a database. The protein whose theoretical peptide masses best match the experimental ones is identified as the source.
Scoring algorithms for peptide mass fingerprinting?
MOWSE
Mascot
Explain the MOWSE algorithm
Simplest algorithm in PMF. It takes into account the fact that:
Some peptides are very common across proteins, while some are rare and unique to specific proteins.
The larger a protein is, the more peptides it will generate when digested, and the more we increase the chances of random matches between the experimental and theoretical results in the database.
How?
In silico digestion of the protein database
Every protein in the database is "digested" computationally using trypsin rules (cleaves after K and R).
For each resulting peptide, its molecular weight (MW) is calculated and rounded to the nearest integer Da.
This results in a list of ALL peptide masses across ALL proteins in the database.
Frequency histogram of peptide masses:
A histogram is built of how frequently each peptide mass occurs across the database.
These frequencies are then normalised by dividing them by the total number of peptides in the database.
This normalised value reflects the likelihood of observing that peptides mass by random chance.
Matching experimental masses to database
You have a list of experimentally obtained peptide masses from unknown protein (using mass spectrometry).
Algorithm compares each measured peptide mass to the theoretical peptide masses from each protein in the database.
If a measured mass is withing the error tolerance of a theoretical mass, it is considered a match.
Score proteins: for each protein:
Find how many of the measured peptide masses match its theoretical peptide.
For each matching mass, get its normalized frequency.
Multiply all the normalised frequencies obtained together → Pn
Calculate MOWSE score:
Score = 50000 / (Pn x H)
H is the molecular weight of the protein
The higher the score, the more likely this protein is the correct match.
Mascot algorithm
Extends on the MOWSE algorithm for peptide mass fingerprinting.
It has additional parameters:
Missed cleavages
Post-translational modifications
Mass tolerances
Optimising parameters of Mascot algorithm.
Select a purified protein of which the identity is known.
Digest with trypsin and perform mass spec to get the peptide mass list.
Find parameters that give the best score for the target protein.