16. Intracellular Messengers - Part 1 "Diversity of Second Messengers"
1/15
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
16 Terms
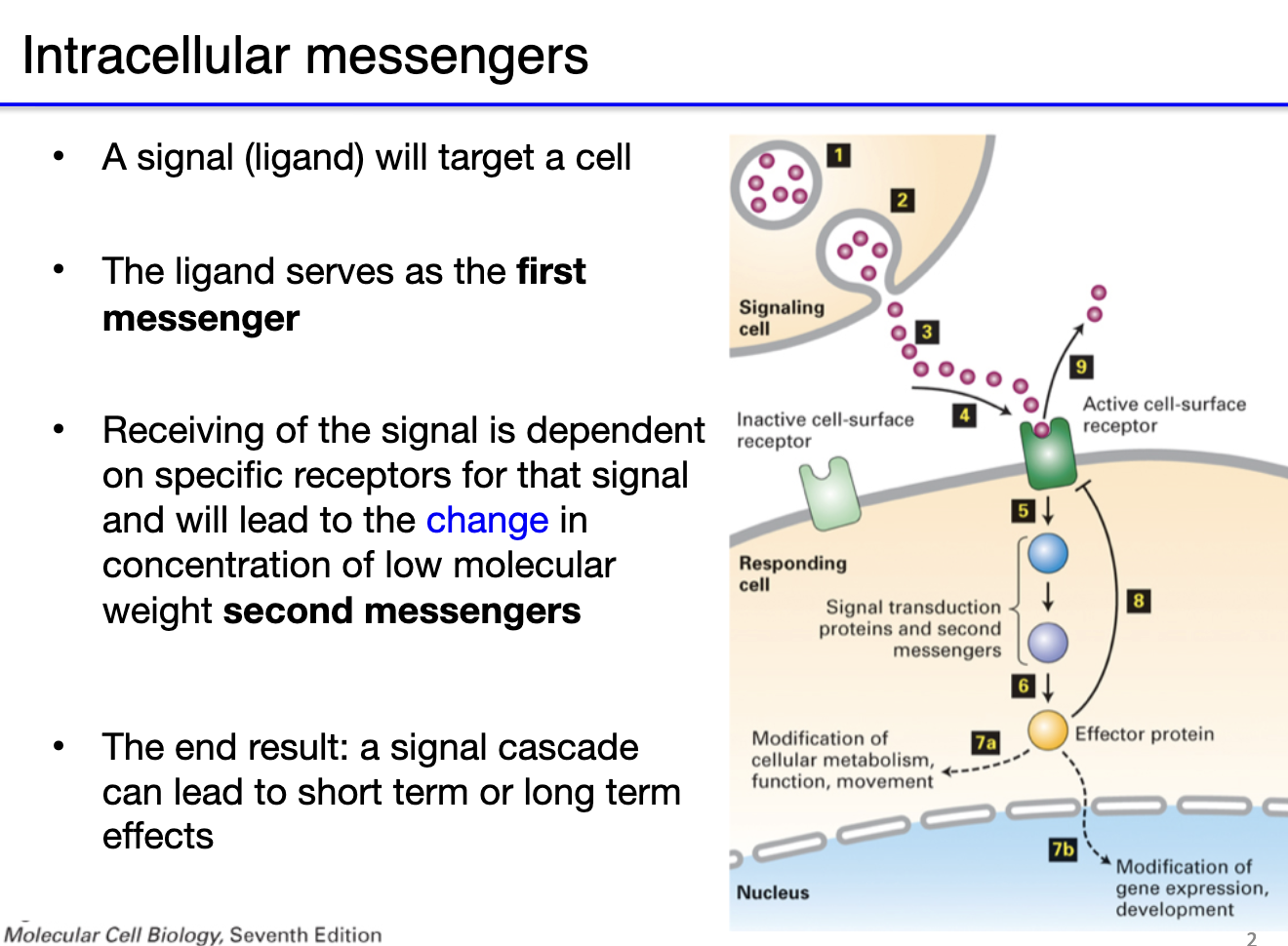
What are first and second messengers in neurophysiology?
First Messenger:
Ligand binds receptor on target cell (e.g., neurotransmitters, hormones)
Initiates signal at cell surface
Direct effect possible (ionotropic receptors open → no second messenger)
Second Messenger:
Intracellular molecule that transduces the signal
Often triggered by metabotropic receptors
Activates signaling cascades → short- or long-term effects (e.g., metabolism, gene expression)
Flow:
Ligand (first messenger) → receptor → change in concentration of second messengers → signal cascade → cellular response
What’s the difference between direct and indirect neurotransmission?
Direct transmission: NT binds ionotropic receptor → receptor itself is the channel → fast, direct gating.
Indirect transmission: NT binds metabotropic receptor (e.g., G protein-coupled receptor, tyrosine kinase) → receptor activates a signaling cascade → channel affected indirectly → slower response.

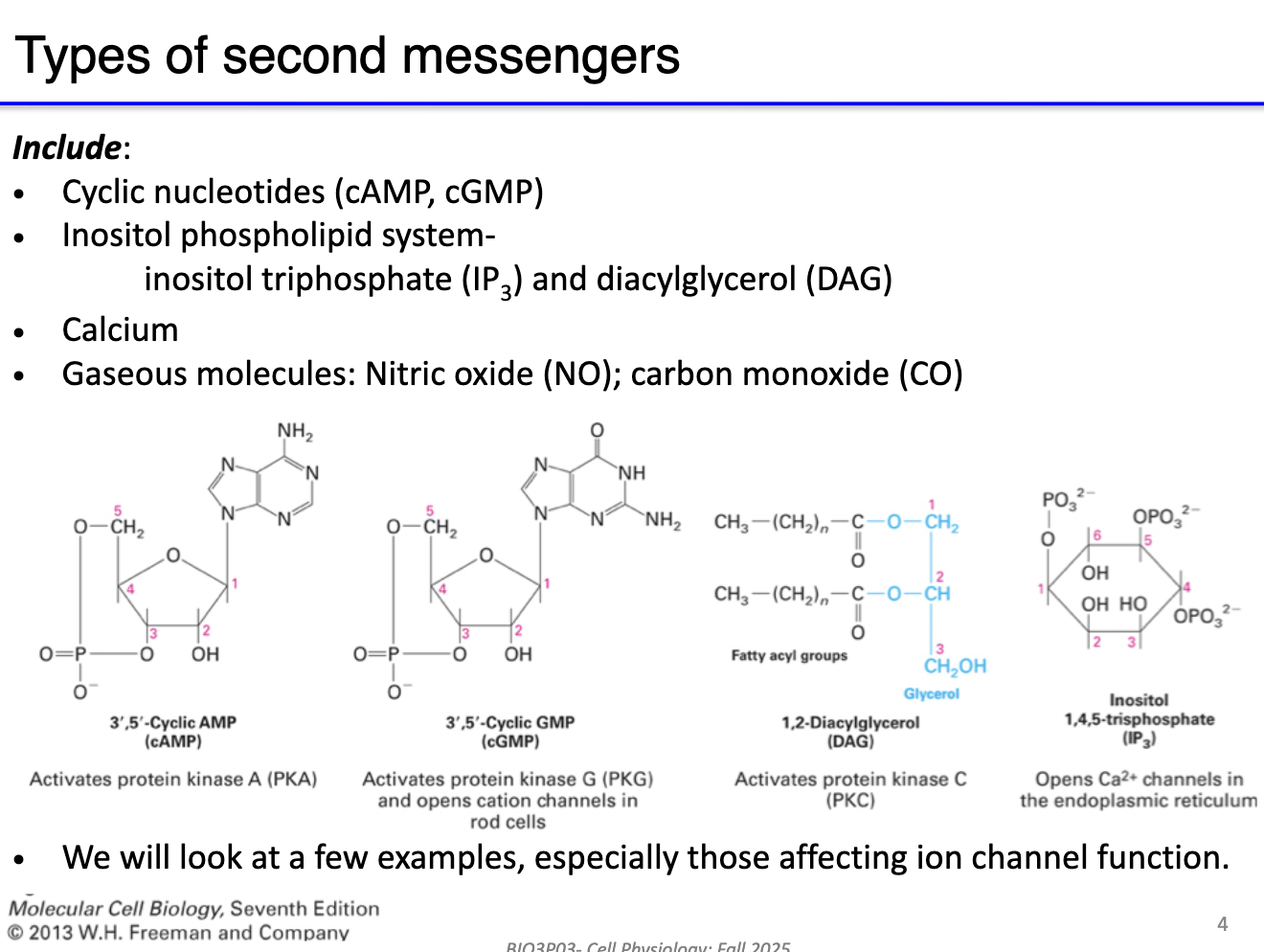
What are the main types of second messengers and their role?
Second messengers: Intracellular molecules that transduce signals from receptors to intracellular targets.
Key examples:
cAMP, cGMP – cyclic nucleotides
IP3 – inosital trophosphate signaling
Calcium (Ca²⁺)
Gaseous molecules – nitric oxide (NO), carbon monoxide (CO) - NOT TESTABLE
Function: Initiate signaling cascades in muscle and neurons; amplify or propagate extracellular signals.
What are the main types of cell surface receptors and key features of G protein-coupled receptors (GPCRs)?
Channel-linked receptors: Ionotropic (e.g., nicotinic AChR) → direct gating, fast signaling.
G protein-coupled receptors (GPCRs): Indirect, receptor separate from channel → triggers signaling cascade.
Catalytic receptors: Activate enzymes → amplify signal through second messengers (involved in cell proliferation and differentiation).
Intracellular/nuclear receptors: Alter gene expression (not a focus in this course).
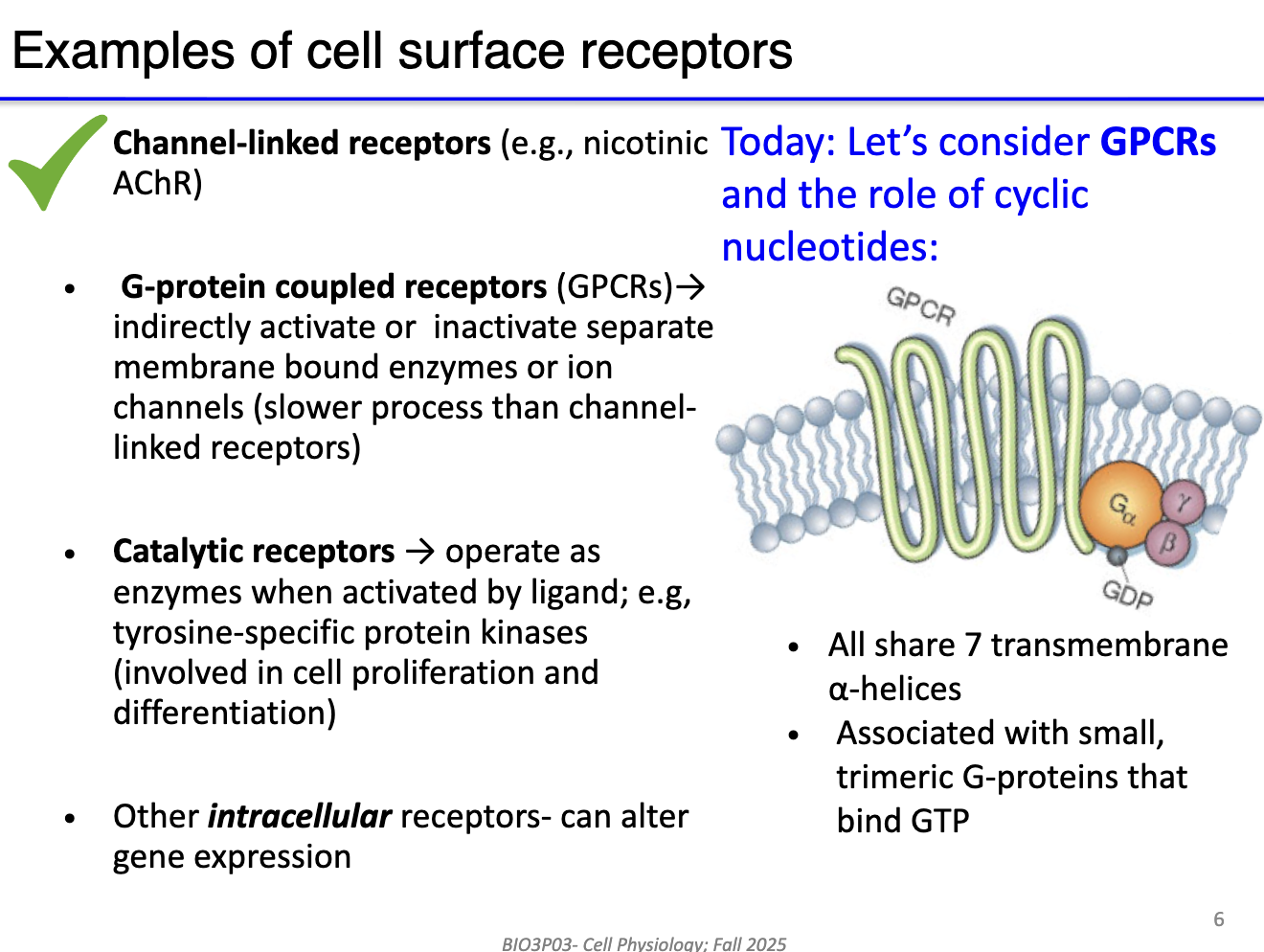
What is the structure of the GPCR?
7 transmembrane helices (“serpentine”)
Coupled to trimeric G protein (α, β, γ subunits)
G protein binds GDP (inactive) or GTP (active)
Can amplify signals via second messenger production.
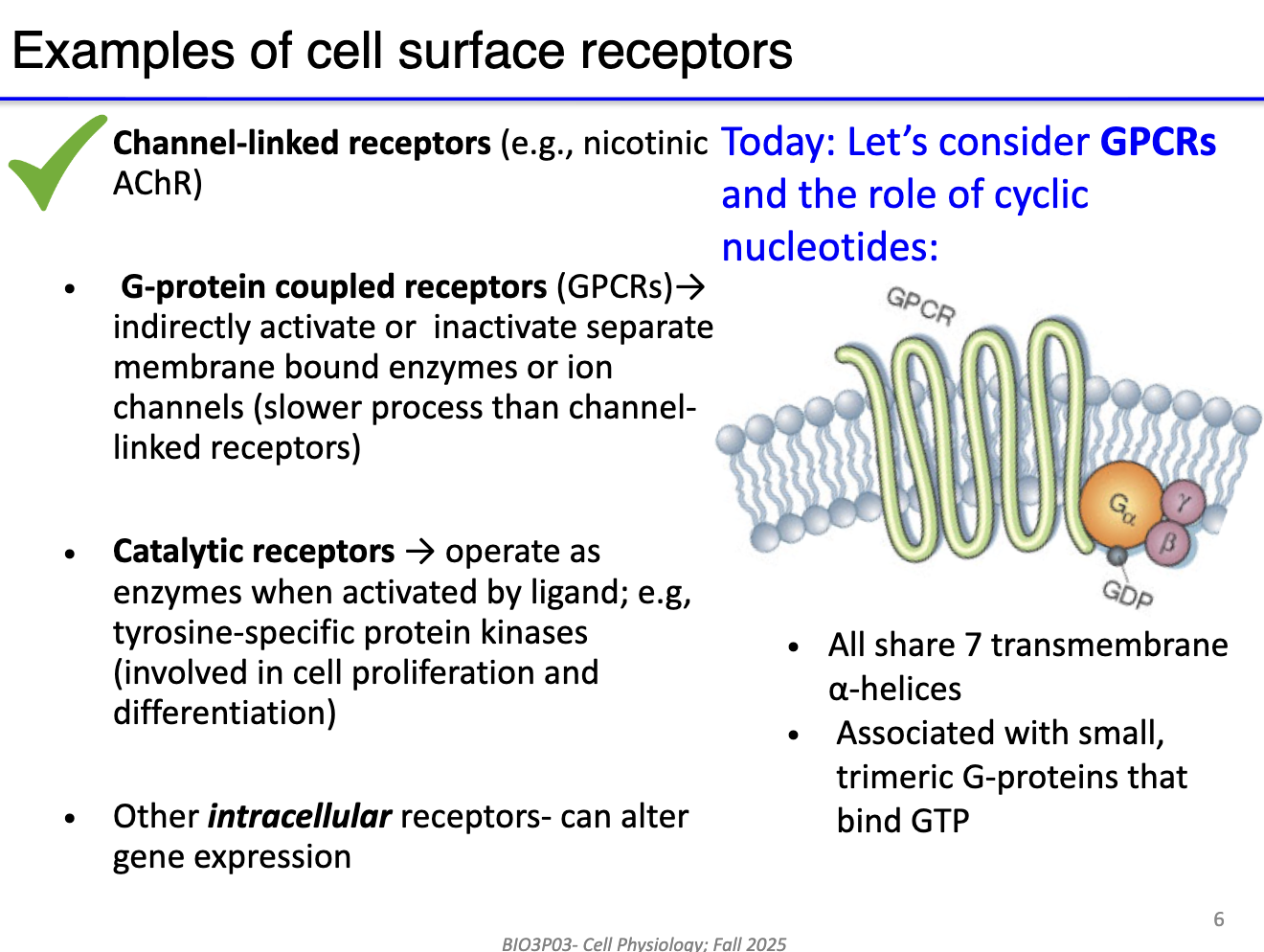
How do G-proteins transmit signals from GPCRs to target proteins?
Unbound state: GPCR not bound to agonist; G protein complex associated with GDP.
Agonist/NT binding: NT binds → conformational change → GDP released, GTP binds α-subunit.
Subunit activation: α-subunit (or βγ complex) separates to modulate effector proteins (e.g., channels).
Signal duration: Active until α-subunit hydrolyzes GTP → GDP; subunits reassemble with GPCR.
Key point: One agonist binding can activate the G protein complex for a duration determined by GTP hydrolysis.
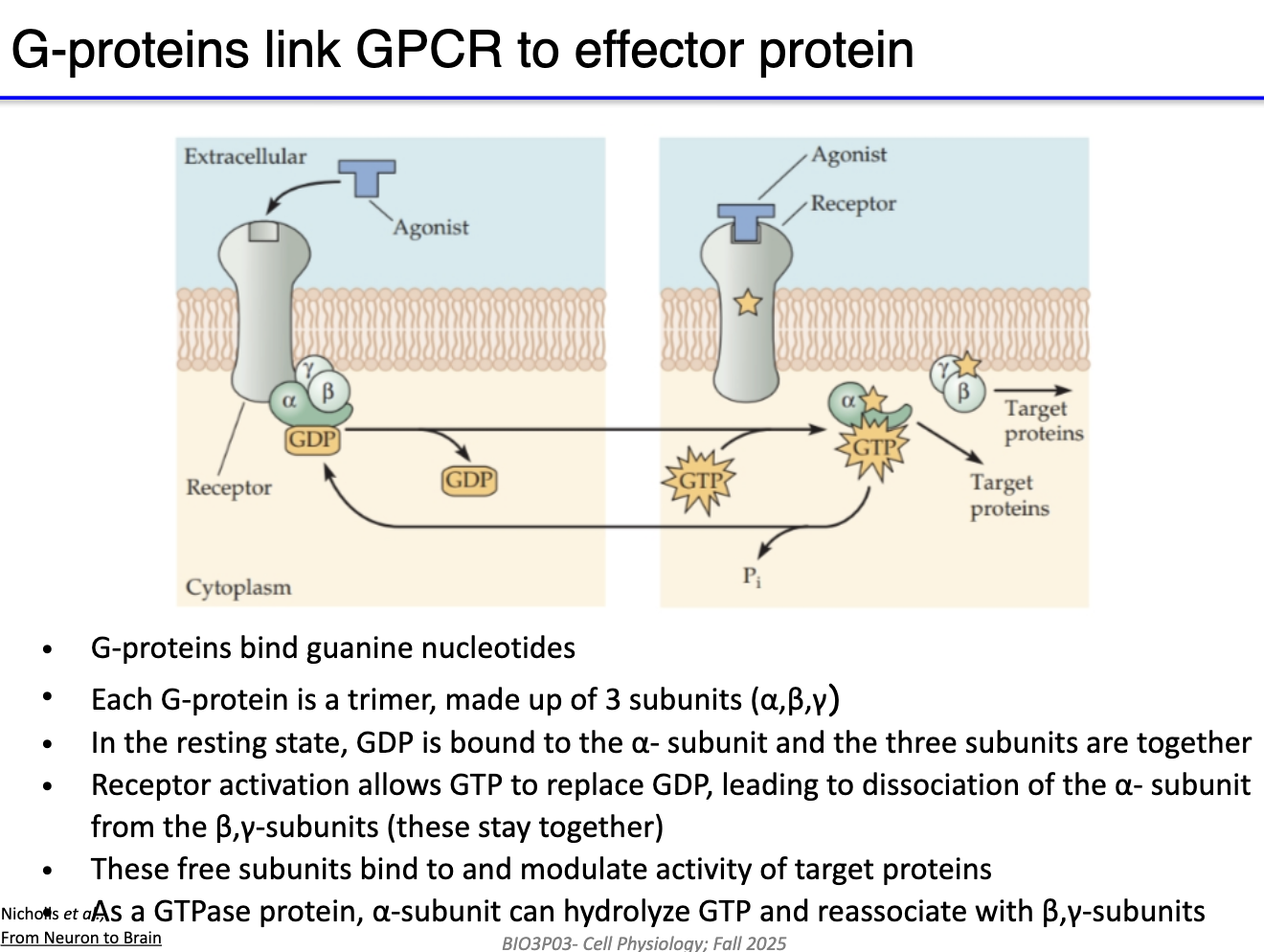
What is the diversity and role of G protein subunits in GPCR signaling?
Human genome ~23,000 genes → ~3.5% related to GPCR pathways.
GPCR inactive when unbound or bound to antagonist.
Agonist binding → activates G protein complex (GTP binds α-subunit).
27 Gα subunits, including:
Gs → stimulatory → activates adenylyl cyclase → ↑cAMP (second messenger)
Gi → inhibitory → inhibits adenylyl cyclase → ↓cAMP
Gq → involved in calcium signaling
G12/13 (not important for this course)
Beta & gamma subunits: mostly regulatory; sometimes directly modulate channels.
All G-protein can facilitate cellular signaling.
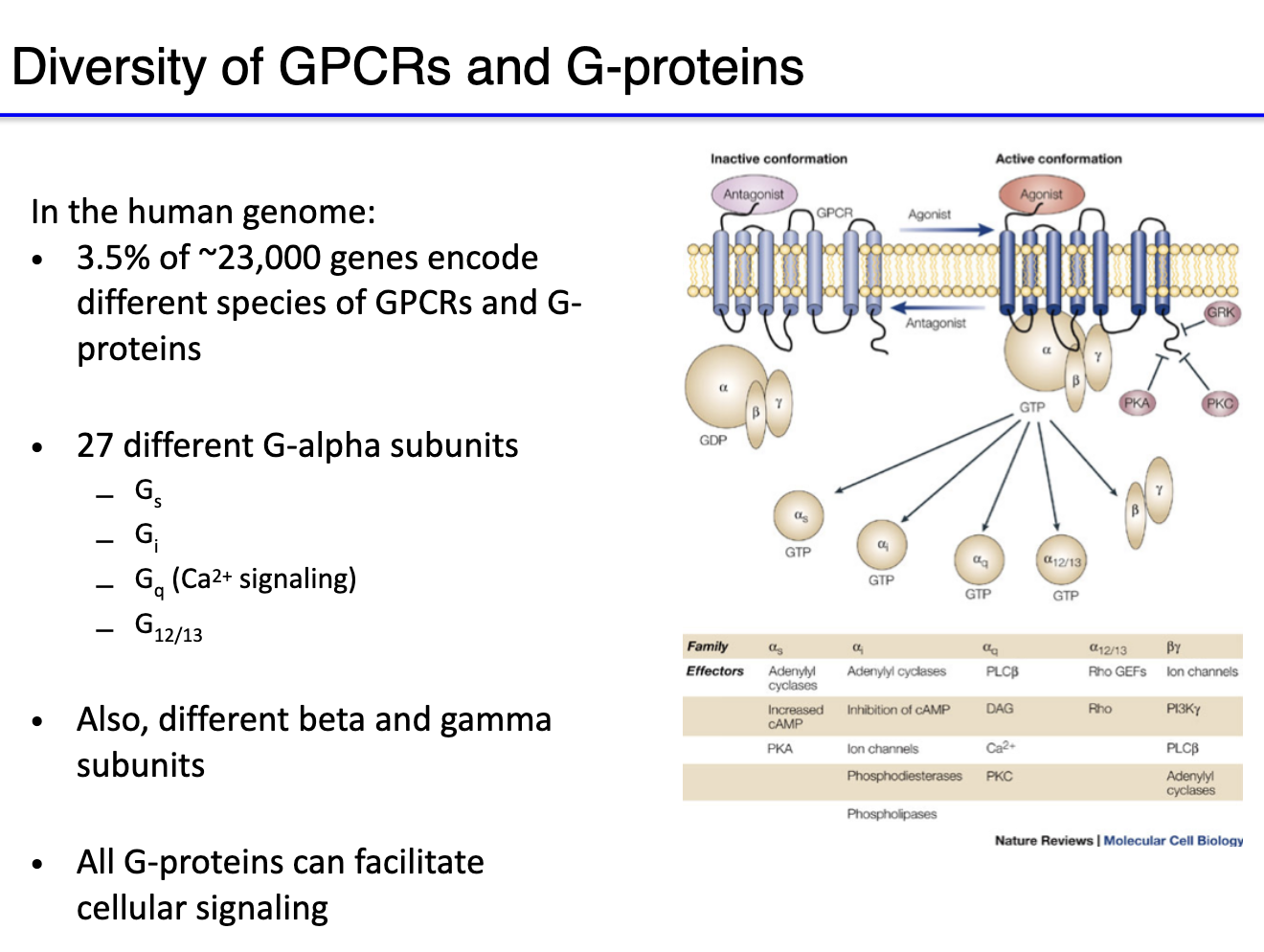
How does norepinephrine activate the beta-adrenergic receptor and downstream cAMP signaling?
Norepinephrine binds beta-adrenergic receptor (a GPCR).
GPCR-bound G protein sheds GDP → alpha subunit binds GTP.
Beta & gamma subunits activate adenylyl cyclase.
Adenylyl cyclase converts ATP → cAMP (second messenger).
cAMP activates Protein Kinase A (PKA).
PKA opens voltage-activated calcium channels.
Overall effect: stimulatory, increasing cellular activity.
How does cAMP activate Protein Kinase A (PKA)?
PKA = tetramer → 2 regulatory (R) subunits + 2 catalytic (C) subunits.
At low cAMP: R subunits bind and inhibit C subunits.
When cAMP increases: cAMP binds R subunits → causes conformational change.
R subunits release the C subunits.
Free C subunits = active PKA.
Active PKA phosphorylates target proteins, including channels → promotes channel opening.

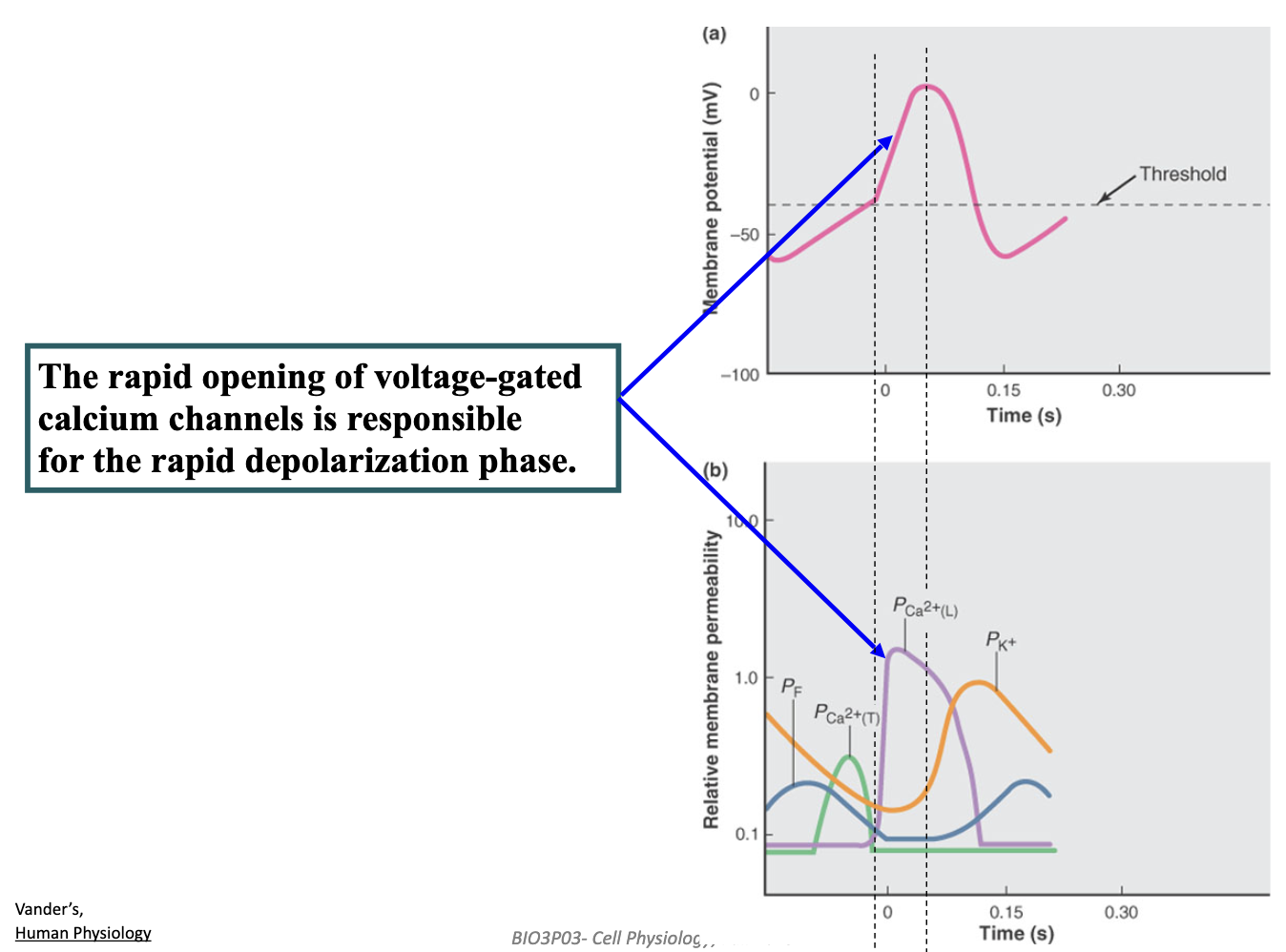
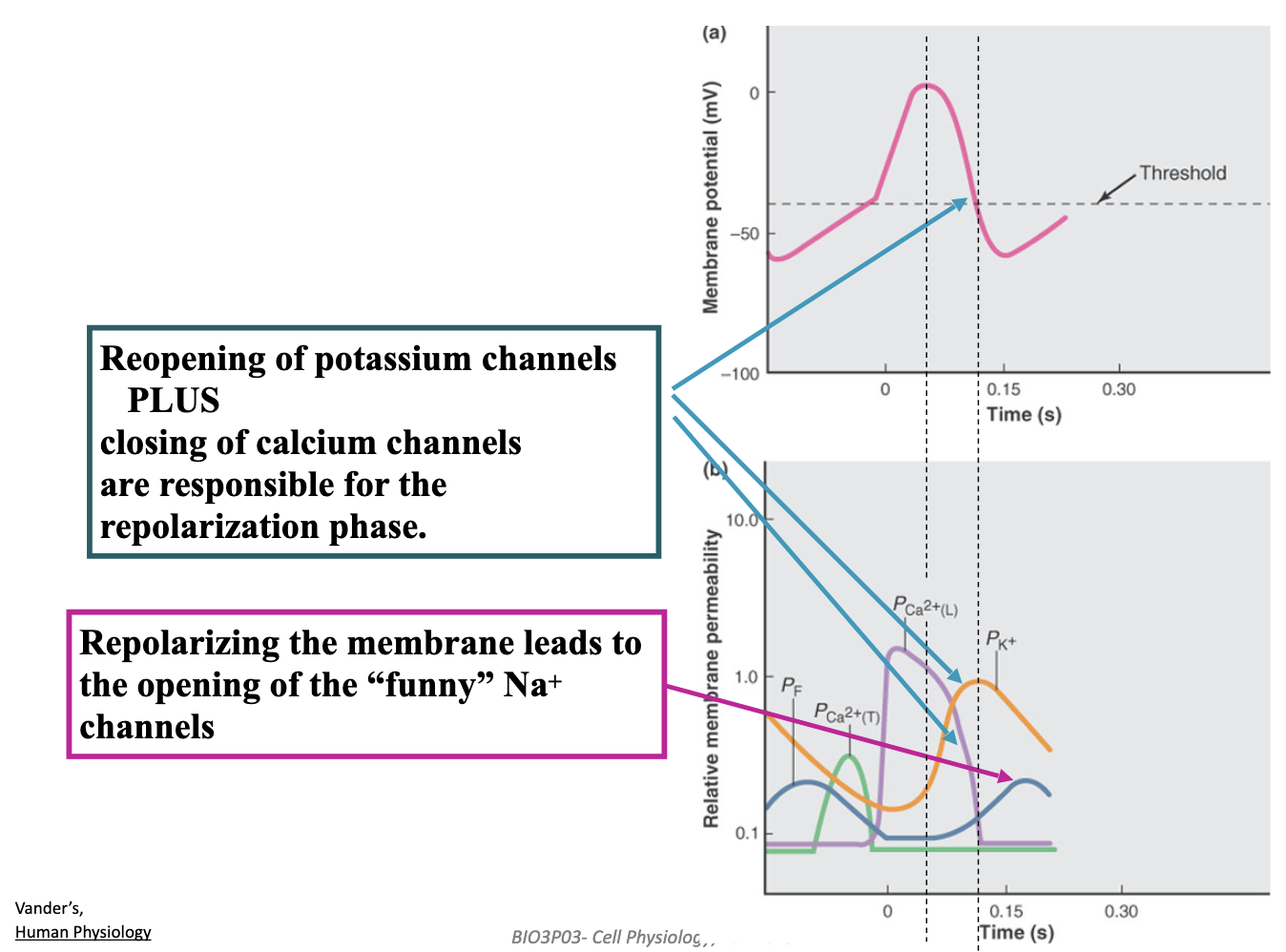
How do pacemaker cells in the heart generate an autonomous rhythmic heartbeat?
Hyperpolarization opens HCN (“funny”) channels, causing slow Na⁺ influx → start of the pacemaker depolarization.
Rising depolarization then opens T-type Ca²⁺ channels → pushes cell to threshold.
L-type Ca²⁺ channels open at threshold → action potential upstroke.
K⁺ efflux + Ca²⁺ channels closing → repolarizes the cell.
Resulting hyperpolarization reopens HCN channels → cycle repeats → automatic rhythm.

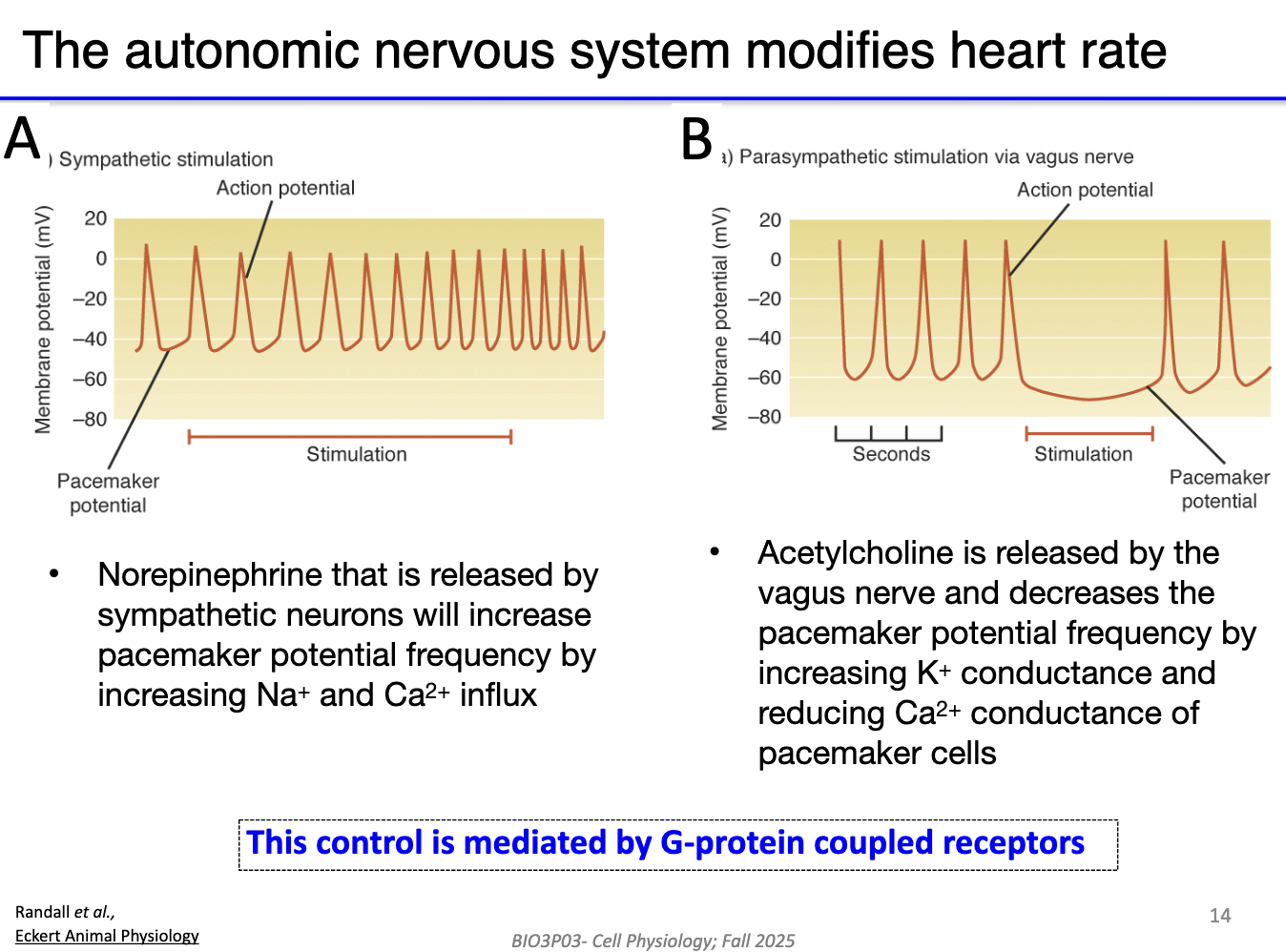
How do the sympathetic and parasympathetic nervous systems regulate heart rate in pacemaker cells?
Sympathetic (fight-or-flight): Norepinephrine ↑ funny current + Ca²⁺ currents → steeper pacemaker depolarization → faster heart rate (action potentials closer together).
Parasympathetic (vagus nerve): Acetylcholine ↑ K⁺ efflux + ↓ funny current + Ca²⁺ currents → slower depolarization → slower heart rate.
Heart rate is controlled by a balance of “gas” (sympathetic) and “brake” (parasympathetic), acting simultaneously on pacemaker cells.
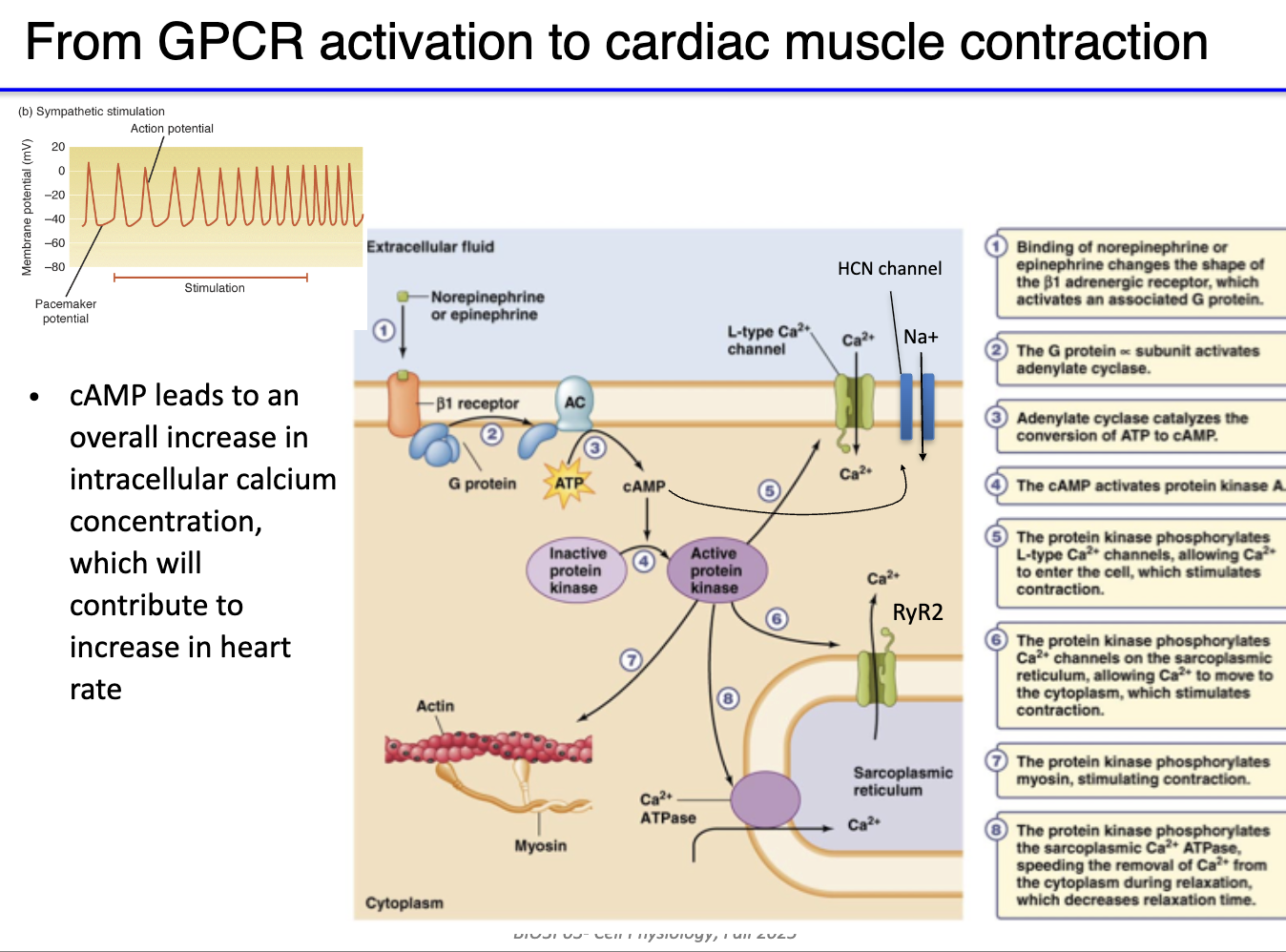
How does sympathetic stimulation (NE) increase heart rate in pacemaker cells?
NE binds β₁-adrenergic GPCR on pacemaker cells
Gs protein activated → α-subunit stimulates adenylyl cyclase
Convert ATP → cAMP production
cAMP + PKA both increase activity of L-type Ca²⁺ channels
Channels open more easily → more Ca²⁺ influx
Faster depolarization → higher action potential frequency
Result: ↑ heart rate
How does parasympathetic stimulation (ACh) decrease heart rate in pacemaker cells?
ACh released from vagus nerve → ACh binds muscarinic M₂ GPCR on pacemaker (SA node) cells
GPCR activates G protein → βγ subunits dissociate
βγ subunits open voltage-gated K⁺ channels → K⁺ efflux
Hyperpolarizes membrane potential → slower depolarization toward threshold
Slower pacemaker potential → decreased action potential frequency → lower heart rate
Gi subunit role: Inhibits adenylyl cyclase → reduces Ca²⁺ current
Overall effect: Acts as a “brake” on sympathetic stimulation → balances heart rate regulation
**In lecture he kept saying vagal nerve.

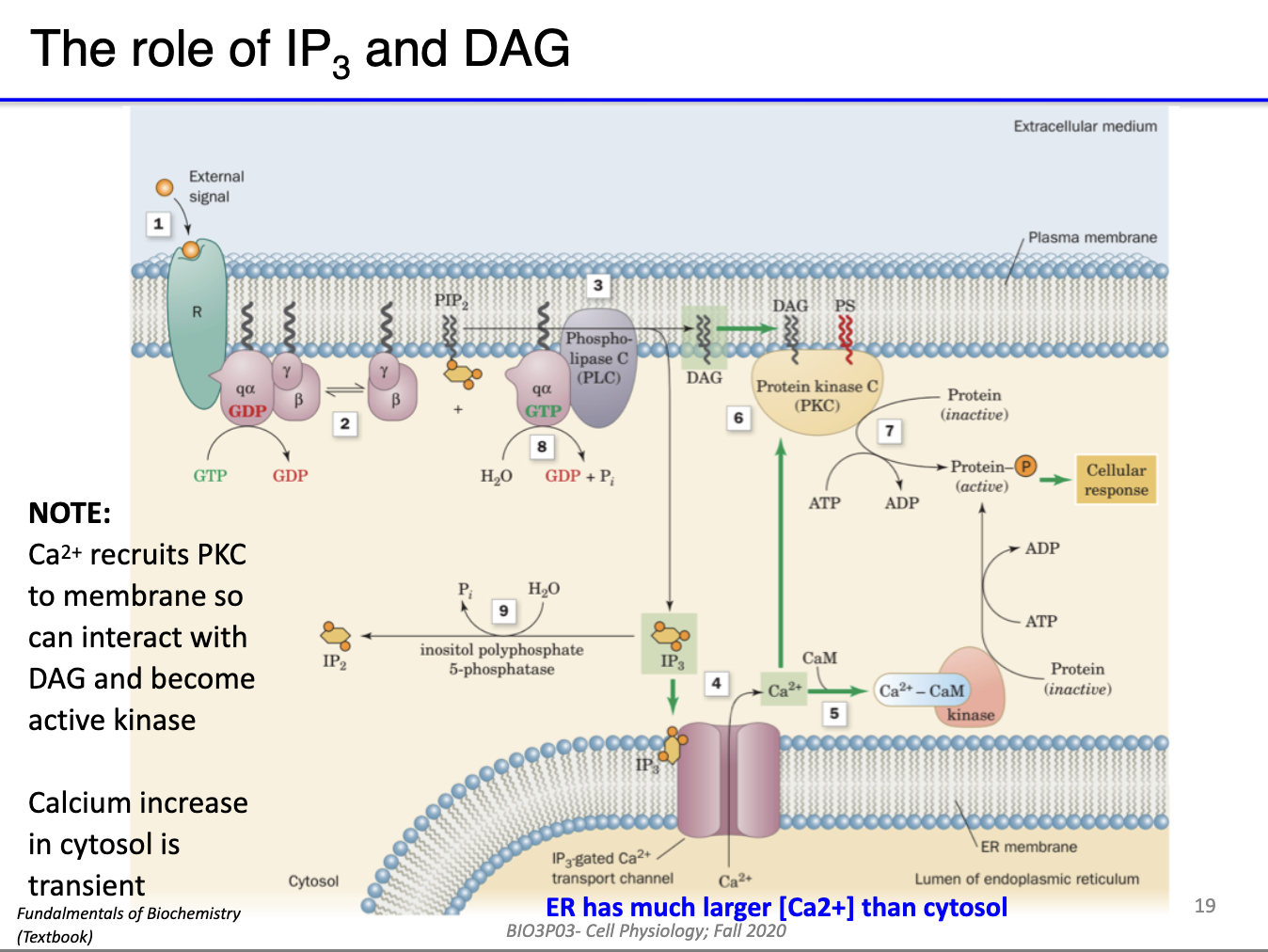
How does the GPCR-mediated inositol phospholipid (IP3/DAG) signaling pathway work?
GPCR activation: Ligand binds GPCR → α subunit of Gq protein separates from βγ subunits
Gq protein: α subunit activates phospholipase C β (PLCβ)
Substrate: PLCβ catalyzes PIP2 → DAG + IP3
Second messengers:
IP3: Diffuses through cytoplasm → interacts with Ca²⁺ channels on ER/SR → Ca²⁺ release
DAG: Stays in inner membrane leaflet → activates protein kinase C (PKC) and D
Outcome: Initiates intracellular signaling cascades → downstream effects (e.g., calcium signaling, protein phosphorylation)
Notes:
PLCγ/tyrosine kinase pathways not tested in this course
Distinct from Gs/cAMP pathway; uses Gq/PLCβ → IP3/DAG instead of adenylyl cyclase

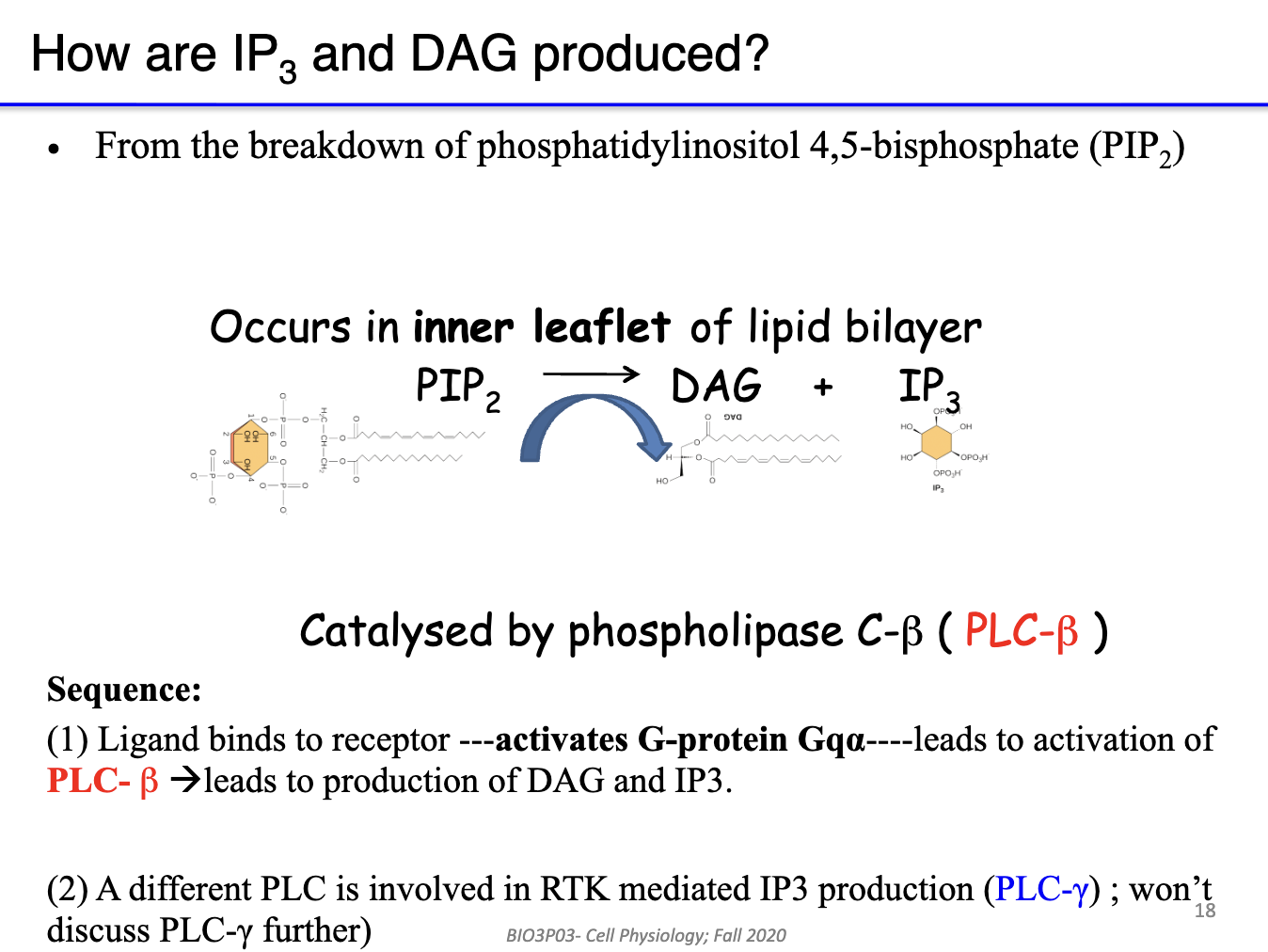
How does PIP₂ produce DAG and IP3 in Gq GPCR signaling, and what are their roles?
PIP₂ (phosphatidylinositol 4,5-bisphosphate) is the substrate
GPCR ligand binds → Gq α-subunit activates PLC-β
PLC-β cleaves PIP₂ → DAG + IP3
DAG: lipid-based, stays in inner leaflet of membrane → activates PKC locally
IP3: soluble, diffuses through cytoplasm → binds ER/SR → releases Ca²⁺
Outcome: DAG & IP3 act as second messengers for downstream signaling
Note: PLC-γ (tyrosine kinase-associated) not relevant here
How does Gq GPCR signaling lead to second messenger responses?
GPCR activated by ligand → Gq α-subunit exchanges GDP → GTP
Gq α-subunit activates PLC-β
PLC-β cleaves PIP₂ → DAG + IP3
DAG: stays in inner membrane → activates PKC
IP3: diffuses through cytoplasm → releases Ca²⁺ from ER/SR
Outcome: DAG & IP3 serve as second messengers, initiating downstream cellular responses