22. Autophagy 2
1/21
Earn XP
Description and Tags
complete
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
22 Terms
Autophagy in neurodegeneration
Why does autophagy play such a crucial role in maintaining neuronal health?
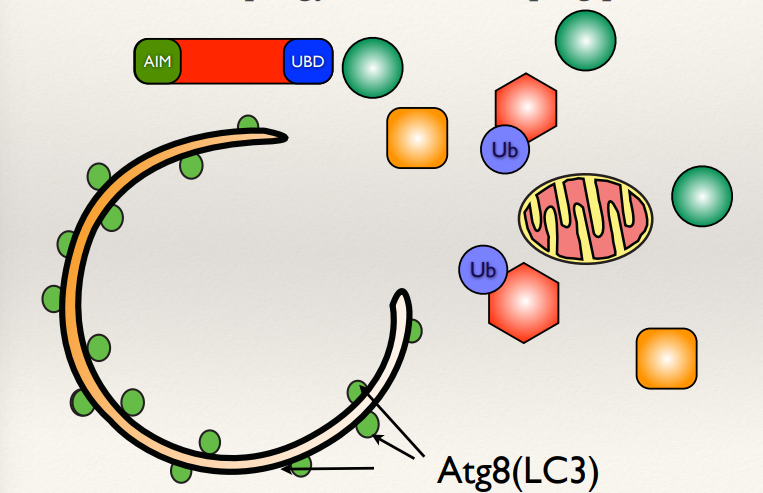
What is Atg8 (LC3)?
What is the AIM and UBD motif?
Acts as a cellular housekeeping mechanism, which is particularly vital in neurons, as they are non-dividing, long-lived cells with limited regenerative capacity.
Atg8 in yeast (LC3 in humans): A ubiquitin-like protein that facilitates membrane elongation and autophagosome formation.
AIM (Atg8-Interacting Motif) and UBD (Ubiquitin-Binding Domain) are motifs on cargo proteins or receptors that interact with Atg8/LC3, enabling the selective sequestration of target materials into autophagosomes.
Autophagy in mitochondrial health
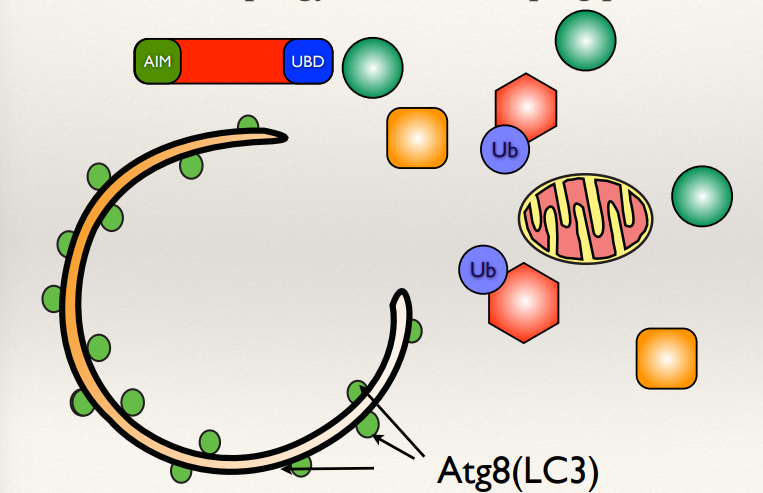
How does autophagy contribute to mitochondrial health?
What chemical can damage mitochondria?
What targets the damaged mitochondria and removes it?
Autophagy selectively removes damaged mitochondria to maintain cellular health.
CCCP can chemically damage mitochondria, depolarising mitochondria, triggering autophagy.
The damaged mitochondria is targeted by LC3 which is on the autophagosome membrane. Autophagosomes form around the mitochondria to remove it.
Lacking functional autophagy
What accumulates in cells lacking functional autophagy?
Which molecules evidence this?
Cells that lack autophagy accumulate protein aggregates, leading to cellular dysfunction and contributing to neuronal death.
This is evidenced by markers such as ubiquitin and GFP-tagged ATP18, which indicate the presence of undegraded protein aggregates.
Neurons are particularly sensitive
Why are neurons highly sensitive to cellular damage?
What happens when autophagy is specifically disrupted in neurons?
What are the consequences of long-term damage accumulation in neurons?
Neurons are sensitive due to their large size and the fact that they do not regenerate over a lifetime. Damage accumulates over time, which can eventually reach a pathogenic threshold, leading to neurodegeneration.
Disruption leads to the accumulation of ubiquitinated protein aggregates and increased apoptosis. This results in a very rapid onset of neurodegeneration, highlighting the critical role of autophagy in neuronal health.
Over time, accumulated damage can cross a pathogenic threshold, leading to the formation of toxic aggregates, cellular dysfunction, and ultimately, neurodegeneration.
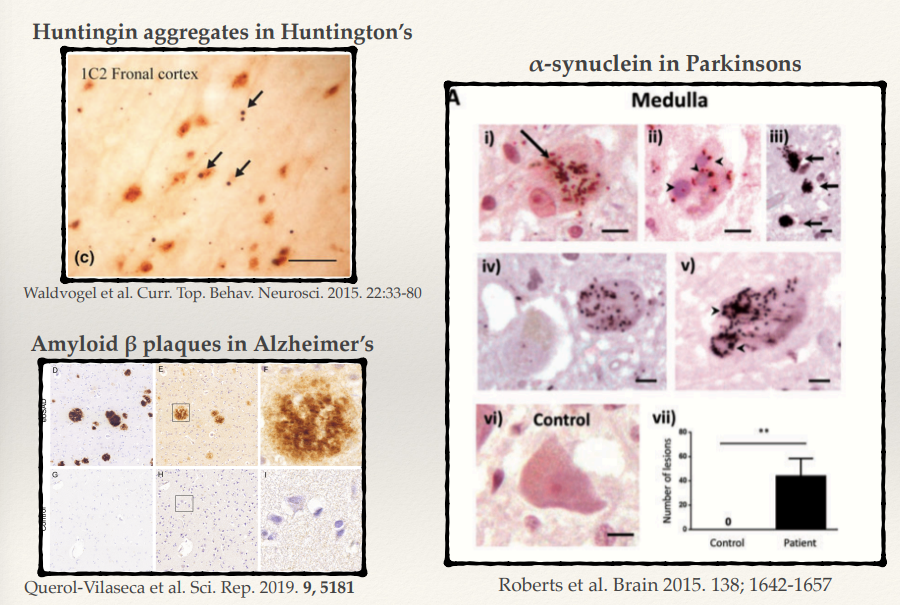
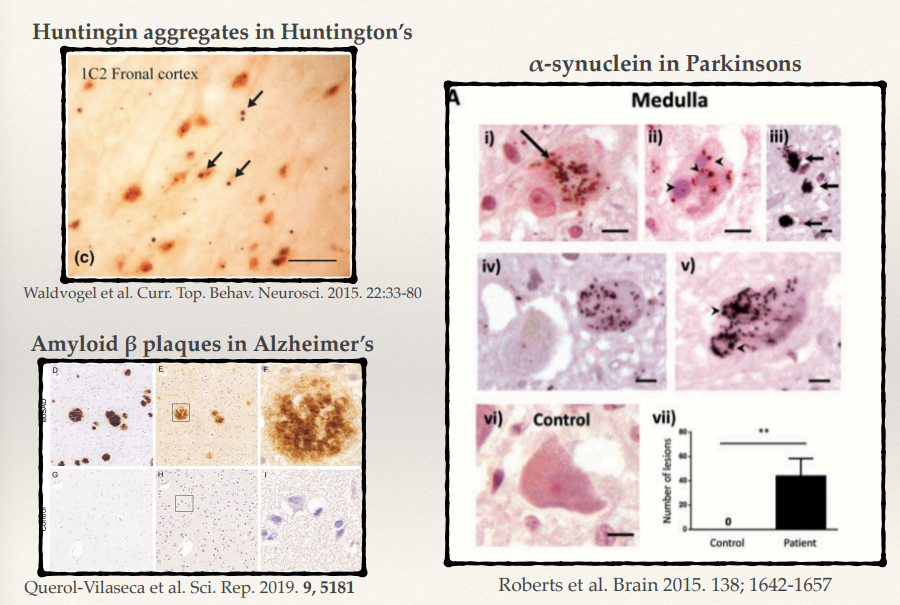
Proteinopathies in neurodegeneration
What are proteinopathies, and how are they linked to neurodegeneration?
What is the key protein involved in Huntington’s disease, and how does it contribute to pathology?
What protein aggregates are found in Parkinson’s disease, and what do they form?
What are the hallmark protein aggregates in Alzheimer’s disease (2)?
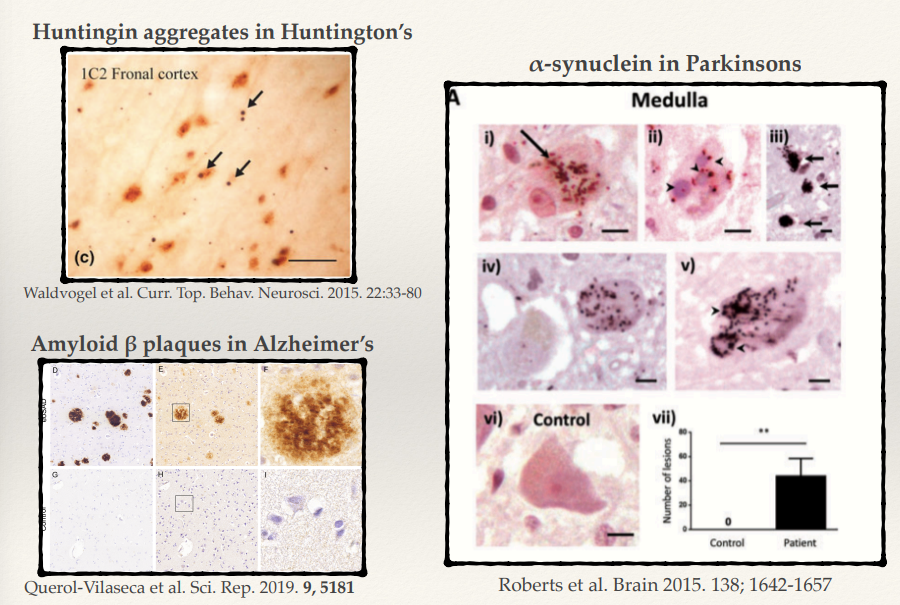
Proteinopathies are diseases characterised by the misfolding and accumulating of specific proteins aggregates. These aggregates disrupt cellular function and are central to the pathology of many neurodegenerative diseases.
Huntington’s disease involves huntingtin protein with expanded polyglutamine (CAG) repeats. Misfolded huntingtin forms toxic aggregates, leading to neuronal dysfunction and death, particularly in the striatum and cortex.
In Parkinson’s disease, α-synuclein aggregates form Lewy bodies. These aggregates disrupt cellular homeostasis, impairing synaptic function and mitochondrial health, particularly in dopaminergic neurons.
Alzheimer’s disease is characterized by:
Amyloid β plaques: Extracellular aggregates of amyloid β peptides.
Tau tangles: Intracellular aggregates of hyperphosphorylated tau protein. These disrupt synaptic communication and lead to neuronal death.
Despite involving different proteins, what common pathological feature is shared across Huntington’s, Parkinson’s, and Alzheimer’s diseases?
All these diseases involve the same phenotype: accumulating in big ubiquitin aggregates:
Huntingtin in Huntington’s disease.
α-synuclein in Parkinson’s disease.
Amyloid β and tau in Alzheimer’s disease. These aggregates impair cellular function, leading to neurodegeneration.
Huntington’s, Parkinson’s, and Alzheimer’s all have the a common pathological feature, what about symptoms?
They have different kinds of symptoms
Different types of neurones are particularly sensitive to different types of proteins
Could be a combination of how heavily these proteins are expressed in different regions of the brain or how highly expressed the genes that are causing the underlying diseases are.
Huntington’s Disease
What is the genetic mutation responsible for Huntington’s disease?
How does the length of polyQ repeats determine Huntington’s disease?
What correlation does the proportion of glutamine in this region have with the onset and severity of the disease?
Huntington’s disease is caused by a polyglutamine (polyQ) expansion in the huntingtin (HTT) protein, resulting from an increase in CAG repeats in the HTT gene.
PolyQ sequence length varies from person to person:
Q < 18: Normal, healthy protein.
Q > 35: Disease-causing, with severity increasing as the repeat length grows.
A direct correlation between proportion of glutamine in the region with onset and severity of the disease- so you can predict quite accurately a person’s susceptibility to Huntington’s.
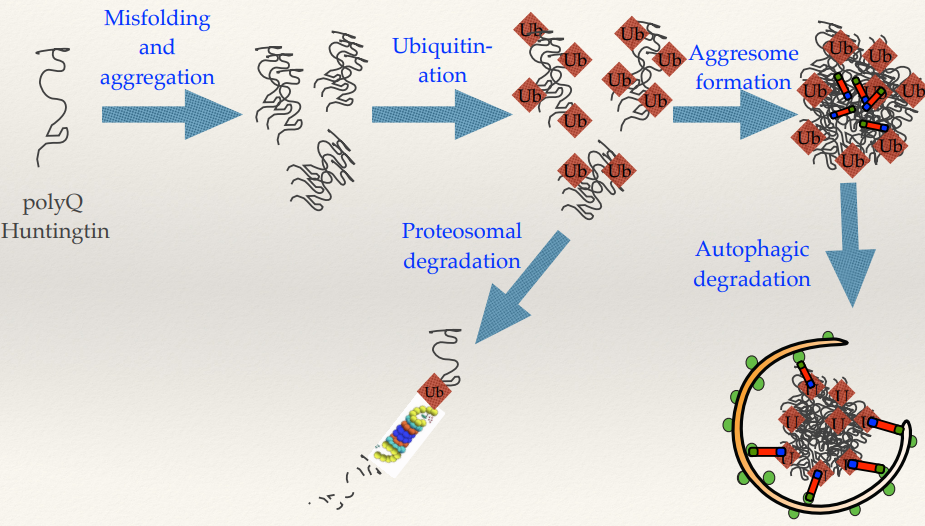
Huntington’s disease protein misfolding and aggregation pathway
Name the order of the steps in the pathway (5).
What are the cellular pathways for managing misfolded proteins, what happens?
What is an aggresome?
(SEE IMAGE) Misfolding and aggregation --> ubiquitination (also then --> proteosomal degradation) --> aggresome formation --> autophagic degradation.
Misfolded and aggregated polyQ Huntingtins are:
Ubiquitinated: Tagged with ubiquitin for recognition.
Proteosomal degradation: Directed to the proteosome for breakdown.
Aggresome formation: If proteasomal degradation is insufficient, aggregates are sequestered into an aggresome.
Autophagic degradation: Aggresomes are cleared via autophagy.
Aggresome: very large aggregates of lots of misfolded proteins lumped together. Theory is that sequestering them away from the body/rest of the cell, the large aggresomes can then be targeted by autophagy.
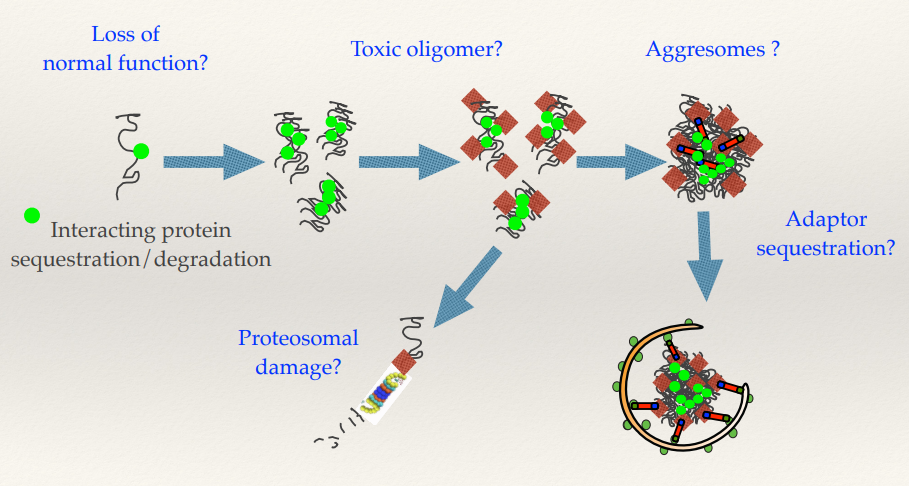
Multiple mechanisms of toxicity through the misfolding and aggregation pathway in Huntington’s disease
What does the proteins misfolding lead to?
What do the oligomers (micro aggregates) become?
What happens with aggresomes and adaptor sequestration?
Misfolding proteins means you’re losing the active protein - Loss of normal function. Losing the Huntingtin protein, affecting the cell’s function.
The oligomers (micro aggregates) can also be toxic, as they actually damage the proteosome. The proteosome feeds in one at a time and will degrade each one individually. Micro aggregates can clog up and damage the proteosome. This affects the cell’s ability to use the proteosome to regulate other signalling pathways - highly deleterious for the cell.
You can sequester proteins within the aggresome - anything that binds to Huntingtin will also potentially be sequestered in these aggresomes and removed from its normal functions within the cell.
Autophagy and Parkinson’s disease
How common is Parkinson’s disease in the UK?
What is the main neuropathological features of Parkinson’s disease (2)?
How does genetics contribute to Parkinson’s disease?
Parkinson’s disease affects 1–2 per 1000 people in the UK, primarily in older adults.
The key feature is the loss of dopaminergic neurons, accompanied by the formation of α-synuclein aggregates (Lewy Bodies).
Parkinson’s has complex genetics, only 5–10% of cases are familial and α-synuclein itself is rarely mutated.
Parkinson’s: α-synuclein mutations (rare)
What is the main pathways for α-synuclein degradation?
How does this pathway degrade α-synuclein?
Name the example α-synuclein mutation.
What impact do α-synuclein mutations have on CMA?
How would α-synuclein then be degraded?
Why is this a problem?
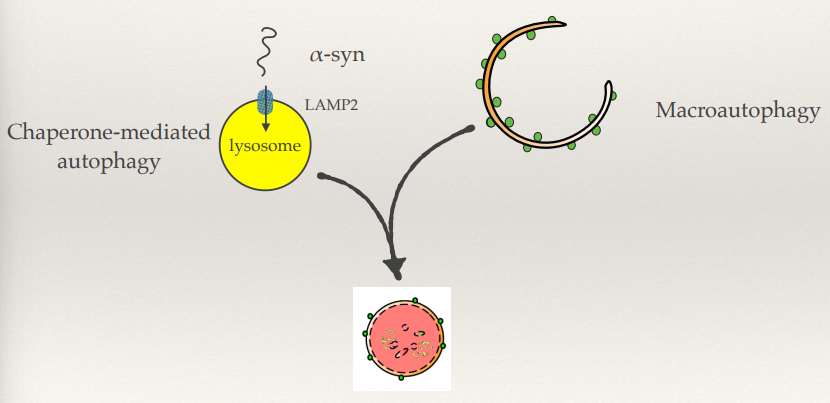
Pathway for degradation:
Chaperone-mediated autophagy (CMA): α-Synuclein is recognized by chaperones and transported into the lysosome via LAMP2A receptors.
Chaperones bind α-synuclein and guide it to LAMP2A receptors on lysosomes. α-Synuclein is unfolded and translocated into the lysosomal lumen for degradation.
A53T
Mutated forms of α-synuclein, like A53T, bind to LAMP2A but fail to translocate into the lysosome. This blocks the CMA pathway, leading to a build-up of toxic aggregates and increased cellular stress.
The accumulated proteins then need to be degraded by macroautophagy as well.
Macroautophagy is a backup pathway and its a matter of capacity - as you get older, the rate of being able to produce macroautophagy decreases. This disrupts the balance of damage removal and damage being produced.
Parkinson’s and mitochondrial disfunction
How are mitochondria involved in Parkinson's disease?
What role do mitochondria play in producing Reactive Oxygen Species (ROS)?
What hypothesis has been derived due to the role of mitochondrial damage in Parkinson’s?
Parkinson’s is also caused by dysfunction in mitochondria. Damaged mitochondria accumulate, impairing cellular energy production and contributing to neuronal death.
Mitochondria are the primary source of reactive oxygen species (ROS). Under stress or dysfunction, mitochondria generate excess ROS, which can damage cellular components like lipids, proteins, and DNA.
Hypothesis: Parkinson’s may be caused by mitochondrial-derived oxidative damage more so than by α-synuclein mutations.
Parkinson’s: PARKIN and PINK1
What do PARKIN and PINK1 do?
What is PINK1, and where does it accumulate?
What % of LoF mutations in PINK1 are associated with sporadic early-onset Parkinson’s?
What is PARKIN, and when is it recruited?
What % of autosomal recessive diseases is PARKIN mutated in?
What % of sporadic early-onset Parkinson’s cases is PARKIN mutated in?
PARKIN and PINK1 regulate mitophagy
PINK1 is a mitochondrial kinase, it accumulates on the outer membrane of damaged mitochondria.
Loss of function mutations in PINK1 are associated with 5–10% of sporadic early-onset Parkinson’s cases.
PARKIN is a cytosolic E3 ubiquitin ligase that is recruited to the mitochondria when PINK1 accumulates on it.
50% of autosomal recessive disease cases.
10-15% of sporadic early-onset Parkinson’s cases.
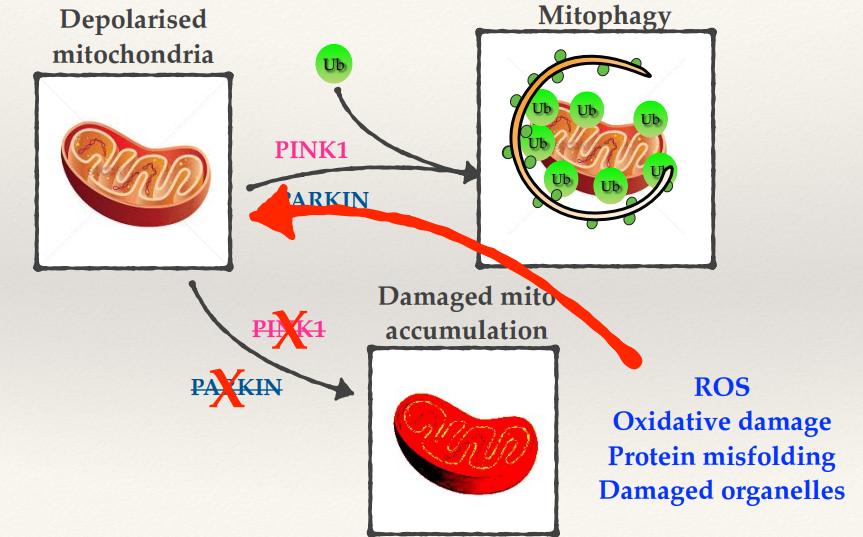
Parkinson’s: Mitophagy and Neurodegeneration
What do PINK1 and PARKIN do to damaged mitochondria?
What happens if you have mutations in PINK1 and PARKIN?
What is linked with the progressive nature of diseases such as Parkinson’s?
If you have a damaged mitochondria PINK1 and PARKIN work together to ubiquinate it and then target that mitochondria specifically to mitophagy.
Mutations in PINK1 and PARKIN cause accumulation of damaged mitochondria and its no longer able to be removed, this will generate high levels of ROS. This will cause oxidative damage, protein misfolding and damaged organelles.
Once you start experiencing some accumulated mitochondrial damage, you will then produce more ROS, which will damage more of your mitochondria which would make more damage. This spirals into damage and dysfunction within your cells.
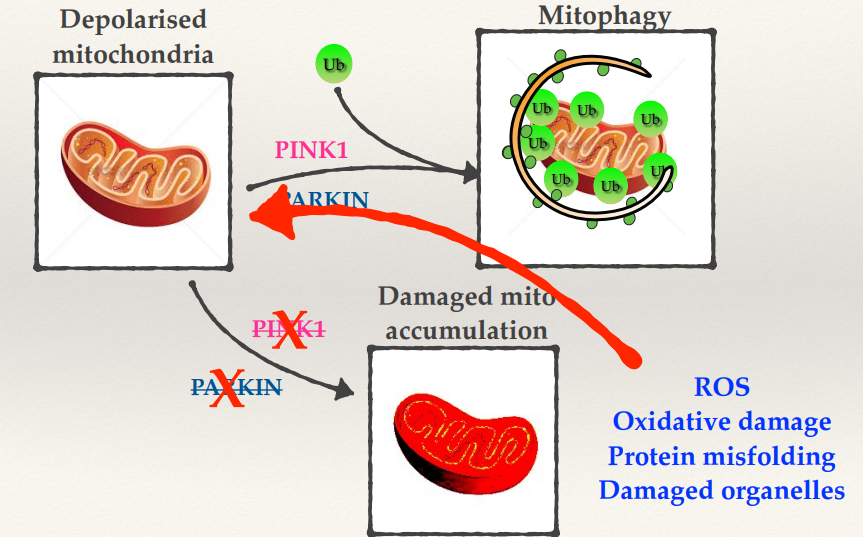
Explain some other mutations affecting autophagy in neurodegeneration.
Impaired autophagosome formation
Disrupted lysosomal function
Disruption of cargo recognition
Mutations in the ability to target autophagic cargo - e.g tauopathies
Inhibited autolysosome formation
None of the mutations completely block autophagy - that would be fatal.
Autophagy and cancer
What is cancer caused by?
What impact does autophagy have on cancer?
Explain how it has this impact.
In cancer cells what observation is often found?
What role does Beclin1 (Atg6) play in cancer?
Cancer is caused by accumulation of DNA damage
Autophagy is tumour suppressive
Autophagy can prevent cells from accumulating damage, so you don’t get as much damage to organelles, ROS and protein toxicity, thus reducing oxidative stress, DNA damage and tumourigenesis.
Autophagy is often downregulated in those cells.
Beclin1 is monoallelically deleted in 40-75% of ovarian, breast, and prostate carcinomas. This deletion impairs autophagy and may contribute to tumourigenesis.

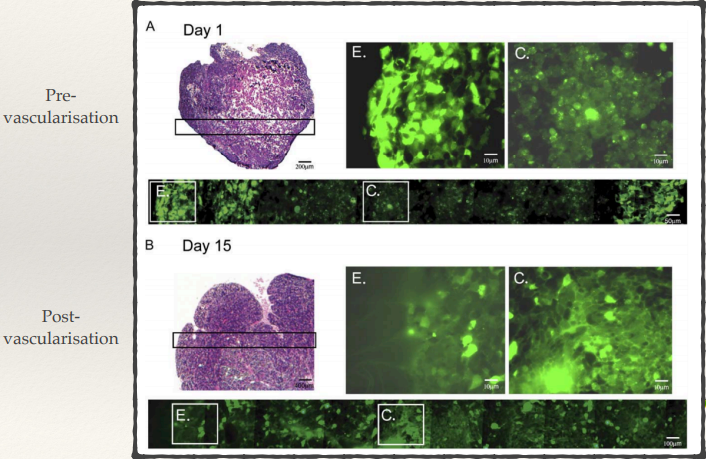
Autophagy and cancer
How does autophagy behave in hypoxic, nutrient-poor tumour regions?
How does autophagy differ between pre-vascularisation and post-vascularisation?
What would blocking autophagy do?
Hypoxic, nutrient-poor regions:
Autophagy is upregulated, as cancer cells adapt to low oxygen and nutrient conditions by breaking down internal components to provide energy and maintain survival.
This pro-oncogenic autophagy supports tumour growth by helping cells survive under stressful conditions.
Pre- and post- vascularisation:
Pre-vascularisation tumours have lots more autophagy occurring in the centre of the tumour since there’s no vascularisation there. Tumours in early stages depend on autophagy to survive limited nutrient supply (starving).
Having autophagy is only really important in the early stages because as tumours develop you have things like angiogenesis (new blood vessels developing), so good blood supply reaches the centre of the tumours - autophagy returns to normal.
If autophagy is blocked (particularly in early tumours) it can cause the centre of the tumour undergo apoptosis or necrosis - so they won’t be able to survive in the centre and tumour can die.
Autophagy also inhibits apoptosis
Is autophagy pro-survival or pro-death?
What protein generates autophagosome formation?
What protein suppresses apoptosis?
What interaction occurs with these proteins?
What happens if you activate autophagy?
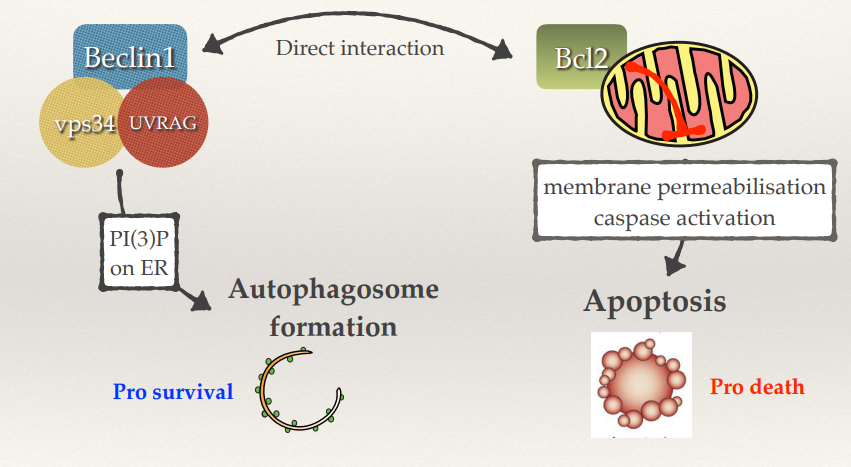
Autophagy tends to be pro-survival (e.g trying to survive starvation).
Beclin1 is part of a complex that generates autophagosome formation.
Bcl2 (on the surface of mitochondria) suppresses apoptosis.
Beclin1 and Bcl2 directly interact, so sometimes you’ll have low autophagy and maximum apoptosis. Bcl2 is unable to suppress apoptosis and Beclin1 is unable to allow autophagosomes to be generated. Cells will go down the apoptotic pathway without trying to survive.
If you activate autophagy Beclin1 goes back to its friendly complex to produce autophagosomes and Bcl2 goes back to the mitochondria, suppressing apoptosis.
Chemotherapy, autophagy and apoptosis
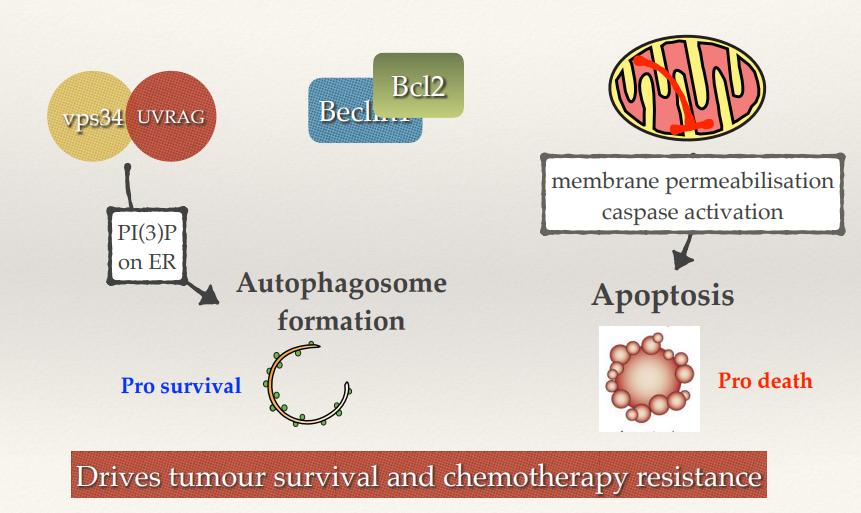
By having autophagy activated, how does it impact tumour cells?
What is the theory in inhibiting autophagy during chemotherapy?
By having autophagy activated it allows the tumour cells to survive and suppress apoptosis. Drives tumour survival and chemotherapy resistance. Not all the cells within the tumour will undergo apoptosis (chemotherapy resistance).
Theory is that by inhibiting autophagy whilst undergoing chemo you might be able to push more cells into the apoptotic pathway and prevent the likelihood that some of them will survive and go on to cause secondary tumours.
Autophagy’s double function with cancer
When is autophagy anti-oncogenic?
Generally autophagy is anti-oncogenic:
Cell Homeostasis
Damage removal
Reduced ROS/genotoxicity
Reducing inflammation
Pro-oncogenic - once you have cancer, the cancer cells then use autophagy in order to try and survive harsh conditions:
Survival during oxygen or nutrient shortage
Prevention of apoptosis
Survival during chemotherapy
Describe 3 strategies for autophagy therapeutics in cancer.
Blocking survival to metabolic stress with autophagy inhibitors (prevent survival through early-tumour starvation).
Inhibit autophagy to increase apoptosis during chemotherapy.
Elevate autophagy to remove damage and prevent cancer.