(pt 1) exam #1 - intro to molecular diagnostics (cls 605)
1/128
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
129 Terms
what makes up a nucleotide? (3)
pentose sugar
nitrogenous base
phosphate group (can have 1, 2, or 3)
free nucleotides in active form typically have 3 phosphate groups attached before they get attached to a DNA/RNA strand (triphosphates)
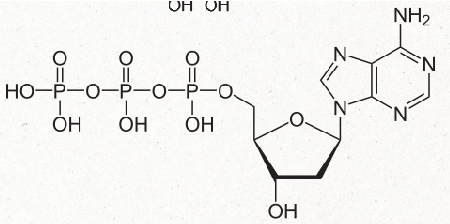
difference betwen RNA vs DNA nucleotide
RNA nucleotide has a OH group at the 2' carbon while a DNA nucleotide does not
carbons are numbered 1'-5' with the nitrogenous based attached to carbon at the 1'
nucleoside
has no phosphate groups attached
still has pentose sugar and nitrogenous base
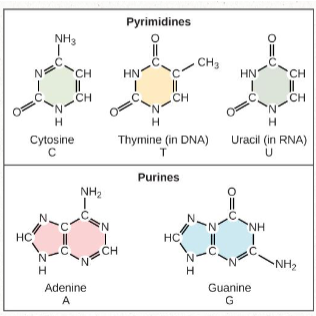
nitrogenous bases
includes:
Pyrimidines: cytosine and thymine/uracil (one ring)
Purines: adenine + guanine (two rings)
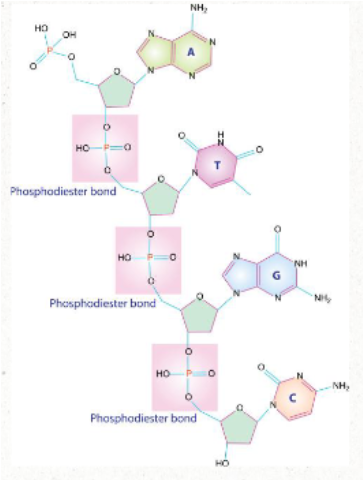
how do nucleotides become nucleic acids?
One strand of nucleic acid is generated when nucleotides attach to each other by their phosphate group
This creates what is called the sugar-phosphate backbone of the molecule
bond is created via a condensation rxn
bonding gives the molecule directionality--new nucleotides are added to the 3' end of a DNA strand
ATTACHED VIA PHOSPHODIESTER BONDS
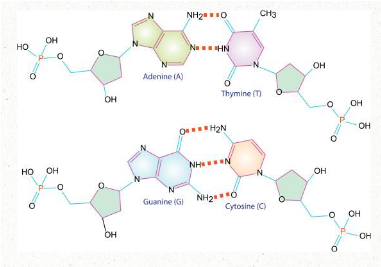
how does DNA become double stranded?
Nitrogenous bases attach via HYDROGEN bonds
Adenosine pairs with thymine
2 hydrogen bonds
Guanine pairs with cytosine
3 hydrogen bonds
For proper base pairing to occur the two DNA strands must run in opposite directions (antiparallel)
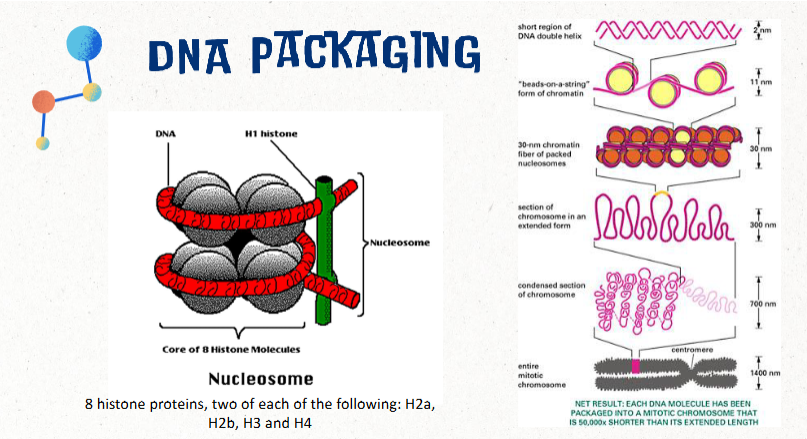
DNA packaging
If DNA was not packaged in any way, the DNA from a single cell would be about 2 meters (6.6 ft) long
This would be approximately the equivalent of packing 24 miles of thread into a tennis ball
packaged into a nucleosome made of 8 histone proteins
two of each: H2a, H2b, H3, H4
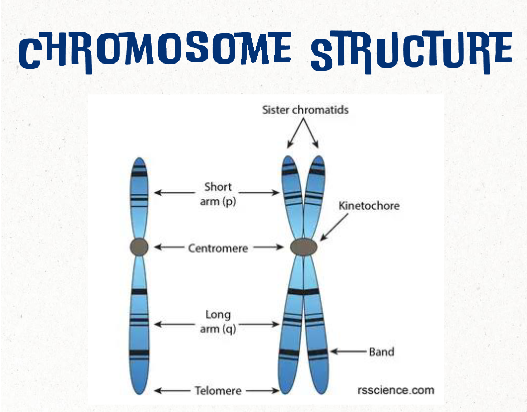
chromosome
threadlike structure of nucleic acids and protein found in the nucleus that carries genetic information (genes)
most human cells contain 23 pairs of chromosomes (46 total)
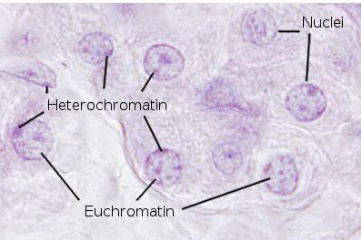
chromatin
less condensed form of DNA and proteins; less organized compared to chromosomes
found in interphase nuclei
euchromatin vs heterochromatin
euchromatin: relaxed chromatin that is transcriptionally active
heterochromatin: more condensed--no transcription (NOT active)
nucleosome
structural unit of a eukaryotic chromosome; consists of approx 150 bp of DNA wrapped around 8 histones
chromatid
one of two identical halves of a chromosome in preparation for cell division
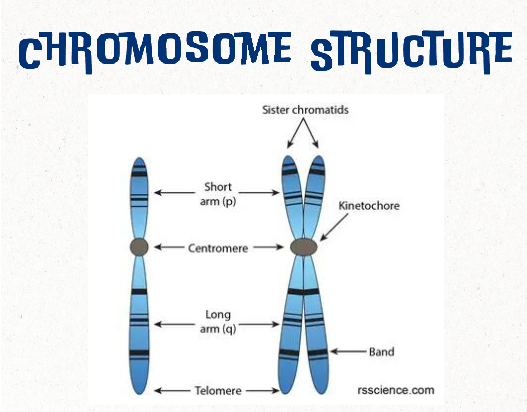
human karyotype (general)
Karyotype is the direct observation of metaphase chromosome structure
Karyotyping can be used to see large changes in chromosomes such as:
Aneuploidy
Translocations
Large insertions or deletions
Inversions
karyotype presentation
Cultured cells are arrested at metaphase
cells are most condensed and easiest to identify
Arrested cells are broken open
Metaphase chromosomes are fixed and stained
Chromosomes are digitally imaged through microscope
Digital images of chromosomes are arranges to form a karyotype diagram
gene (definition)
units of information about heritable traits
each gene has a particular locus
locus
specific spot/location on a chromosome
alleles
different forms of genes that arise through mutation
A diploid cell contains 2 alleles at each locus
Alleles on homologous chromosomes may be the same or different
exons vs introns
exon: coding regions of a gene; included in the final mRNA transcript and translated into protein
intron: non-coding sequences within a gene that are transcribed into RNA but are removed/SPLICED before translation into a protein
genotype vs phenotype
genotype: genetic DNA composition of an organism
phenotype: observable traits
dominant vs recessive allele
dominant allele: allele that affects/masks the other allele with which it is paired
recessive allele: allele whose effect is masked by the other allele with it is paired
homozygous vs heterozygous
homozygous: genetically identical pair of alleles
heterozygous: a pair of alleles for a trait that are not genetically identical
nucleic acid isolation (general)
Goal: isolate DNA from inhibitors and impurities that can interfere with testing
direct interference: things like enzyme inhibitors
indirect interference: impurities that bind to DNA and make it unavailable for a reaction
samples that can be used for isolating nucleic acids
Any sample containing the targeted nucleic acid
Whole blood / buffy coat
Bone marrow
Solid tissue
Lavage fluids
Bacteria, viruses, fungi
Organelles, mitochondria
best type of fixed specimens/fixatives for PCR
Acetone
10% buffered neutral formalin
FFPE specimens (formalin fixed, paraffin embedded)
mid/meh fixatives for PCR (not as good)
Zambonis, clarkes
Paraformaldehyde
Formalin-alcohol-acetic acid
Metharcan
less desirable fixatives for PCR
Carnoys, Zenkers, Bouins, B-5
general steps of DNA isolation (3)
Cell lysis
Disrupt membranes
Denature or degrade proteins
Extraction
Separate DNA from proteins and other impurities
Can use a solid or liquid phase
Precipitation
Further purifies DNA
Removes residual contaminants
(DNA isolation) cell lysis—disrupting membranes
Detergents work by disrupting membranes
Ex: sodium dodecyl sulfate (SDS); cetyltrimethylammonium bromide (CTAB)
Chaotropic salts work by disrupting hydrogen bonds
Ex: urea; guanidium hydrochloride
Results in a "soup" of cellular debris and the contents of the cytoplasm
(DNA isolation) cell lysis—degrading proteins
Proteases--enzymes that degrade protein
Work to:
Degrade proteins that bind DNA
Degrade nucleases (enzymes that degrade nucleic acids)
Detergents and chaotropic salts
Denature proteins which:
Disrupts function
Makes more susceptible to proteases
(DNA isolation) alternative methods of cell lysis
Sonication
Freeze-thaw
Physical disruption
Manual grinding
(DNA isolation; extraction) liquid phase
Phenol/chloroform--denatures proteins, doesn't mix with water so DNA in upper liquid phase (NOT preferred method)
Procedure
Add phenol/chloroform to sample
Mix thoroughly to form emulsion
Separate phases by centrifugation
DNA in upper aqueous phase
Precipitated proteins rest on top of lower phenol chloroform layer
Upper layer containing DNA is pipetted into a new tube for further purification
disadvantages to liquid phase DNA extraction
Lengthy
Labor intensive
Requires further purification
Difficult to automate
Safety issues
Phenol chloroform carryover
(DNA isolation; extraction) solid phase
DNA binds to silica (solid) in aqueous buffers w high salt concentrations (PREFERRED method)
Procedure (what we did in lab)
Lyse cells in high salt buffer
Add silica beads OR pass lysate through a column with silica filter (DNA will stick to silica)
Wash off impurities with high salt buffer
Wash with alcohol to remove salt
Elute DNA (remove from solid) with low ionic strength aqueous solution
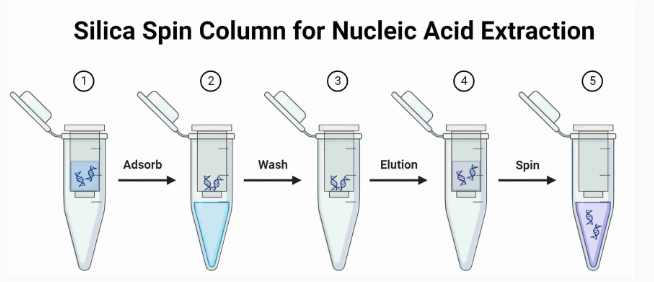
advantages to solid phase DNA extraction
Generally DNA is pure enough so that further cleaning (precipitation) is not needed
Quick, easy, safe, kit available (expensive though)
(DNA extraction) automated silica extraction
Silica coated magnetic beads
Increased binding kinetics/efficiency
Enhanced removal of contaminants
Commercially available

(DNA isolation steps) precipitation
Add salt and alcohol to “clean” DNA
Salt
Cations bind to phosphate groups of DNA and neutralize charge
Now DNA molecules won't repel each other
Alcohol
DNA Is insoluble in alcohol
Adding alcohol makes DNA precipitate out of solution
Precipitated DNA is stringy and can be pelleted or "spooled"
modifications to DNA extraction method for RNA extraction
RNA very susceptible to degradation
Bench/equipment—keep separate, clean with RNase inhibitors
Disposables—certified RNase free, rinsed in 0.1% diethyl pyrocarbonate (DEPC)
Reagents—purchased RNase free or treated with DEPC, Trizol (guanidium, phenol, chloroform)
Reactions—add RNase inhibitor
mRNA isolation
Total RNA can be obtained with either:
A silica-based binding technique or
Purification procedure with TRIzol reagent
From total RNA, mRNA purification is best accomplished via binding to Oligo dT fixed on a solid medium or column
methods for DNA quantification
spectrophotometry
fluorometry
qPCR
gel electrophoresis
methods for assessing purity of isolated DNA
spectrophotometry (purity)
methods for assessing quality of isolated DNA
gel electrophoresis
DNA quantification by spectrophotometry
Absorbance at 260 nm—BASES absorb strongly at this wavelength
An OD (optical density) of 1 corresponds to:
50 ug/mL for dsDNA
37 ug/mL for ssDNA
40 ug/mL for ssRNA
20 ug/mL for oligonucleotides
how to calculate concentration of DNA by spectrophotometry
conc of dsDNA (or whatever) in ug/mL = OD (at 260 nm) x 50 ug/mL x dilution factor
You are trying to find the concentration of your isolated RNA. You run 1 ul of sample on a spectrophotometer and the instrument returns an OD value of 0.5. What is your concentration?
0.5 x 40 ug/mL = 20 ug/mL
You have 100ul of isolated RNA of the concentration 20ug/ml. The protocol you need to run requires 5ug total of RNA. Do you have enough?
(20 ug/mL) / 1000 = 0.02 ug/ul
0.02 ug/ul x 100 ul = 2 ug
no u don't have enough
DNA purity by spectrophotometry (280 nm)
Abs at 280 nm--peptide bonds and some organic solvents absorb at this wavelength
Ratio of 260/280 is a measure of purity
Pure DNA 260/280 ratio is 1.8 (range 1.6-2.0)
Pure RNA 260/280 ratio is 2.0 (range 1.8-2.2)
Contamination by protein or solvent (ethanol or phenol) results in low 260/280 ratios
**If contaminated 260 nm readings cannot be used to estimate concentration
DNA purity by spectrophotometry (230 nm)
Absorbance at 230 nm--peptide bonds, carbohydrates, phenol, thiocyanates, and other organics absorb at this wavelength
Ratio of 260/230 is a secondary measure of purity
Pure DNA ratio should be >1.6
Pure RNA ratio should be >1.8
Contamination results in low 260/230 ratios and may interfere with downstream applications
nanodrop (spectrophotometry)
Sample size = 1-2 uL
Quantitation range = 2 ng/uL tp >3 ug/uL
No cuvettes
Evaluates purity and quantity
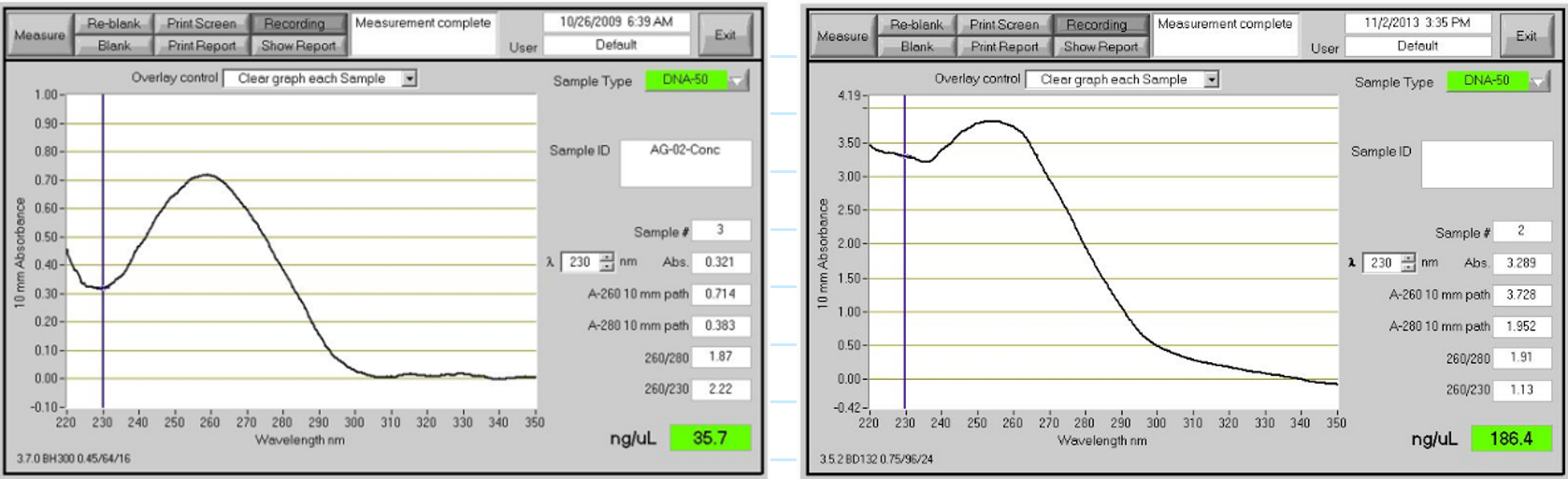
DNA quantitation by fluorometry
Mix DNA with a dye and then measure fluorescence—increased fluorescence is emitted when dye binds to dsDNA
Hoechst 33258
Quantitate to 10 ng/mL
Picogreen
Quantitate to 25 pg/mL (less sensitive to contaminants)
DNA quantitation by qPCR
Compare Ct for specimen to standard curve for human genomic DNA of known concentration
Amplify standard "housekeeping genes" such as
18S rRNA
Beta Actin
G6PD
purity vs quality
Purity: free from contamination
Quality: is the DNA actually DNA and if the product is intact
what method assesses DNA quantity and quality?
electrophoresis
what method assesses DNA quantity and purity?
spectrophotometry
DNA quantification/quality by gel electrophoresis (general)
DNA is negatively charged
When placed in an electric field, DNA will migrate towards the positive pole (anode)
An agarose gel is used to slow down the movement of DNA and separate by size
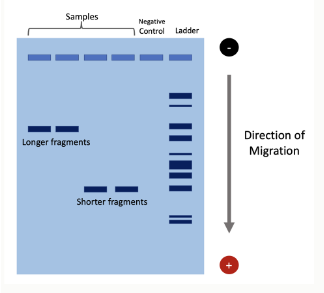
speed of DNA migration in gel electrophoresis depends on what?
Strength of electric field
Buffer
Agarose density
Size of DNA
Small DNA molecules move faster
basic equipment for gel electrophoresis
power supply, gel tank, cover, electrical leads, casting try, gel combs
(eletrophoresis steps/equipment) agarose preparation
Agarose powder is mixed with a buffer and boiled until it's clear
Agarose solution should be allowed to cool slightly and then poured into a casting tray
(eletrophoresis steps/equipment) gel preparation
Gel combs are added before the gel solidifies, this creates wells where samples can be added
When completely cooled, the agarose polymerizes which forms a flexible gel and the combs can be removed
Add enough electrophoresis buffer to cover the gel so that it is submerged with at least 1mm of liquid covering the gel
Make sure each well is filled with buffer
(eletrophoresis steps/equipment) sample preparation
DNA sample is mixed with loading buffer—serves two purposes:
Contains glycerol: adds weight to the sample to help it sink to the bottom of the wells
Contains a tracking dye (bromophenol blue) so that you can monitor where the DNA is on the gel
(eletrophoresis steps/equipment) loading the gel
Samples are loaded into wells
Hold the pipette tip in the liquid right at the top of the well and expel the sample
Be careful not to puncture the gel with the pipette
(eletrophoresis steps/equipment) running the gel
Place cover on electrophoresis chamber, connecting the electrical leads to the power supply
Be sure the leads are attached correctly--DNA migrates toward the anode (red)
When the power is turned on, bubbles should form on the electrodes in the electrophoresis chamber
After the current is applied, make sure the gel is running in the correct direction
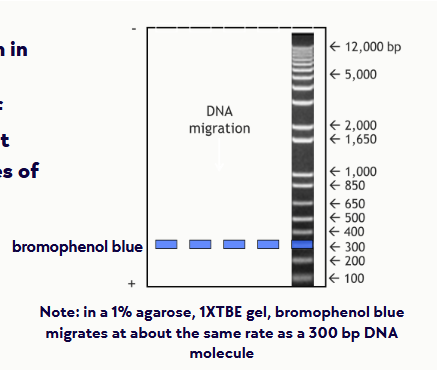
Bromophenol blue will run in the same direction as the DNA
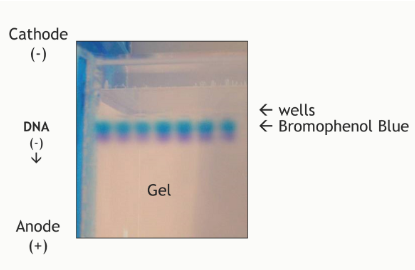
(eletrophoresis steps/equipment) DNA ladder standard
contains fragments of DNA of known sizes so that you can determine the sizes of your DNA fragments
(eletrophoresis steps/equipment) staining the gel
Ethidium bromide
Binds to DNA, fluoresces under UV light
Can be added before gel is poured or gel can be stained after running
Ethidium bromide is a powerful mutagen!!!—NEVER EVER HANDLE WITHOUT GLOVES
(eletrophoresis steps/equipment) visualizing product (pic)
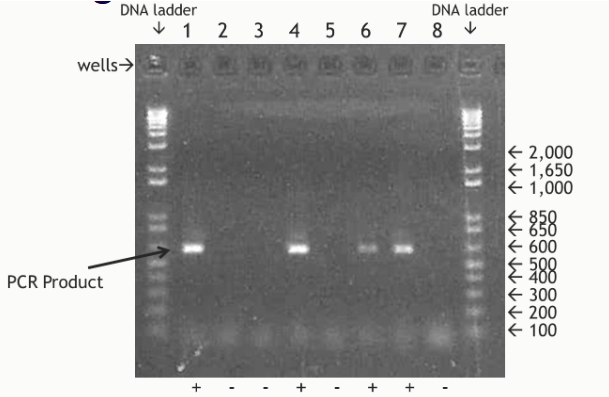
DNA quantity by gel electrophoresis
Fluorescence intensity is proportional to the total mass of DNA
Fluorescence of the sample is compared with DNA standards of known concentration

(electrophoresis practice example) You mix 2ul of your sample with 6 ul water and 2 ul loading dye and load the resulting mixture into lane 1
What is the size of the product?
What is the approximate concentration of your sample?
1000-1500 bp (1250)
40 ng/uL (take 80 ng and divide by 2)
only factor in the 2 uL of sample when deciding conc
DNA quality by gel electrophoresis
high quality nucleic acid appears as a tight band
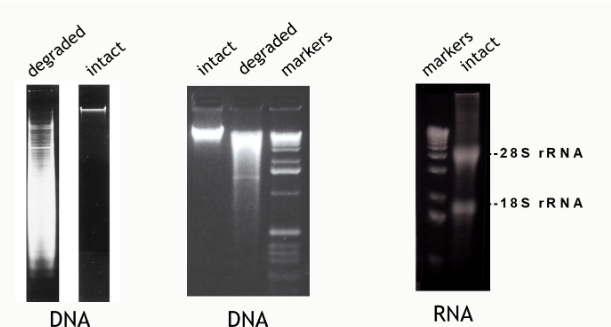
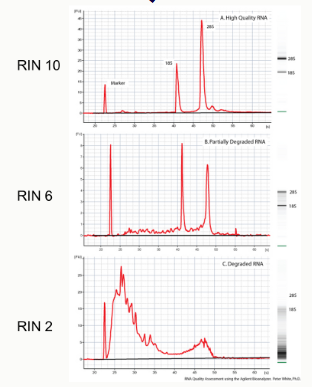
RNA integrity number (RIN)
assesses RNA quality throughout the electrophoretic profile, not just the 28S/18S ratio
RIN > 8 = acceptable indicator of total RNA quality
28S/18S rRNA mass ratio is about 2.5
A 28S/18S absorbance ratio > 2.1 is an indication that the total purified RNA is intact and HAS NOT degraded
human genome
Current tally--21,000 protein coding genes
Genes (25%)
24% intron sequences
1.1-1.4% exon sequences
Intergenic sequences (75%)
45% transposon-derived repeats (moveable gene regions)
5% duplications
3% repeats (ex: STRs)
22% other (ex: spacer)
99.9% identity between individuals
About 1 difference every 1250 bases between randomly selected haploid genomes
(human genome) sequence variation (general)
Single nucleotide polymorphisms (SNPs) are the most common type of sequence variation
DNA chances involving 1 base pair
~98% occur within non-coding sequences
~2% occur in exons
SNPs vs mutations
Mutation: any change in the "normal" human DNA sequence; mutation changes the sequence to something rare/abnormal
Single nucleotide changes ocurring in <1% of population = mutations
Single nucleotide polymorphism: a DNA sequence variation that is common in the population; no single allele is regarded as the standard sequence
single nucleotide polymorphisms (SNPs)
Over 100 million have been identified in the human genome
Some are common in the population with allelic frequencies of 0.1-0.5 (i.e. present in 10 to 50 of every 100 human genomes)
On average, occur once every 1,000 bases in a person (~4 million in every person's genome)
Distributed throughout genome
Major cause of genetic diversity among different (normal) individuals, e.g. drug response, diseases susceptibility
Can detect using a variety of molecular methods
significance of SNPs
Most do not change protein synthesis nor cause disease directly
Few associated with disease (ex: sickle cell anemia)
SNPs can serve as landmarks, since they may be physically close to a disease-associated gene on the chromosome
Because of this proximity, SNPs may be shared among groups of people with a disease (inherited as a haplotype)
haplotype
alleles that are close together on a chromosome and are usually inherited together
projects to understand human genetic variation
HapMap project
1000 genomes project
disease causing mutations
70% SNPs
49% missense
11% nonsense
9% splicing
<1% regulatory
23% small insertions/deletions
7% gross changes (large insertions/deletions, duplications, rearrangements etc)
**point mutations do NOT always have a phenotypic effect
chart of genetic mutations
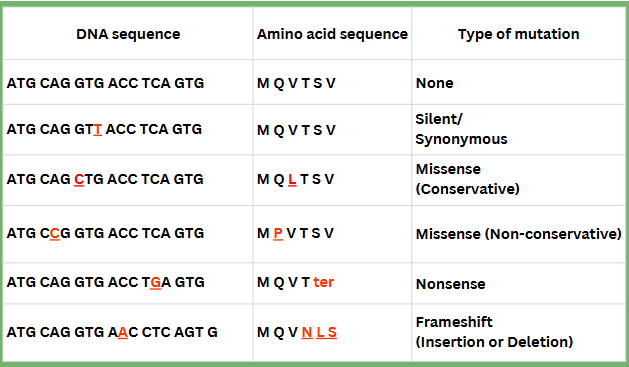
mutations that produce small/little changes
silent/synonymous
missense (conservative & non-conservative)
splice mutations
unstable trinucleotide repeats
nonsense
small insertions/deletions
frameshift
silent/synonymous mutation
point mutation which does not change the amino acid
missense mutation
point mutation which results in a different amino acid being inserted
Conservative: similar amino acid is substituted for original (ex: hydrophobic AA for a hydrophobic AA)
Non-conservative: the changed amino acid is significantly different from the original which may affect protein structure/function
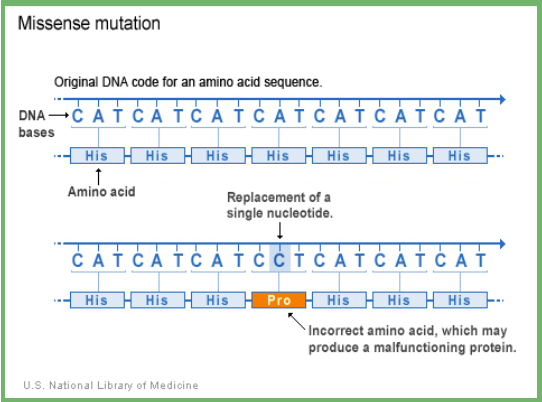
disease caused by missense mutations
Hemoglobinopathies
Cystic fibrosis
Sickle cell anemia
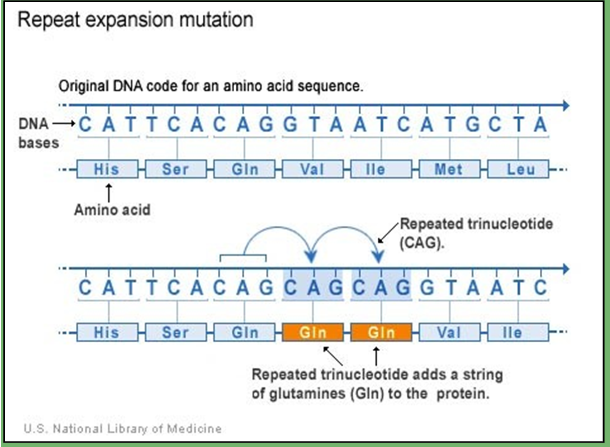
unstable trinucleotide repeats
areas of DNA that have repetitive sequences are prone to expand or contract during DNA replication
Diseases caused by unstable trinucleotide repeats:
Fragile X syndrome--(CGG)n in 5' UTR of FMR-1 gene
Normal: 5-44
Intermediate: 45-54
Premutation: 55-200
Full mutation: >200
Other examples: Huntington's disease, SCA (spinocerebellar ataxia)
splice mutations
disruption of existing splice sites (intron is not removed from mRNA)
Creation of new splice site in exon
Ex: HbE missense mutation and splice error
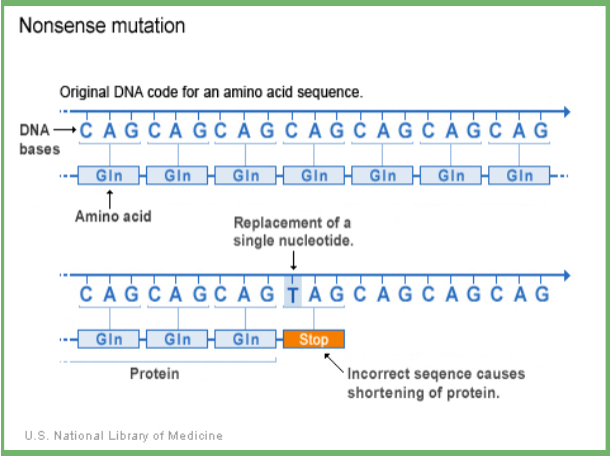
nonsense mutation
point mutations which replaces a codon that codes for an amino acid with a stop codon
small insertion/deletion mutation
adds or removes bases; may or may not cause a frameshift
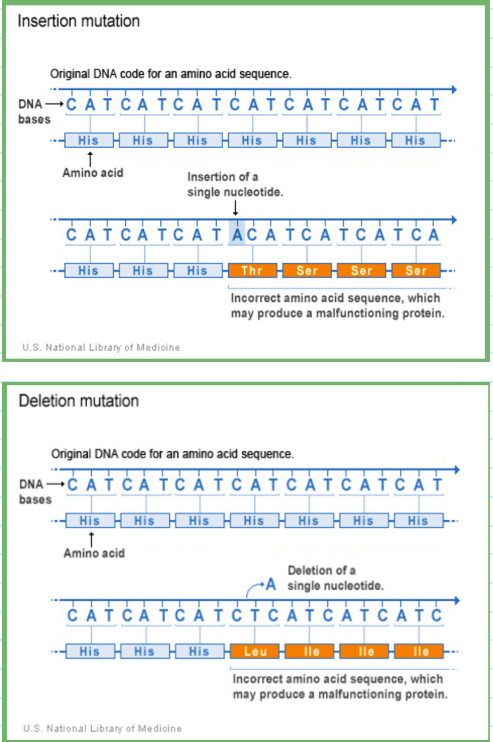
frameshift mutation
codon reading frame altered by insertion/deletion (occurs if the number of bases inserted or deleted is not a multiple of 3)
types of mutations that cause big changes (on chromosome level)
duplication
deletion
inversion
translocation
nondisjunction
isochromosome
duplication & deletion chromosomal mutation
Duplication: gain of genetic information
Ex: MECP2 syndrome
Deletion: loss of genetic information
Ex: Prader-Willi Syndrome
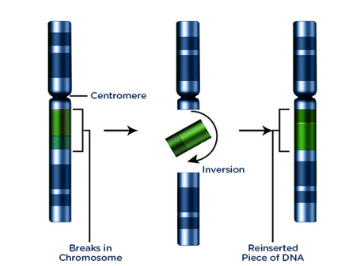
inversion chromsomal mutation
no gain or loss of genetic information
Ex: hemophilia A (FVIII gene), Hunter syndrome (lysosome storage defect, IDS gene)
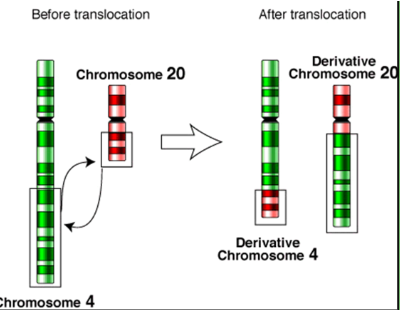
translocation chromosomal mutation
no gain or loss of information; breakage of chromosome and rejoining
Reciprocal or nonreciprocal
Ex: CML (t:(9:22)), AML
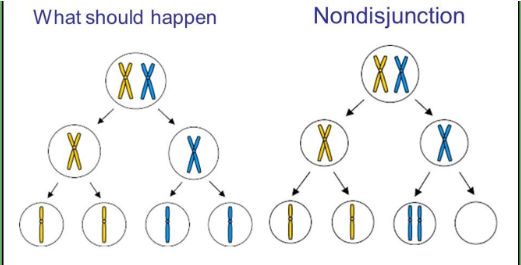
nondisjunction chromosomal mutation
failure of homologous chromosomes/sister chromatids to separate properly during meiosis
Causes aneuploidy (too many or too few chromosomes)
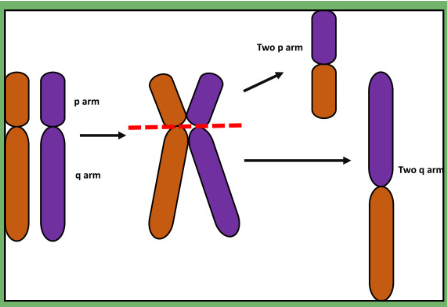
isochromosome
mis-division of the centromere results in duplication of one arm and loss of another
Found in some cancers
mitochondrial genome
Circular chromosome
Maternally inherited
16,569 bp; encodes 13 proteins
Involved in oxidative phosphorylation
Mutations in mitochondrial DNA → disruptions in energy production
epigenetics
changes in gene expressions that do not involve changes to nucleotide sequence; can be heritable changes in some cases
two major epigenetic processes
Chromosome structure changes
Affects gene availability (ex: histone modification)
DNA methylation
Can block transcription
DNA methylation
Primarily cytosine methylation in the context of CpG dinucleotides (islands)
CpG islands are stretches of DNA (500-1500 bp) with more than 60% GC
Found at promoters and contain the 5' end of the transcript
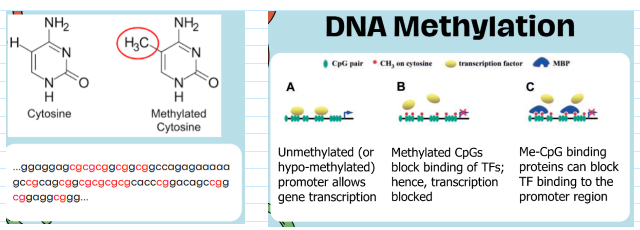
(epigenetics) transcriptional silencing
hypermethylation of DNA in the promoter region can inhibit gene transcription
(epigenetics) transcriptional activation
hypomethylation of DNA in the promoter region can activate gene transcription
(epigenetics) genomic imprinting
one of the alleles of a gene is silenced (no transcription), depending on the parent of origin
X inactivation--one of the X chromosomes in individuals with two X chromosomes is inactivated by methylation (no transcription)
restriction enzymes
In bacteria, part of protection from bacteriophages
Cleave nucleic acid at specific nucleotide sequences
Bacteria protect their own DNA by methylation at sequence recognized by the restriction enzyme
Are thousands of restriction enzymes:
AluI - AG↓CT - Arthrobacter luteus
BamHI - G↓GATCC - Bacillus amyloliquefaciens H
EcoRI - G↓AATTC - Escherichia coli RY13
HindIII - A↓AGCTT - Haemophilus influenzae Rd