Looks like no one added any tags here yet for you.
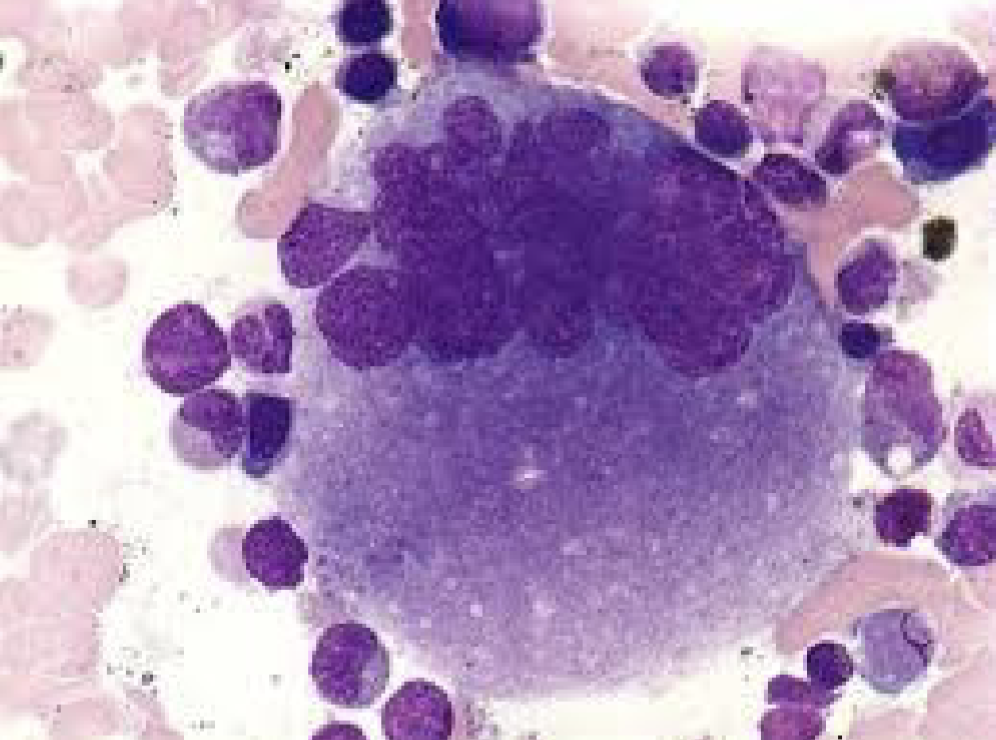
The Megakaryocyte
Megakaryoblast:
20 - 50 microns
Cytoplasm dark blue with blebs or pseudopods and non granular
Nucleus: round to oval centrally located, may contain nucleoli
Promegakaryocyte:
Size 20 - 80 microns
Undergoes mitotic divisions (endomitosis)
Granulation starts to appear
Nucleoli are usually visible
The Platelet:
Small disc shaped cellular fragments
Megakaryoblast develops into the megakaryocyte which
gives rise to the thrombocyte
Life span of a platelet?:
10 days (9.5 days)
Platelet production:
Thrombopoietin (TPO) hormone primarily produced by liver stimulates stem cells to differentiate into the megakaryocytic lineage
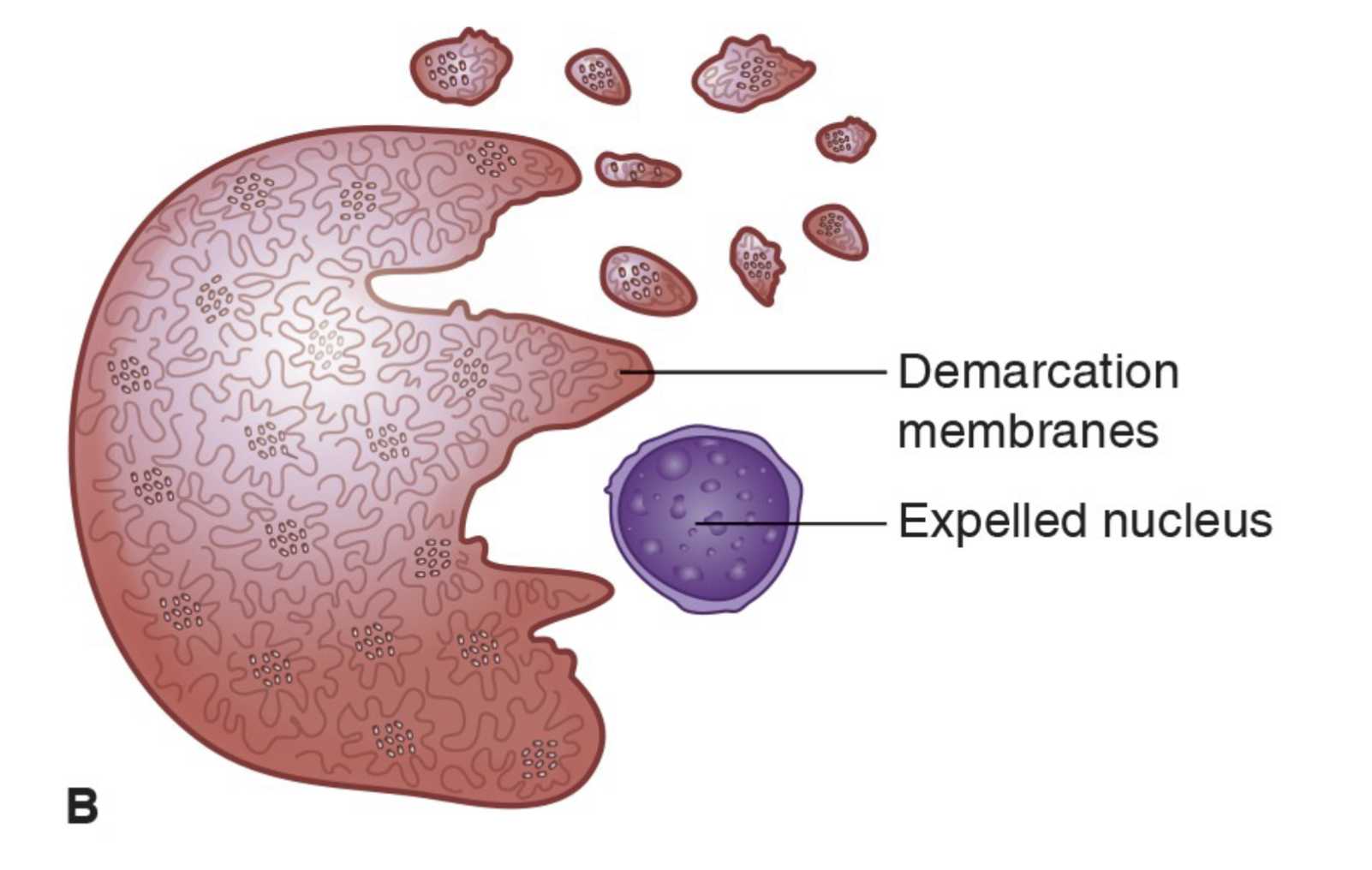
Megakaryocytic cells undergo multiple mitotic divisions without cytoplasmic division (called endomitosis)
Generates giant multinucleated cells
Platelets are produced directly from the megakaryocyte cytoplasm
As the megakaryocyte matures the granules in the
cytoplasm begin to cluster into small groups.
Thrombopoiesis:
The megakaryocyte membrane ruptures and the shedding of platelets occurs.
Each megakaryocyte produces 2,000 to 4,000 platelets
Platelet release from Mature Megakaryocyte
Platelet characteristics:
Younger platelets are larger than old platelets
Approximately 2/3 of platelets produced are in the peripheral blood and 1/3 are in the spleen
The platelets in the spleen are interchangeable with the peripheral blood
Platelet turnover is 35,000/mm per day
Platelet Estimations:
In a field of 100 RBC’s 1 platelet is equal to 20,000 platelets
Large platelets
Diameter greater than 4 microns
Seen during increase turn over
such as with immune thrombocytopenic purpura (ITP)
Giant platelets
Diameter greater than 7 microns
Size of a RBC
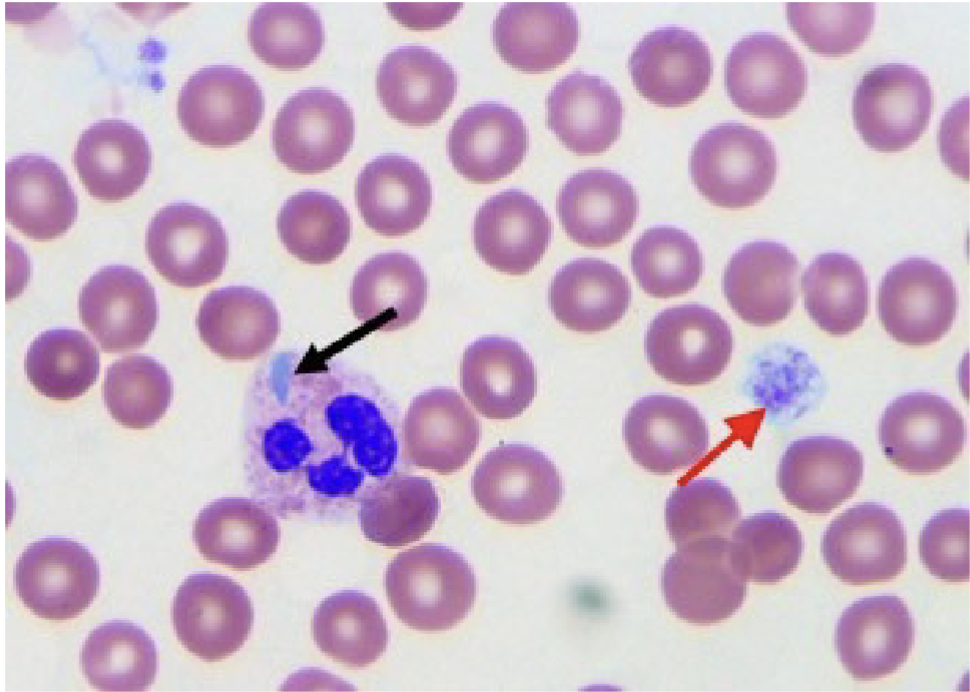
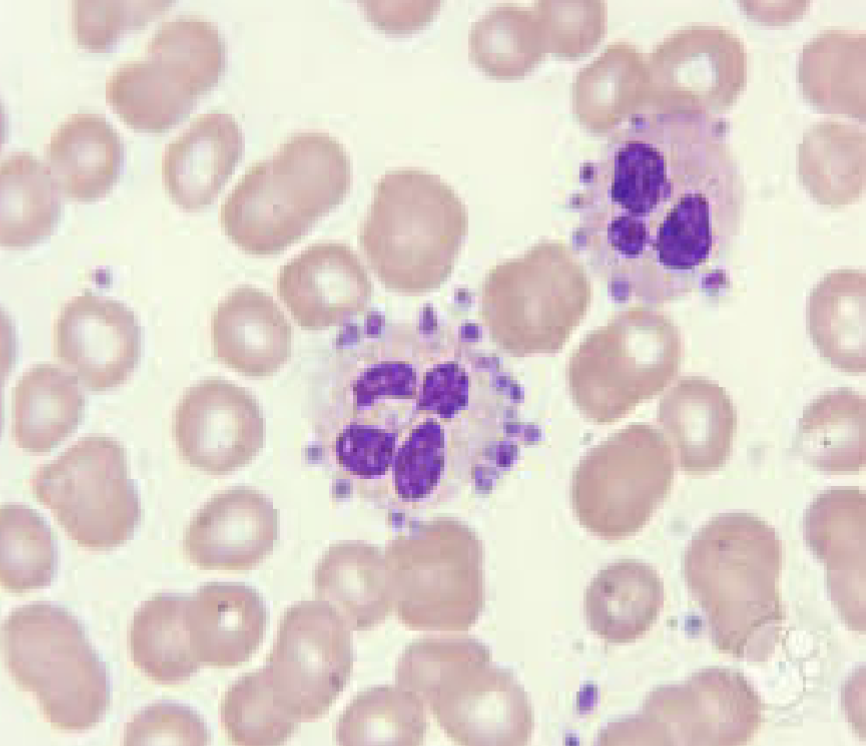
Giant platelets and May Hegglin:
Red arrow points to giant platelet
Black arrow points to May-Hegglin body. Note
the neutrophil with the blue inclusion
If large quantity of giant platelets exist, the
automated platelet count might be falsely decreased
Size falls outside upper threshold of automated analyzers
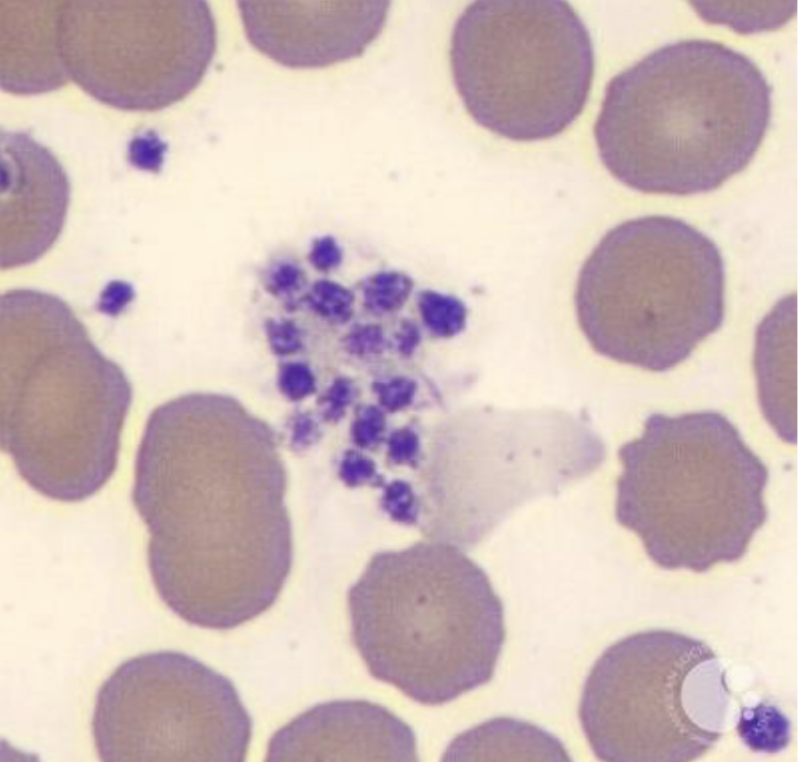
Platelet Clumping:
EDTA can cause platelets to surround the neutrophil – called satellitism
EDTA dependent antibodies react with platelet glycoprotein IIb/IIIa
Platelet clumping may also be caused by traumatic venipuncture
Decrease PLT count on automation
WBC count flagged on automation
Corrective Action of Platelet Clumping:
redraw patient using sodium citrate tube (only time you would use this instead of EDTA)
Correction PLT count x 1.1 (to compensate for sodium citrate)
Platelet clumping
Platelet Satellitism
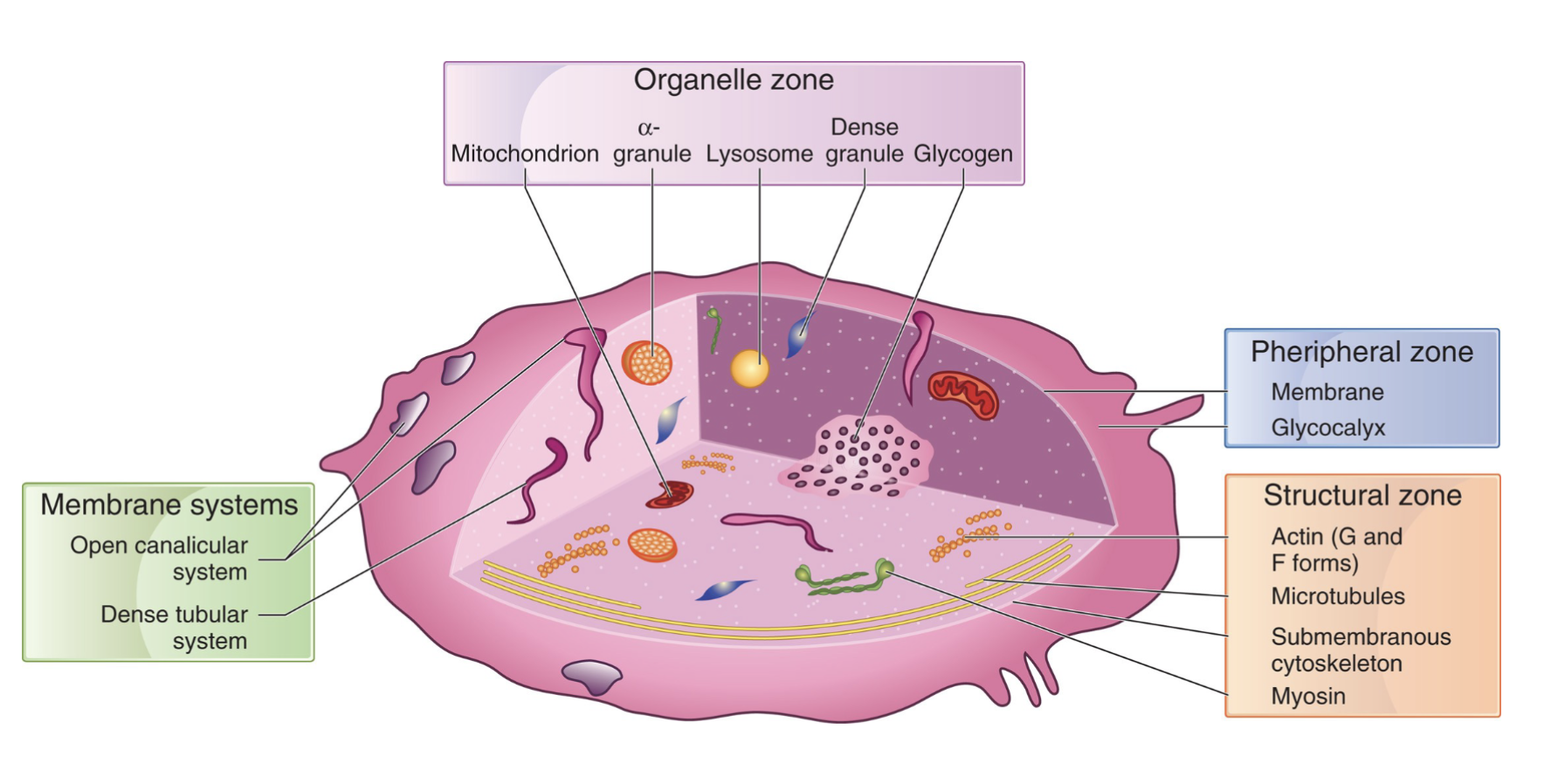
Platelet structure:
may be very small, but they are actually very complex and metabolically active
Four layers of Platelet structures:
Peripheral Zone
Structural Zone (Sol-gel Zone)
Organelle Zone
Platelet Membrane system
Peripheral Zone:
Composes the surface coat
Responsible for adhesion which is the attachment of the platelet to a foreign surface
Responsible for aggregation which is the attachment of platelets to other platelets
Important for coagulation
Sol Gel Zone:
Sometimes referred to as structural zone
Matrix of platelet cytoplasm
Contains several fiber systems in various states of polymerization (cytoskeleton)
Composed of microfilaments and microtubules
Provides the cytoskeleton and contractile system
Needed for platelet shape
*Platelets flat biconcave disc until activated, once
activated change shape “inside out” signaling
Organelle Zone:
Storage and secretion of substances essential for platelet function
Contains mitochondria, alpha granules, dense bodies and lysosomal granules
Important for metabolism, hemostasis, and vessel repair
Platelet Membrane System:
Important regulators of intracellular calcium
concentration
Calcium is important for platelet metabolism and
activation of the coagulation process
Production of prostaglandin synthesis, needed for platelet aggregation
Platelet Ultrastructure and Functions
Platelet structure and physiology:
Size: 2 to 3 micrometers in diameter
Dense granules and alpha granules
Platelets are biconvex in circulation
When platelets are activated, they change shape and become tiny spheres, forming pseudopodia
Normal platelet count:
150,000 to 450,000
Where is one-third platelet volume?
Spleen
Rest is in circulation
Vascular System
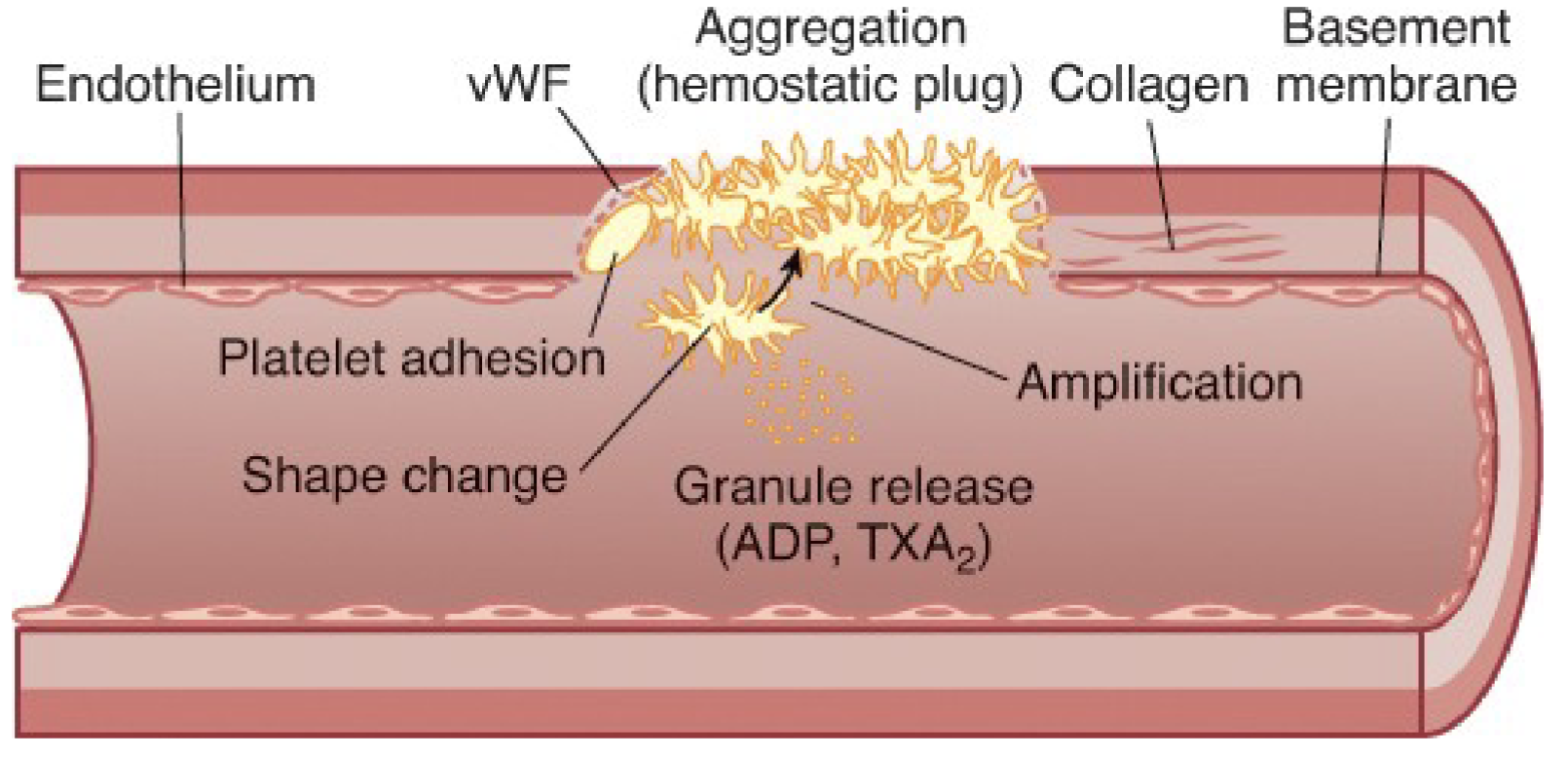
Primary hemostasis:
The process of platelet plug formation
Adhesion
Shape change
Secretion: discharge of alpha and dense granules
Amplification
Aggregation
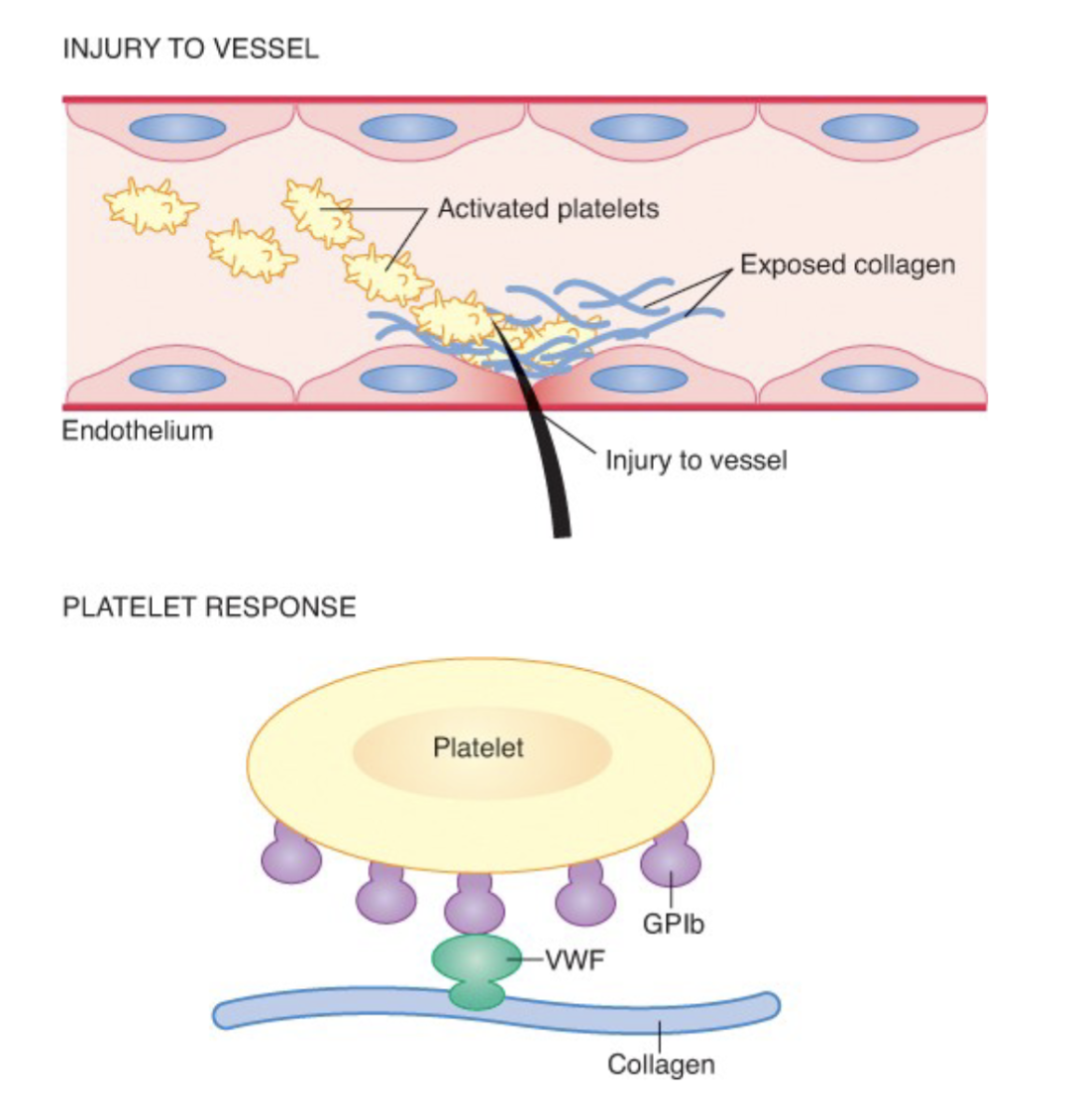
Platelet kinetics: Adhesion:
Exposed collagen and subendothelial cells initiate the
first step of the platelet plug formation.
Platelets reach the surface and change shape from
discs to spiny spheres
Platelet kinetics: Granule Release:
The contents of the dense bodies and alpha granules are released to regulate clot formation
Platelet kinetics: Aggregation:
Platelets stick to each other to form stronger platelet plug along with the coagulation proteins
What does platelet adhesion require
Occurs when platelets attach to collagen in the exposed basement membrane
gpIa/IIa on the platelet surface
vWF
gpIb/IX binding site
Platelet amplification requires:
Secreted substances, such as TXA2, recruits more platelets to the site of injury
Platelet aggregation requires:
The use of fibrinogen as the mediator protein
Fibrinogen attaches to platelets at the site of gpIIb/IIIa
Platelet plug is formed to stop blood loss
Platelet Granules: Alpha:
Clot-activating granules
Glycoproteins IIb and IIIa
Fibrinogen
von Willebrand Factor (vWF)
Factor V (5)
Factor VIII (8)
Platelet Granules: Dense:
Platelet-activiating substances
ADP
Serotonin
Calcium
Thromboxane A2 (TXA2)
*****Blockage of TXA2 impairs platelet function
Aspirin (and specifically the acetylation of
aspirin inactivates cyclogenese, which blocks
TXA2 production
Von Willebrand Factor:
can be found in megakaryocytes, it’s a component of alpha granules and made by megakaryocytes as they mature
Platelet function in primary hemostasis
Types of Platelet Disorders:
Quantitative
Qualitative
Platelet disorders leading to bleeding disorders are a result of?:
Quantitative abnormality of platelets
Quantitative Disorders:
Thrombocytopenia — most common cause of abnormal bleeding
What is thrombocytopenia caused by?:
A decrease or ineffective production of platelets
An increase in platelet destruction or utilization
An increase in platelet sequestration (removal) by the spleen
Loss from the body
Dilution factors
Congenital Hypoplasia:
Decrease cell (platelet) production in the bone marrow
Fanconis Syndrome:
a chromosomal defect where the bone marrow fails to produce RBC’s, WBC’s and platelets. (Pancytopenia, decrease in all cells). Found in children
Other reasons for decreased production:
Infection
Newborns infected with virus, such as Rubella
Intrauterine exposure to certain drugs that are capable of damaging production sites
Acquired Hypoplasia:
a result of the action of chemical or toxic drugs to the bone marrow
Radiation or chemotherapy: the platelets are the last
cell line to return to normal and are first to be affected.
Replacement of the bone marrow by neoplastic diseases (like leukemia)
Decrease in proliferation of megakaryocytes caused by marrow replacement (aplastic anemia)
Ineffective Thrombopoiesis:
the bone marrow contains normal or increased numbers of megakaryocytes but the number of platelets in peripheral blood are decreased
What is ineffective thrombopoiesis seen with?:
Megaloblastic anemia
Ethanol abuse
Pre-leukemia
Hereditary Thrombocytopenia:
Inherited disorders where the platelet number is decreased
Increased Destruction:
Immune related factors
Idiopathic Thrombocytopenia Purpura (ITP)
Idiopathic Thrombocytopenia Purpura (ITP)
Unknown origin
Patients often have very big bruises
Acute condition (resolves in a few weeks)
Found predominately in children (2-6 years)
Majority of cases develop after recovery from viral infection
66% of cases are antibody directed
Usually self limiting, spontaneous remission in about 80% of cases
platelet destruction is increased
Idiopathic Thrombocytopenia Purpura (ITP) causes:
Causes excessive bleeding
Unusually low level of platelets
Sometimes referred to as immune thrombocytopenia
After infection such as measles or chicken pox, immune system produces IgG antibodies which coat platelets.
Leads to filtering out by spleen
Can treat with steroids if doesn’t resolve on its own
Steroids “turn off” or slow down immune activity
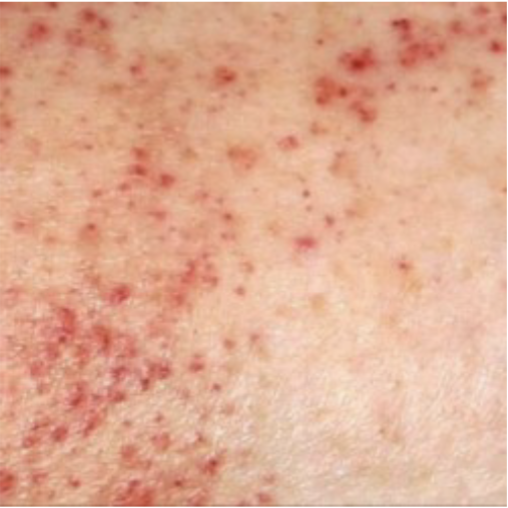
Idiopathic Thrombocytopenia Purpura (ITP): Laboratory Findings:
Platelet count <20,000
Sudden onset of petechiae and bruising
Image shows petechiae
Frequent nose bleeds
Idiopathic Thrombocytopenia Purpura (ITP): Treatment:
Usually last 3 weeks – 6 months
Immunosuppressants are sometimes given
Chronic ITP:
Can be seen at any age but more often found in women between ages 20-50 years old
Platelets are sensitized by platelet antibodies (usually IgG type antibodies)
The clinical course of CITP is some patients consists of alternate periods of remission and relapse (periods where platelet count is almost normal and then drops again)
Chronic ITP: Laboratory and Clinical Findings:
Platelet count 30,000 – 60,000
Platelets are large and abnormal in appearance
Petechiae and bruising present
Chronic ITP: Treatment:
Keep platelets above 40,000
Steroids to reduce spleen removal
prednisone
Splenectomy may be advised
Transplacental or Neonatal disorders:
A self limiting form of thrombocytopenia
Caused by maternal IgG platelet antibody crossing placenta
IgG only antibody subtype that crosses placenta
Normally good for IgG to pass placenta as it gives the
newborn circulating protecting IgG while newborn forms own immune system
Causes destruction of the newborn’s platelets
Greatest danger for the fetus is during delivery and immediate post partum
Danger of central nervous hemorrhage
Can have intercranial bleeding
Neonatal Increased Destruction Laboratory Findings:
Platelet count less than 30,000
In then decreases further during the first few hours of
life
Neonatal Increased Destruction: Treatment:
Usually last an average of 3 – 4 weeks
Recovery follows clearance of antibody from circulation
Must evaluate severity
May require platelet transfusion
Self-limiting once antibody is gone
Drug Induced:
An immune reaction with destruction of platelets results after exposure to certain drugs
Most common quinidine, heparin (HIT) and some antibiotics
Caused by drug-dependent antibodies
Usually seen 1-2 weeks after patient is on drug treatment
Treatment includes removing the offending drug
Post transfusion Purpura:
Occurs one week after blood transfusion
Antibodies are stimulated from foreign antigen and cross react with platelets
Disappears 2-5 weeks without treatment
Treat with intravenous immunoglobulin (IVIG)
(uncommon problem, rare, but serious)
Other causes of decreased platelet counts:
HIV
Hepatisis C Virus
Helicobacter pylori
Some infections may lead to decreased platelet count as disease progresses
Though to occur as a result of immune reaction
Non-Immune: Thrombotic Thrombocytopenia Purpura (TTP):
Intravascular platelet aggregation
A rare disorder which the exact cause is unknown
Characterized by low platelet count and hemolytic anemia (red blood cells are damaged)
Formation of platelet clots in capillaries and arteriole
See schistocytes (RBC fragments) in peripheral blood
Due to red blood cell injury from damaged epithelium
As blood flow through the RBC’s become damaged and an anemia results
Schistocytes
TTP affects all ages, more commonly women in child bearing age
Non-Immune: Thrombotic Thrombocytopenia Purpura (TTP): characteristics & findings:
Platelet counts average about 20,000
PT and aPTT are normal
not involving coagulations factors
Platelets clot, not fibrin
Increased lactate dehydrogenase (LDH) common for TTP
TTP characterized by Macroangiopathic Hemolytic Anemia (MHA)
Intravascular hemolysis
Decrease haptoglobin
Neurological complications
Etiology of Thrombotic Thrombocytopenia Purpura (TTP):
Deficiency of ADAMTS-13
Molecular test looks at vWF attached to factor 8
The protein has a relation wit vWF (Von Willebrand Factor)
Function to break this large molecule into small pieces
Large vWF multimers tend to increase platelet adhesion
Supposed to be small but high in thrombocytopenic purpura (TTP)
vWF is important in adhesion of platelets for normal hemostasis
Non-Immune: Throbotic Thrombocytopenia Purpura (TTP): Treatment:
Anti-platelet agents, plasma exchange transfusion, steroids
Can be fatal due to formation of clots
Disseminated Intravascular Coagulation DIC:
Results from a major tissue injury or infection
Thrombotic occlusion of the microcirculation
Consumption of platelets and clotting factors
RBC fragments and hemolytic anemia
Decreased platelet count
Abnormal coagulation testing
DIC involves coagulation factors
Hemolytic Uremic Syndrome (HUS):
Predominates in children age 6 months to 4 years (although any age can contract)
Usually self limiting
High occurrence with E. Coli 0157:H7 infection
Can be caused by other infections
Often follows an acute viral infection and is associated
with vomiting and diarrhea
Platelet consumption occurs in the kidneys
Renal damage – abnormal kidney tests
Signs are hemolytic anemia, (presence of schistocytes, thrombocytopenia and renal failure)
Mechanical Destruction of Platelets:
Artificial heart valves
ECMO: extracorporeal membrane oxygenation
Blood pumped outside the body to heart-lung machine that removes CO2 and sends oxygen back to blood
Increased splenic sequestration:
When spleen becomes enlarged, the number of
platelets sequestered is increased
Example: Hodgkin’s lymphoma, cirrhosis of liver
Dilution:
Patients who experience massive hemorrhage
requiring blood replacement
No viable platelets in transfusion units
Thrombocytosis:
Overproduction of platelets
Usually platelet count over 400,000 (or 450,000)
Relative Thrombocytosis or Primary
Uncontrolled proliferation of platelets both in
peripheral and bone marrow
Example:
Splenectomy, acute blood loss, acute and chronic
inflammation, myeloproliferative disorders (CML), or
increased hematopoiesis
Thrombocythemia:
Platelet count >1-2 million
Platelets are clumped, bizarre shape and size
Function may be abnormal
Usually secondary to another disorder
Qualitative reasons for platelet disorder functioning:
Glanzmanns
Storage pool
Bernard-Soulier
Von Willebrand’s disorder
Aspirin
Qualitative: Glanzmanns:
Aggregation disorder
Qualitative: Storage Pool:
Secretion and release action disrupted
Granules not released
Qualitative: Bernard-Soulier:
Adhesion disorder
Missing membrane receptors for vWF
Qualitative: Von Willebrand’s Disorder:
Adhesion disorder
Receptor there but not functional
Qualitative: Aspirin:
Loss of aggregations (prostaglandin synthesis)
Patients who present with bleeding disorders (due to platelet issue) will typically present with?:
Mucosal Bleeding