BIOL 325 GENOME/EXOME/RNA SEQUENCING STUDY GUIDE
1/63
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
64 Terms
explain how genetic information is stored within the double helix (written response question)
Genetic information is stored within the double helix through the specific sequences of the four nucleotide bases that complementary pair together. Adenine goes with Thymine and Cytosine goes with Guanine. The specific sequences that these pairs form encode the genetic instructions for building and maintaining an organism
what does every sequencing method/process needed?
needs a labeled primer
primer needs to hit right before the sequence of interest to prime and start the sequencing process
needs high quality template
isolated DNA or RNA
needs dideoxynucleotides
dATP, dCTP, dGTP, dTTP
added separately to each of the four tubes
what is sanger sequencing?
DNA replication in a test tube that terminates a growing chain through dideoxynucleotides
what are dideoxynucleotides? how does the removal of the -OH group impact the addition of nucleotides?
dideoxynucleotides remove one of the hydroxyl (-OH) groups to not allow a phosphodiester bond to form which in turn terminates the growing chain
the removal of an -OH does not allow to chain to continue because the next nucleotide needs the -OH
what is the purpose of adding an enzyme into the sanger (chain terminating) sequence method?
with addition of enzyme (DNA polymerase), the primer is extended until a dideoxynucleotide (ddNTP) is encountered which will end the chain
how is ratio important in sanger sequencing?
with the proper dNTP:ddNTP ratio, the chain will terminate throughout the length of the template
how do you set up chain termination? what would be an example of it?
in four different tubes they will have the DNA polymerase, a mix of dideoxynucleotides and standard nucleotides
ex. for the adenine tube —> 4 dNTPs + 1 ddATP = would want more normal ATP than the dideoxy so some normal will get incorporated into the sequence and eventually get an A but when the ddATP gets added = chain ends at ddA
all chain will end with whatever dideoxy was added
ddCTP = ddC
ddGTP = ddG
ddTTP = ddT
name and describe the standard method for the determination of nucleotide sequences (written response question)
The standard method for determination of nucleotide sequences is the sanger sequencing method. The sanger sequencing method is a chain termination sequence where a DNA polymerase or an enzyme will extend the primer until a specific dideoxy is encountered which will terminate the sequence of the specific dideoxynucleotide. The ratio of the dideoxynucleotides and the standard nucleotides is crucial in the Sanger sequencing method and each dideoxynucleotide is labeled with a fluorescent dye for specific identification of each of the nucleotide bases. By resolving the terminated chains through electrophoresis, a sequencing ladder occurs where an individual can determine the sequence by reading the gel from bottom to top/smallest fragment to largest fragment.
what is cycle sequencing?
is a chain termination sequencing done in a thermal cycler
done in a thermal cycler to cycle through the replication process thousands of times to hit every NT in that specific tube
it is amplification based son heat-stable DNA polymerase is needed because if a non heat-stable polymerase is used, it is killed during the denaturing step
what are fluorescent dyes? what is fluorescently tagged in sequencing methods?
are multicyclic molecules that absorb and emit fluorescent light at specific wavelengths
the primer OR nucleotides are fluorescently tagged and could be tagged with hundreds of different types of dyes
what are fluorescent dyes based on? what is covalently attached to the molecules?
a lot of fluorescent tags/dyes are based on fluorescein (green) or rhodamine (red) derivatives
for sequencing applications, these dyes/molecules can be covalently attached to molecules
what is dye primer sequencing?
primer is tagged with fluorescent due conjugated nucleotides at the 5’ end of the chain
the 5’ end that was sequenced will be fluorescent s the size based output will tell what the end nucleotide is
what is dye terminator sequencing? what is needed for each of the four dideoxynucleotides?
fluorescent dye molecules are covalently attached to the dideoxynucleotide at the 3’ end of the chains
since nucleotide is tagged —> need four color for the different nucleotides to detect them
an instrument will detect all the different emissions which will help detect all the four nucleotides that will have distinct dyes/colors
all four reactions can be dine in a single tube because of the different colors used
what is the DNA ladder for DNA terminator sequencing read on?
electropherogram
what is automated sequencing?
uses a gel capillary tube with an instrument/electropherogram to detect different emissions
dye primer or dye terminator sequencing on capillary instruments
sequence analysis software provides analyzed sequences in text and electropherogram form
peak patterns reflect mutations or sequence changes
understand how to read data from an electropherogram (written response question)
To read data from an electropherogram, the different nucleotides will have different peaks and colors that are specific to the nucleotide bases. The heights of the nucleotide peaks will not always be perfect compared to each other however they should be fairly close. no peak for a nucleotide means that there was an error in the read. The order of the nucleotides can be read through their different colors and heights which will help in determining the sequence but also determine if there is a single mutation present.
what is a homozygous mutation?
T/T or A/A mutation = one allele is mutated and the other stays normal and happens quite often in mutations especially cancer
can tell because a peak of a T or an A is similar to the other peak of the same nucleotide = both alleles have a T or an A
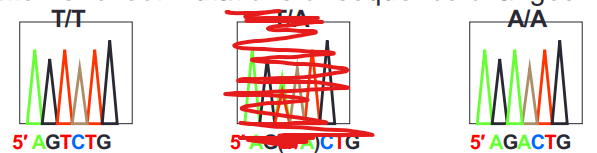
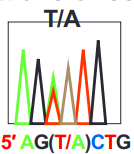
what is a heterozygous mutation?
mutation that has another peak of a different nucleotide in another nucleotide
what would be considered good sequence data? what would be considered an ugly piece of data and what would be a bad read?
both strands are sequenced and should both give a complementary output
sometimes the peaks are not perfect so that is why the other strand is sequenced to confirm what nucleotide the non-perfect peak is
sometimes there are compressions where the nucleotide is pushed up against a secondary structure in the DNA
like it is looped out or loop in and it cannot be read meaning that it is an ugly piece of data
if the other strand looks good and the other strands nucleotide is not clear but can be inferred then you can trust the sequencing data
if there are values or peaks on both of them at the same location then it is a bad read so you cannot trust the sequence data
what are other instances of bad sequence data and what would their solution be?
problem = overloaded/too much DNA
solution = check concentration of template
problem = dirty sample —> protein or lipid contamination
solution = clean up DNA/use DNA clean up kits
problem = too much voltage applied to the capillary gel/slowly gets higher and higher throughout the assay
solution = check voltage on the machine
problem = short reads (more common and usually the ratio of the nucleotides are messed up)
solution = too much ddNTPS to dNTPs or low processivity enzyme so fix ratio/add more processivity enzyme
too much DNA or too little DNA in comparison to nucleotide ratio —> will not read will
what is RAS mutant? how was an electropherogram used to sequence and detect RAS mutant? how can you confirm the mutant?
RAS mutant = oncogene/mutated gene that can cause cancer and commonly mutated in LOTS of cancers
in a normal codon sequence, there can be two peaks that are the same height if they are the same nucleotide but they are still unique peaks
electropherogram detects the heterozygous mutation in the RAS gene of G>T in exon 12
can confirm them mutation by reading the other strand because the other strand will have similar peaks in the same location as the other strand
what is BRCA1? how was an electropherogram used to sequence and detect BRCA1 mutation?
BRCA1 = breast cancer susceptibility gene
supposed to help control the cells but once mutated, the cells grow uncontrolled
many mutations in the BRCA1 gene but top 10 is deletion of two nucleotides, A and G, next to each other (frameshift mutation)
electrophreogram detects the frameshift of the BRCA1 mutation and the peaks will start getting messed up and the peaks will begin to go inside each other and there will be gaps
cheap assay/test
define and describe the purpose of bisulfite sequencing (written response question)
Bisulfite sequencing is used to detect methylation in DNA. Bisulfite deaminates/removes amine group from cytosine making uracil. The methylated cytosine stays as cytosine, while the unmethylated cytosine turns into uracil. A methylated cytosine is NOT changed by bisulfite treatment. The bisulfite-treated template is then sequenced and a highly methylated sequence is a sign of the region being silenced.
what is high-throughput or next gen sequencing? what does it apply to?
high demand resulted in low cost and fast methodologies
high-throughput sequencing includes next gen “short-read” and third generation “long-read” sequencing method which applies to
genome sequencing
exome sequencing
transcriptome profiling (RNA sequence)
DNA-protein interactions (ChIP-sequence)
epigenome characterization
what are the five sequencing approaches for next gen sequencing?
pyrosequencing (2nd gen)
ion cunductance
reversible terminator
ligation mediated (SOLID)
real time or phospholinked nucleotides
what is pyrosequencing?
when a nucleotide is added to the growing reaction, light is emitted and can determine what nucleotide was added by detecting the light
each nucleotide is added one at a time and only one of the four will generate a light signal and it is washed away and repeated again
light signal is recorded on a pyrogram
what is the process for pyrosequencing?
only one out of the three phosphate groups in the added nucleotide is needed to hook it to the next nucleotide so the other two phosphate groups are released (PPi is removed)
experiment part = use an enzyme with a substrate that turns the diphosphate into ATP
when the substrate luciferin in the enzyme luciferase, it will break down ATP and produce light
detector needed to detect light
what is ion semi conductor sequencing?
whenever a nucleotide is added, a proton (H+) gets released
based on the detection of hydrogen ions released during DNA polymerization
H+ released changed the pH of the solution which is detected by an ISFET
the unattached dNTP molecules are washed out before the next cycle
how is the signal detected in ion semi conductor sequencing?
start with micro wells (96 well plate) and when the nucleotide is added, a proton (H+) is released
the well is sitting on a ion sensitive layer so when the ion is released, it gets excited
below the ion sensitive later is an ISFET ion sensor that reads the excitement and creates a signal to the computer
all are contained within a semiconductor chip which is similar to what is used in the electronics industry
what are the pros and cons for ion semi conductor sequencing?
PROS
rapid sequencing speed
low operating costs
only sensor is needed
CONS
short read length
what is ion semi conductor sequencing best suited for?
small applications like
nucleic acid mutations
microbial genome
transcriptome sequencing
targeted sequencing
what is illumina dye sequencing aka bridge amplification or reversible dye termination?
automated, simple and efficient
can sequence multiple strands at once = longer reads
each NT is labeled with a different fluor
what is the summarized version of the process of illumina dye sequencing?
adapters are added to the ends of the DNA fragments and through reduced cycle amplification, additional motifs are introduced like sequencing binding site, indices, and regions complementary to the flow cell oligos
the oligos are complementary to the adapters on the DNA fragments
illumina dye sequencing produces clusters
basically one strand is read then it recreates the second strand so the second strand is sequenced
both strands need to be sequenced to confirm if you have a correct sequence
what are the uses for illumina dye sequencing?
uses reversible dye-terminators which enables the ID of single nucleotides as they are washed over DNA strands
used for
whole genome sequencing (WGS)
regional
transcriptome
metagenomics
small RNA discovery
methylation profiling
protein-nucleic acid interaction analysis
name two technological updates that have greatly improved DNA sequencing (written response question)
one technological update that has greatly improved DNA sequencing is pyrosequencing. pyrosequencing provided a bright way to determine a DNA sequence by emitting light whenever a certain nucleotide was introduced. another technological update that has greatly improved DNA sequencing is illumina dye sequencing. illumina dye sequencing allows multiple strands to be sequenced at once and is automated.
how has NextGen sequencing revolutionzed the process? distinguish and describe these subtypes: pyrosequencing, ion semiconductor, illumina dye (written response question)
NextGen sequencing has revolutionized the process as it now allows for high-throughput sequencing, which allows the process to be low cost and faster. Pyrosequencing is a second-generation sequencing method where a nucleotide emits light as it is added to the growing reaction. Ion semiconductor method is where, instead of light, a proton is released when a nucleotide is added. Illumina dye sequencing is when fragmented DNA is attached through a flow cell and forms a bridge to another adapter, and is amplified to form clusters. Each nucleotide on the forward and reverse strands is read through reversible dyes and should be complementary to each other.
what is genomics?
attempts to study entire genomes and their organizations
provides contextual framework for genome information
what was the first genome sequenced in 1984?
EBV (epstein barr virus)
what is human genome project (NIH)?
started with known sequences and used the known sequence to walk and prime into unknown sequences
published in 2000 a week apart from celera but incomplete
what is ventner’s group, celera?
ventner sequenced his own genome
whole genome shotgun which cut genome into fragments and sequenced —> used computer to overlap the fragments
assembly is HARD, often leaves gaps
published in 2000 a week apart from HGP but incomplete
how much did the first genome cost at first, in 2006, and 2014?
first human genome sequenced = $2 billion
2006 = $50 million
2014 = $1,000
what is the 1000 genomes project from 2008?
sequenced 1000 genomes to identify variations in the human genome
national human genome research institute (NHGRI), wellcome trust sanger institute, and the beijing genomics institute
what is workflow of whole genome shotgun sequencing (WGSS)?
entire genome is sheared randomly into small fragments by enzymes, sonicator, or mechanically
how long the genome is sheared will determine how big the fragments will end up being
the reasonable fragment sizes are primed off of and sequenced
a computer is used to realign/overlap the sequenced fragments but it is slow
what is workflow of hierarchical shotgun sequencing?
genome is broken into thirds then further sheared into reasonable fragment sizes and their order is deducted
the further sheared DNA fragments is sequenced and overlapped to construct the genome consensus
how are entire genomes sequenced today? (written response question)
Entire genomes are sequenced through whole genome shotgun sequencing. Whole genome shotgun sequencing shears an entire genome and the reasonable fragments are sequenced and primed off of. The sequenced fragments are then overlapped using a computer to construct the genome.
what are the mutants to wild type comparisons?
cancer genetics
take area of patients tumor that seems to be behaving differently than another area so the entire genome of each of them is sequenced and see how they compare to each other
antibiotics or drug resistance
targeted therapies
what is ligation sequencing (ABI SOlID)?
sequence method that reads two nucleotides at a time
what is the process for ligation sequencing?
magnetic bead(s) is created with an adapter sequence on it and primers complementary to the adapter sequence
dye nucleotide sequences are labeled
every combination of a nucleotide sequence is on the probe, and each has a different color in it
add a big mix of probes and let them adhere to whatever the correct complementary sequence is
needs LIGASE to hold the probes to the sequence
ligase adds the probe to the nucleotides and gives it color
now know what the two nucleotides are
wash everything away and add probes back to the sequence to find the next two nucleotides
what is the NGS workflow for nucleic acid extraction?
isolate nucleotides first and it could be from any sample like solid tissue or fluid/plasma
use kit to extract NA from sample
do quality control to make sure it is clean and not degraded
what is the NGS workflow for library prep?
ligate adapter on the DNA
for genome sequencing, size select the DNA that was sheared
probably will use spin column
amplification/prep
quality control
what is the NGS workflow for sequencing and analysis?
sequence
data analysis
base calling
read alignment
variant calling
variant annotation
human will look at mutation location and loading it into other data bases
what is DNA barcoding? how does this relate to when a new outbreak occurs?
it is when a sequence that is unique to an organism is scanned and created into a unique ID/barcode
the genome that is unique to the organism tends to be the mitochondrial gene which makes cox genes
if a new outbreak occurs, the organism is sequenced and compared to barcodes of known organisms to have an idea to what is causes the new outbreak
name/describe the three ways to prepare a template for sequencing (written response question)
emulsion PCR: amplifies DNA in small water-in-oil droplets while keeping similar sequences apart to avoid formation of artifacts
rolling circle PCR: amplifies the template by adding complementary adapters onto sheared DNA, making them overlap each other and creating a circle. A polymerase is added to make a really long repetitive copy of that DNA fragment and is primed and sequenced.
solid-phase amplification: amplifies the template by forming bridges on the flow cell by complementary binding to adapters and forming clusters of identical strands
what are the run times of NGS, their read lengths, and their error rates?
run times range from 10 hrs to 5 days
read lengths are 35-400 bases
error rates are worse but the output in gigabases are larger
what is massive parallel sequencing analysis?
input samples must be a library of random amplified DNA or sequences attaches to adapter sequences
each fragment is amplified on a solid sequence — each cluster then is one set of sequencing reactions
since humans share 99.9% of their genome, what is the 0.1%? what is the cause of 80% of rare disease?
0.1% of genes trigger the difference in our development
over 80% of rare disease are caused by genetic mutations in that tiny difference, affect ~8% of the population (1 out of the 1000 disease)
detecting such diseases is challenging
what does NGS not require for genome sequences?
does not require a capture step for genome sequences which offer coverage across the entire genome
what is the exome and what is exome sequencing?
exome = expressed part of your DNA
exome sequencing = capture-based method, targets coding regions of the genome
what is the transcriptome and what is transcriptome sequencing?
transcriptome = the sun of all the mRNA expressed from the genes of an organism
transcriptome sequencing = capture-based method, detects coding plus multiple forms of RNA
how is RNA sequenced? (written response question)
Before, RNA used to be sequenced by converting the mRNA into cDNA through reverse transcriptase. By converting the mRNA to cDNA, it can cause errors in two areas, making it error prone. Now, RNA is sequenced through the ion-proton system to detect proton that is released. It directly sequences RNA to remove an error-prone step. It has the potential to find cancer mutation that affect intron removal and to compare affected person to normal transcriptome.
what is bioinformatics?
biology working with pure science, technology, and statistics and work in silico (computer)
created barcoding database
what are the 6 consensuses sequence data comparisons?
tumor genomes
evolution
creating phylogenetic tree
species ID
environmental response
putative protein structure
epeigenetics data
what are computers required to do?
align the sequences
identity: identical residues (DNA, RNA, or AA)
similarity: results in AA with same property
homology: a numerical value of how conserved a sequence is
what is BLAST?
basic local alignment search tool
blasts and searches for things
NIH funded website
put info into website and it spits out all the possibilities
anything that has been sequenced by a funded research lab puts their sequences in BLAST