Lecture 3 - Sequences and Database
1/44
Earn XP
Description and Tags
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
45 Terms
What are biological databases
A computerized archive of biological information (often specialized*) in which
• Data is stored and organized
• Sometimes further analyzed, annotated, and visualized
The goal : Must provide methods for researchers to find what they are looking for
• Queries with search criteria
• Downloads
What are primary databases
contain raw, original data, usually generated from experimental results. These databases are repositories for unprocessed or minimally processed data such as nucleotide sequences, protein sequences, and gene expression data
Relatively little to no curation for some
Another name: core data
What are some examples of primary databases
GenBank: Stores nucleotide sequences from various organisms (genes/genomes) (Apart of NCBI under national institute of health)
European Nucleotide Archive (ENA): Provides nucleotide sequences submitted by the scientific
community. (Similar to GenBack, european version)
Protein Data Bank (PDB): Contains 3D structural data of proteins and nucleic acids. (4 unique letter code within database to identify them)
UniProtKB (Swiss-Prot/TrEMBL): Stores protein sequence
All have highly variable quality, does not guarantee all data is high quality
What is the purpose of primary databases and when would you use it
To serve as repositories of original experimental data, allowing researchers to access and share biological data. These databases are often the starting point for further analysis.
You’d use a primary database if you wanted to search for the raw nucleotide sequence of a gene or obtain the protein structure from experimental data
What is the data type/content of primary databases
The data is usually directly submitted by researchers, often without any extensive analysis or interpretation. It’s raw or annotated only with basic information like species, sequence, or structure
Content: Nucleotide sequences, protein sequences, structural data, expression data, mass-spec data, and more…
What is secondary databases
Derived from primary databases but contain curated, processed, and interpreted information. They include data that has been analyzed, refined, and sometimes merged with other data sources to provide more insights.
Highly curated and processed
Another name: annotations
What are examples of secondary databases
Pfam: Stores protein families and domains. (analyze protein sequences and group them into families (evolution, common ancestor)
InterPro: Provides functional analysis of proteins by classifying them into families and predicting domains and important sites. (similar to Pfam ) (Can search by sequence, text, or domain architecture)
PROSITE: Contains information about protein domains, families, and functional sites.(identify key amino acids and functional sites)
KEGG: Provides curated information on biological pathways and molecular interactions. (group enzymes into metabolic pathways)
GTDB: Genome Taxonomy Database; stores a curated set of representative genomes for all available bacterial/archaeal species, taxonomically labeled and phylogenetically analyzed (one genome = each speices, looks at bacteria and archaea. Idea: create giant tree of life)
AnnoTree: Stores functionally annotated genomes from the GTDB
What is the data type/content of secondary databases
The data in secondary databases is curated, analyzed, and interpreted by experts. It often includes functional annotations, structural predictions, evolutionary relationships, and interactions.
Content: Functional annotations, classifications, pathways, and other bioinformatic predictions
What is the purpose and example of usage of secondary databases
To provide added value by analyzing and interpreting the data from primary
databases. This helps users understand the biological significance of raw data.
Example Usage: You’d use a secondary database when you need functional annotation of a
protein, domain predictions, or pathway information for a set of genes or proteins.
Are all databases divided into primary or secondary databases
No, many biological Databases contain both primary and secondary databases (NCBI for example)
What are the types of data annotation in the UniProtKB database
SwissProtDB is a subset of the data base with human experts validating data and making sure it is high quality and correct (star)
TrEMBL is an automatic annotation (no star)
What are flat file databases
They store data in a plain text format with no relationships between data entries
• Data is organized in a simple, sequential manner without complex indexing
What are some characteristics of flat file databases
Human-readable: Files can be opened and viewed in basic text editors.
No relational structure: Each entry is independent; no relationships or links between entries.
Simple storage: Suitable for small to moderately sized datasets
What are the advantages and limitations of flat file databases
Advantages:
• Easy to create and share across different systems.
• Flexible and simple for basic data storage and retrieval tasks.
Limitations:
• Scalability: Inefficient for very large datasets or complex queries.
• Data retrieval: Slow when extracting specific information compared to relational databases.
• No built-in data validation: Errors in data entry or formatting can easily occur
What are example formats in computational biology of flat file databases
GFF/GTF and GenBank (GBK) formats: For genomic feature annotations (e.g., gene positions)
FASTA: For storing DNA, RNA, or protein sequences
CSV/TSV: For tabular data (e.g., gene expression, variant data
Work in “plain text” mode; other text formats (e.g., docx) will not work
What are some characteristics of GenBank
Header: non computational information
Features: gives predictions/annotations
Origin: Primary raw data
Difficult for very large genomes
What are some characteristics of GenBank
Header: title/label most of times either, mySequence or sequence 1, 2, etc…
Sequence: under header
What are relational databases and examples
Data stored within a number of tables linked together by a shared identifier or key
- key must be unique to each record
Handles huge amounts of data
- reducing data in memory
- faster search and retrieval
Examples
- Most large bioinformatic databases use relational DB tools such as MySQL or PostgreSQL
• e.g., AnnoTree DB developed here at UW uses a MySQL database
How are biological sequences represented
Can be represented as “strings”
In the form of DNA, RNA or Protein sequences
What are the six standard nucleotide/nucleotide ambiguity codes
Cytosine - C
Guanine - G
Adenine - A
Thymine - T
Uracil - U
Any (A,G,C,T) - N
What website can you use to translate DNA or RNA sequences into a protein sequence
Expasy operated by the SIB Swiss Institute of Bioinformatics
What do reading frames imply for DNA/RNA sequencing
reading frame ‘5 to ‘3 - 1: start from first letter going forward
reading frame ‘5 to ‘3 - 2: start from second letter going forward
reading frame ‘5 to ‘3 - 3: start from third letter going forward
reading frame ‘3 to ‘5 - 1: start from first letter going reverse
reading frame ‘3 to ‘5 - 2: start from second letter going reverse
reading frame ‘3 to ‘5 - 3: start from third letter going reverse
What are strings in biological sequencing
A sequence of characters or symbols, used in computer science to represent data
Biological sequences are represented as strings of letters, where each letter corresponds to a nucleotide or amino acid
What are DNA/RNA strings in biological sequencing
Composed of the characters A,T (or U), C, G representing nucleotides
What are protein strings in biological sequencing
composed of one-letter codes representing amino acids
Why do we use strings for biological sequences
Efficient Representation: Strings allow biological sequences to be efficiently stored, processed, and
analyzed in computational tools.
Manipulatable: Using algorithms, strings can be searched, aligned, compared, and transformed to
study biological patterns and relationships.
What are some operations we can do on strings
Alignment: Compare two or more sequences (strings) to find similarities.
Search: Locate specific patterns or motifs within a biological sequence.
Mutation: Simulate biological mutations by modifying characters in a string
What are common biological string formats
FASTA (.faa, .fna, .fa., .fasta): A common format for storing biological sequences as strings.
Plain Text: Biological sequences are often stored in plain text as strings for easy manipulation and sharing
What are some advanced search options
narrowing down results based on factors like organism, function, or location
What are controlled vocabularies/Ontologies
standardize terms, helping researchers define and refine their search criteria.
These standardized terms ensure consistent interpretation and improve the
accuracy of search results
What does ‘AND’ mean in terms of logic operators in searches
Retrieves records that match all criteria, representing the intersection of two
sets.
Example: Genes involved in cancer AND immune response (only genes in both categories)
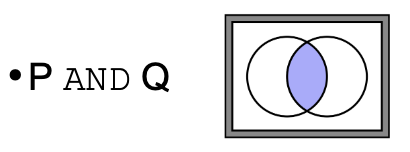
What does ‘OR’ mean in terms of logic operators in searches
Retrieves records that match any of the criteria, representing the union of two sets.
Example: Genes involved in cancer OR immune response (genes in either categories)
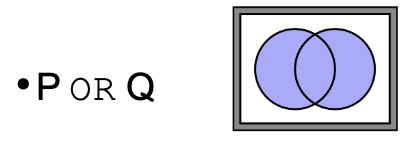
What does ‘NOT’ mean in terms of logic operators in searches
Excludes records that match certain criteria, representing the difference between sets.
Example: Genes involved in cancer NOT in the immune response (only cancer-related genes, excluding immune-related ones)
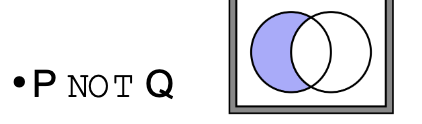
What does ‘NOR’ mean in terms of logic operators in searches
Excludes records matching any of the criteria, representing data outside of the union of two sets
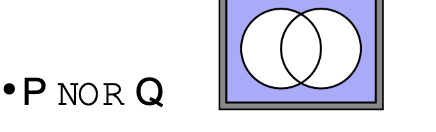
What is the best way to find genes/proteins related to gene/protein of interest
Through homology search (e.g., BLAST), not from text searches
What is sequence redundancy in biological databases
Biological databases (sequence databases in particular) have to deal with
many duplicate (“redundant”) sequences
Can occur due to researcher submissions involving identical sequences
Databases are computationally filtered to reduce or remove redundancy
How can databases be computationally filtered to reduce or remove redundancy
Uniref100 combines identical sequences into a single “cluster”
• identifies 100% identical sequences
Uniref90 combines sequences with 90+% identity
• clusters highly (90% or greater) similar sequences
Uniref50 combines sequences with 50+% identity
• clusters 50% or greater similar sequences
Uniref50 redundancy < Uniref90 redundancy < Uniref100 redundancy
What are some examples of computer error in database entries
Incorrect annotations or predictions
Missed relationships (insufficient information extraction)
What are some examples of human error in database entries
Highly variable quality of deposited sequence data (Vector sequences left in)(PCR chimeras)
Taxonomic misidentifications and mislabeling
Inconsistency in researcher labeling of gene/protein names and functional descriptions
Simple typos
Database propagation of initial human misannotations
What are the three broad classes of errors in database entries
Sequence - errors within the sequences
Metadata - incorrect identification of information/names towards the data entry
Propagation - encouraging ideas from other records/databases. not updating the records when it changes
Why is there no BLAST search capable of searching the SRA
Some bioinformatic databases are growing so large, they are becoming
difficult to analyze
NCBI Sequence Read Archive (SRA) has all raw sequencing data for virtually all types of sequencing projects. IT is the world’s largest repository of sequencing information