BIEB 146 weeks 1-2 material
1/72
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
73 Terms
order:
protein, DNA, RNA, translation, folding, polypeptide, transcription
DNA → RNA through transcription
RNA → polypeptide through translation
polypeptide → (3D) protein through folding
what percent of the human genome/human genome content is genes
1.5% is genes (protein-coding genes)
98.5% is everything else (non-coding DNA)
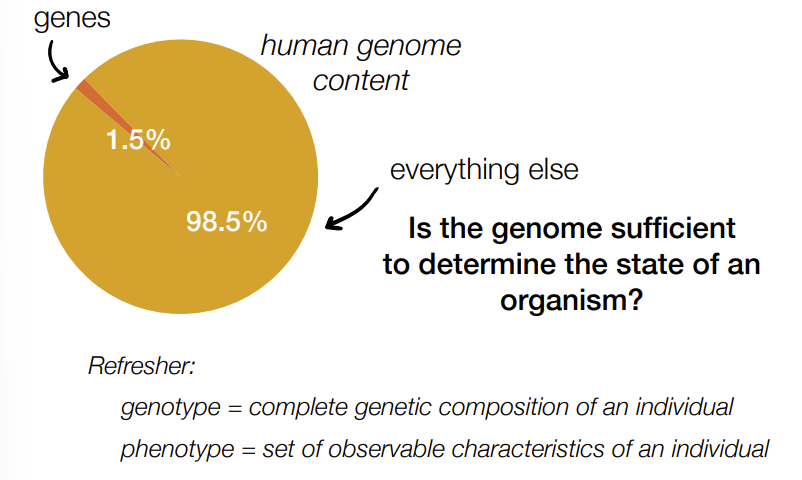
def. genotype and phenotype
genotype - complete genetic composition/makeup of an individual
phenotype - set of observable characteristics of an individual
Is genotype sufficient to predict phenotype?
a) yes
b) no
no
phenotype equation (aka the factors that determine phenotype)

genotype
environment
expression (how genes are expressed to be either ‘on’ or ‘off’)
experience

C, A (could technically also be B)
C b/c often sequence multiple individuals from same species to observe genetic variation and make sure have representative sample
A b/c many organisms aren’t classified/grouped or match any known species, which increase the # of sequences much more than the # of species (aka both #s increase, but # of sequences increase at a faster rate than the # of species)
technically B b/c this can happen & it does increase the # of sequences but not # of species
#of bp in human genome
3 billion bp
If we wrote out the human genome (3 billion bases) inside blank 7-book sets of Harry Potter (6,000,000 characters), about how far would it reach if we lined up one genome’s worth of books in a row? (where 1 genome worth of books 12 inches aka 1 foot)
a) 1 ft
b) 18 ft
c) 180 ft
d) 500 ft
e) 5000 ft
d) 500 ft
3 billion bases/letters divided by 6,000,000 letters per set is 500 sets per genome aka feet
% of identical genomes between humans and -:
humans, chimps, mice
human-human: 99.9%
human-chimp: 96%
human-mouse: 85%
classical genetics aka forward genetics (3 parts/steps)
starts with pheno- that researchers want to study
induce mutagenesis (causing random mutations in organism’s DNA)
mutated organisms studied to identify the genetic basis/gene responsible for that specific pheno-
aka starts with a specific pheno- that you want to observe & you work backwards by causing random mutations in the organism’s DNA to identify the genes responsible for it
mapping def.
finding the # of genes and location of genes
genetic linkage def.
traits that always show up together
b/c of the tendency that genes located close together on the same chromosome are inherited together
AND
traits that usually recombine are further away from one another
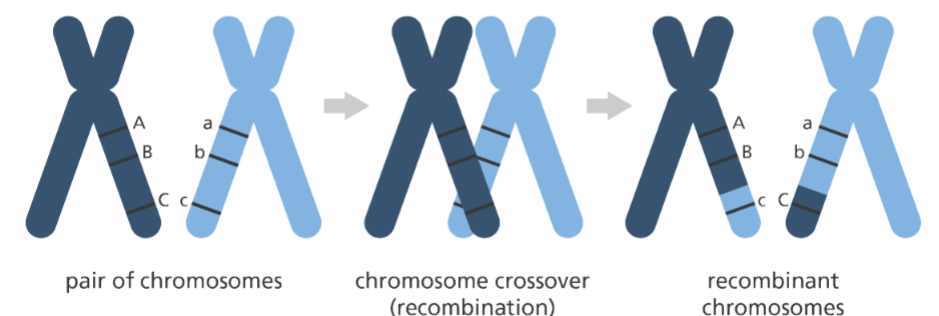
C

A) ABC becomes ABc
ddNTP vs dNTP in Sanger sequencing
dNTP with the 3’ OH group:
facilitate the extension of DNA of the primer/complementary strand (NOT template strand) when matching complementary nucleotide bases to the template strand using RNA polymerase
ddNTP without the 3’ OH group, is broken, has 3’ H (no 3’ O):
are modified nucleotides that act as a chain terminator in Sanger sequencing by preventing extension & used to determine the sequence by fluorescent (not radioactive) labeling to identify the bases at each “termination” (aka fluorescent labels for each base, below:)
ddATP, ddCTP, ddTTP, ddGTP

reading direction of template vs primer strand
temple is 3’ to 5’
primer is 5’ to 3’
how to read the gel electrophoresis for Sanger sequencing
gel electrophoresis is for primer strand
top to bottom of gel electrophoresis is from end of primer reading backwards (aka reading backwards starting from the terminating end)
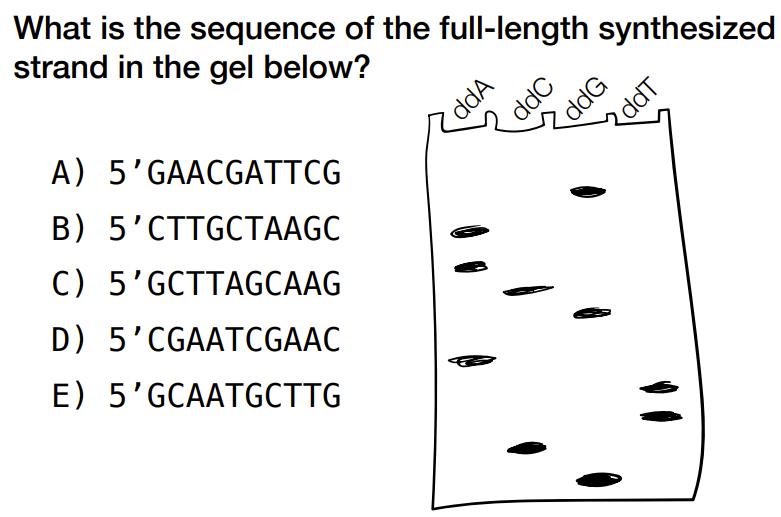
c) 5’ GCTTAGCAAG
(remember that you can read from top to bottom for left to right bases, but remember that the top of gel is 3’ end for the primer & 5’ end of primer/synthesized strand is at the bottom of the gel)
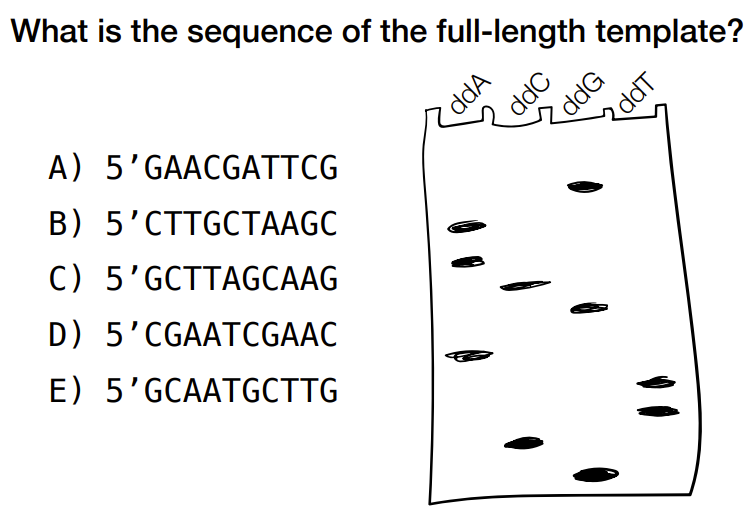
b) 5’ CTTGCTAAGC
identify the primer strand from gel & take complementary bases to find the original/template strand
steps of sanger sequencing (can have both gel and capillary electrophoresis that occur in order OR have either occur separately ← 3 ways)
(sequencing based on DNA replication, 1977
also called dideoxysequencing)
uses RNA polymerase to make complementary primer strand (polymerization)
dNTPs help extend the primer strand when it adds the complementary bases & ddNTPs stop extension and do chain termination that allows us to read what the bases are from the end of the primer chain
uses fluorescent labels (of ddNTPs as ddATP, ddTTP, ddCTP, ddGTP) → into a capillary (use a laser and detector) → to graph of waves of different colors to represent order of different bases
(then, need to find overlap and undergo assembly to form contiguous sequence/contigs)




effect of longer DNA strands on sanger sequencing graphs/electrophoregrams
worse signal aka harder to determine order of bases


E) both C and D
C b/c ddNTPs allow for chain termination of the fragment/sequence that allow for identifying the order of bases of the primer strand (should have sig. more dNTPs than ddNTPs)
D b/c good separation = more accurate reading of bases, poor separation limits accuracy of base reading for long DNA fragments
limits to traditional sequencing technology (sanger sequencing) (2)
struggles with sequencing longer DNA fragments (~1000 or more bp) aka takes a much longer time to sequence large fragments
harder to accurately identify bp in these larger fragments
3 strategies used during the human genome project (~which strategy is it)
primer walking (top-down strategy)
whole-genome shotgun sequencing (genome broken into small fragments)
hierarchical shotgun sequencing (genome broken into large fragments)
(shotgun sequencing is a random strategy b/c specific DNA segments aren’t cloned or targeted before sequencing AKA b/c no targeting or amplification of DNA fragments before sequencing)
steps of primer walking (top down strategy) (3/~4)
1 con
starts with a random primer (primer fragment) that will bind to the complementary part of the template strand
template strand is the single strand of when double-stranded DNA is denatured
do sanger sequencing to get a the sequence of a DNA fragment (that’s attached to the primer)
add new primer [that is the complement of the just produced DNA fragment (aka of the most previously made)] right below & do another sanger sequencing run w/ new primer as the new starting point
repeat
then align, overlap, and assemble the reads (aka synthesized DNA fragments, not including the primer part) to determine sequence of template strand
will get DNA fragments of different lengths
con:
only done 1 at a time, so can’t multiplex (aka can’t do multiple sequencing at once)


D) 3,000,000 days
steps of shotgun sequencing (in general, not talking abt specifically hierarchical or whole-genome yet)
(4-5)
fragment a genome into small fragments through sonication (using sound waves to break DNA strand into random small fragments)
(don’t design primers)
BUT
have to isolate fragments from one another (or else will get multiples bases read at one position for each position) in order to do multiplexing BY amplifying clones (aka make copies for each individual DNA fragment):
put one fragment in a vector (empty circle of DNA) → put into bacteria to transform the vector+fragment that amplifies that piece of DNA by making more copies/cloning
(this isolates fragments from one another & make copies of that fragment when inside bacteria)
then: sequence fragments, find overlaps, assemble DNA fragments by joining the overlaps of bases of 2 DNA fragments
get a contiguous/connected sequence


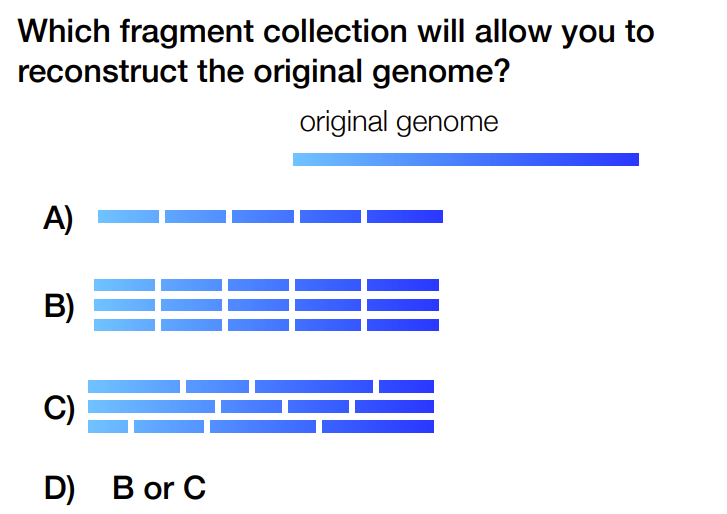
C
Needs random fragmentation, not identical
not A b/c no way to find overlap, need multiples
not B b/c breaks are in same place so can’t find overlap even though there are multiple copies
need to have multiple copies of the genome & fragmented randomly (i.e. with sonication)
2 things needed in order to sequence fragments & do multiplexing
need to have multiple copies of the genome
random fragmentation (after random fragmentation: DNA fragments are isolated from one another)
2 types of vectors & give example(s) of each with size
big vectors aka “ACs” (vectors =artificial chromosomes)
BACs = bacterial AC (<300 Kb aka <300,000 bases)

little vectors
plasmids (<10 Kb)
lambda viruses (<18 Kb)

whole-genome shotgun sequencing steps (4/~5)
fragment genome & randomly into small random DNA fragments
isolate fragments & clone/amplify/make copies in bacteria (using usually small vectors like plasmids and lambda viruses)
sequence (conditions met above allow for multiplexing)
assemble based on overlaps
(aka picking fragments at random)
hierarchical shotgun sequencing (~4)
randomly fragment genome into really random big fragments
put into BACs, undergo cloning
map the genome to find location of fragments (figure what chromosome they correspond to) AKA find the DNA fragments that correspond to parts of one chromosome (mapping) & pick the fragments that cover the whole genome (found through align and assembly)
shotgun sequencing for those corresponding large DNA fragments by → breaking into small random fragments → isolate, put into small vectors, amplify/clone → Sanger sequencing
which strategy sequencing was used for public vs private effort of human genome project
private - whole genome shotgun sequencing
public - hierarchical shotgun sequencing

in next generation sequencing, what are the 3 generations of tech/sequencing
1st gen: Sanger sequencing (1 read at a time, 1977) (if needs to be amplified, then does amplification in vectors)
2nd gen: “short-reads” sequencing (millions of reads at a time, use bridge amplification)
Illumina (2006)
3rd gen: “single molecule” sequencing (no amplification/cloning needed)
PacBio, SMRT (2010)
Oxford nanopore, minION (2015)
Illumina sequencing steps (aka more specific steps of 3 broad steps)
1) fragment:
randomly fragment MULTIPLE copies of the genome
attach adapters to both ends of the DNA fragments (adapters are short pieces of DNA aka “oligonucleotides”)
2) isolate and amplify clones
bind single DNA molecules to surface
adapters hybridize/bind to complementary oligo on lawn of flow cell (lawn of flow cell = surface of flow cell with small DNA fragments w/ oligonucleotides)
polymerase and dNTP make complement strand/primer strand
keep complement strand, get rid of original strand
do bridge amplification (where the complement strand forms a bridge on the lawn of the flow cell) → causing cluster generations to form (aka multiple copies for each complement DNA fragment)
3) sequencing by synthesis
add polymerase and use reversible terminator fluorescent dNTPs (can’t add anything to them, 3’ OH is still present but blocked BUT can be reversed back)
4 of these reversible terminator fluorescent dNTPs flow in (prevent polymerase from attaching more bases)
polymerase will attach the appropriate complementary base (aka will attach the structure of a 3’OH with the fluorophore and terminator), BUT will stop b/c other 3 fluorophores block the 3’ OH
take a picture of color of the first base added
remove the terminator and fluorophore, leaving behind the 3’ OH that is no longer blocked
repeat the whole process starting from adding 4 reversible terminator fluorescent dNTPs
(pictures are converted into a sequence)


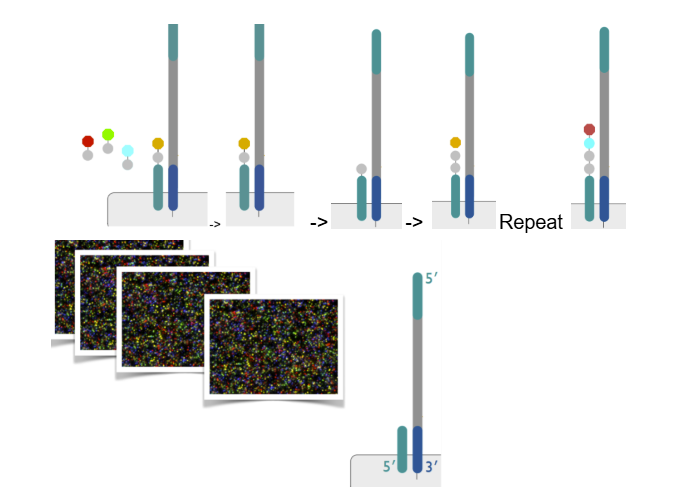
t/f: all of the dNTPs in sequencing by synthesis for Illumina sequencing are reversible terminator fluorescent dNTPs
true
(dNTPs used to make complement right before bridge amplification are not)
dividing steps of Illumina sequencing:
so describe fragmentation steps (2)
1) fragment:
randomly fragment MULTIPLE copies of the genome
attach adapters to both ends of the DNA fragments (adapters are short pieces of DNA aka “oligonucleotides”)
dividing steps of Illumina sequencing:
so describe “isolate and amplify clones” steps (~4)
2) isolate and amplify clones
bind single DNA molecules to surface
the adapters part of the DNA hybridize/bind to complementary DNA oligo on lawn of flow cell (lawn of flow cell = surface of flow cell with small DNA fragments w/ oligonucleotides)
polymerase and dNTP make complement strand/primer strand
keep complement strand, get rid of original strand
do bridge amplification (where the complement strand forms a bridge on the lawn of the flow cell) → causing cluster generations to form (aka multiple copies for each complement DNA fragment)
dividing steps of Illumina sequencing:
so describe “sequencing by synthesis” steps (~5)
3) sequencing by synthesis
add polymerase and use reversible terminator fluorescent dNTPs to make complement of the kept complementary strand (can’t add anything to them, 3’ OH is still present but blocked BUT can be reversed back):
4 of these reversible terminator fluorescent dNTPs flow in (prevent polymerase from attaching more bases)
polymerase will attach the appropriate complementary base (aka will attach the structure of a 3’OH with the fluorophore and terminator), BUT will stop b/c other 3 fluorophores block the 3’ OH
take a picture of color of the first base added
remove the terminator and fluorophore, leaving behind the 3’ OH that is no longer blocked
repeat the whole process starting from adding 4 reversible terminator fluorescent dNTPs
(pictures are converted into a sequence)

b) 5’ ATGAGG
bc “of synthesized strand” aka the strand made
just read from left to right

c) 5’ CCTCAT
“of template DNA”
so take the complement of above (this order for template is in the 3’ to 5’ b/c template always 3’ to 5’ & complement/primer always in 5’ to 3’, so flip order to get 5’ to 3’)
how many reads x bp in — hours for Sanger vs Illumina sequencing
sanger:
384 reads x 1,000 bp in 6 hours
illumina:
25,000,000 reads x 100 bp in 24 hours
how do you assemble Illumina reads
t/f: assemblies are rarely perfect
using the random strategy of shotgun sequencing by joining overlaps of DNA fragments into contiguous sequence(s) aka you have assembled contigs
(read across each position to reconstruct the original sequence)
aka
millions of reads → algorithms → assembled contigs
true

true
t/f: overlap sizes aren’t always the same
true
(ex: look at assembly of fragments with overlap)
4 ways to examine the assembly quality
% of reads assembled
number of contigs
length of contigs
N50
how to determine % of reads assembled
(# of reads that form the contig) / (total # of reads) * 100%
ex:

has 80% of reads assembled
t/f:
Perfect assembly can still have multiple contigs in the case of organisms that have their genomes split up into chromosomes -> in this case, # of contigs should = # of chromosomes of that organism
& most organisms have their genomes split up into chromosomes (except bacteria)
true
how to identify # of contigs and length of contigs
#of contigs: how many contigs there are
length of contigs: usually the length (in bp) of the longest contig unless specified
(below:
contigs are in this form because each contig represents a different contiguous sequence madeup of multiple DNA fragments, but unable to find overlap b/w these contigs)

def. N50
50% of assembly is contained in contigs greater than or equal to this length (length referring to the length of the contig that the N50 values fall in)
how to determine the N50
add all contig lengths to get total bp
find 50% of that total length
find where that value falls when ordering the contigs from longest to shortest
does n50 usually get smaller when assembly quality is worse
yes
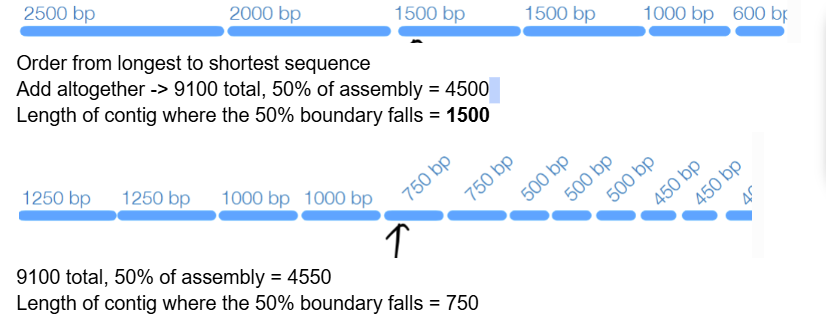

d) 1200
Total 6000, 50% of assembly = 3000, from longest to shortest: at 1200

(assume both have a single chromosome) ← not really relevant to solving this problem(?)
A
N50 of A is more accurate/closer to 50% of ebola assembly compared to a N50 of 10,000 bp for a moon jellyfish with 713,000,000 bp
3 factors that limit/lower assembly quality (w/ brief details)
low coverage (missing sequence, not enough overlaps)
difficult sequences (repeats and heterozygosity ← when mating occurs between diploids)
low accuracy (mistakes in sequencing that lead to bad assembly)
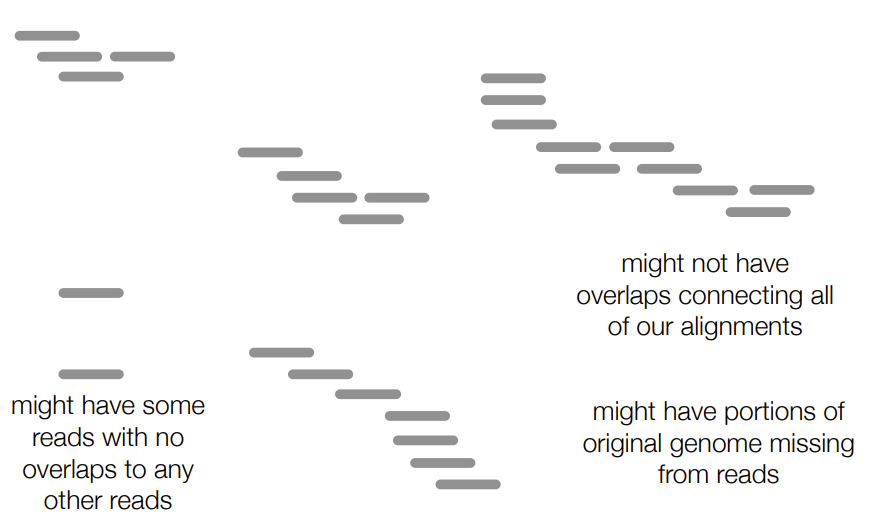
3 possibilities of read assembly/contiguous sequence
might not have enough overlaps connecting all our alignments (aka have the reads BUT don’t have enough overlap to connect them into a contiguous sequence)
might have some reads with no overlaps to other reads
might have parts of the original/template genome missing from the reads (aka the contiguous sequence formed doesn’t include all the parts of the template genome B/C some parts of the original genome weren’t sequenced at all)
for one of the factors that limit/lower assembly quality:
def. coverage
how to determine (depth of) coverage visually
what is low coverage (2 parts explained more as 3 parts)
how do you calculate coverage
coverage - # of reads that support a certain position
depth of coverage is how many reads at a certain position (look vertically)
low coverage → missing sequence & not enough overlaps (aka parts of DNA weren’t initially sequenced, not enough overlap, no overlap)

^ where numerator is the total # of bases sequenced

b) 10x
(2500 x 20)/5000
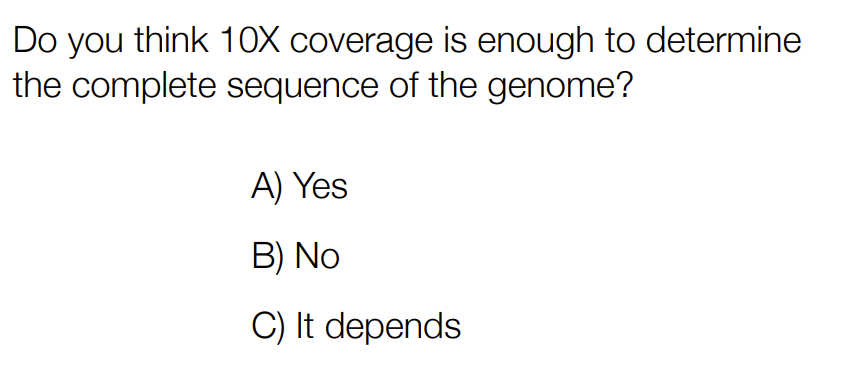
c) it depends
Generally, 30x-50x coverage is recommended
Shorter genomes (ie bacteria) can have less coverage (might have less possible missing pieces)
aka it depends on the size of the genome

c) 2500
50 = 100*x/5000 -> x = 2500
for one of the factors that limit/lower assembly quality:
what is difficult sequences (2 parts)
has repeats
Same sequence motif that’s repeated multiple times (i.e. CATCAT appearing twice that can form one of the 2 possible contigs below)


heterozygosity
for diploids, the DNA fragments could come from either chromosome (father or mother) → causing some reads to have different bases at the same position


a) repeat
for one of the factors that limit/lower assembly quality:
what is low accuracy (2 parts)
have mistakes in sequencing of genome that lead to bad assembly of reads
if have mistake (aka different bases at same position) in high coverage, is rare & likely a sequencing error
if have mistake in low coverage, we don’t know if significant error or just a minor sequencing error

2 errors that can occur in cluster generation or sequencing by synthesis (parts of Illumina sequencing)
incorrect base

missing base

how do we measure sequencing error
collect raw intensities for each fluorescently colored base
convert the intensities into a quality score (aka the probability that the most intense color is the correct base at that position)



Which position has the highest quality score?
Base 3
(Intensity of most intense color) / (total intensity of all 4 colors)
aka has the highest intensity of blue, especially looking at the other 3 bases that are very low in intensity
how do you improve the assembly when there are sequencing errors in low coverage (2, one has 2 parts)
use quality score
scaffolding
scaffolding with paired-end sequencing
scaffolding with long reads sequencing

^ data from sequencing - low coverage

c) the individual being sequenced has a heterozygous allele
b/c “both reads have high quality scores”, aka at this position that has coverage of 2x (from 2 reads at that position) both reads have high quality scores → meaning that both bases are likely to exist → meaning heterozygous allele (i.e. Aa)
Not B b/c would expect cluster of heterogeneous -> low quality
describe scaffolding with paired-end sequencing (~5)
make strand and wash away this strand that was just made
use bridge amplification to make complement strand
remove original strand/template strand, keeping the complement strand
sequence again in the opposite direction (to make a the template strand)
aka sequence from the top to bottom


how does paired end sequencing for scaffolding help with sequencing errors
if base pairs are perfectly complementary for the complement and synthesize “template” strand, you know there were sequencing errors and can remove the mis-matched base pairs from your data
for paired end sequencing for scaffolding:
what happens when each pair is fully overlapping (1)
often fragments are longer than the read length & so only the ends of the fragment are sequenced, what happens (1)
complete overlap (where you would get rid of the overlap/joining of reads if there’s mismatch of base pairing in an assumed perfect overlap)
pairs are separated by an unknown sequence of known length (aka of a long DNA fragment, small portion reads done by forward and reverse reading & if we know the fragment length, then we know about how far apart the forward and reverse reads are from each other)

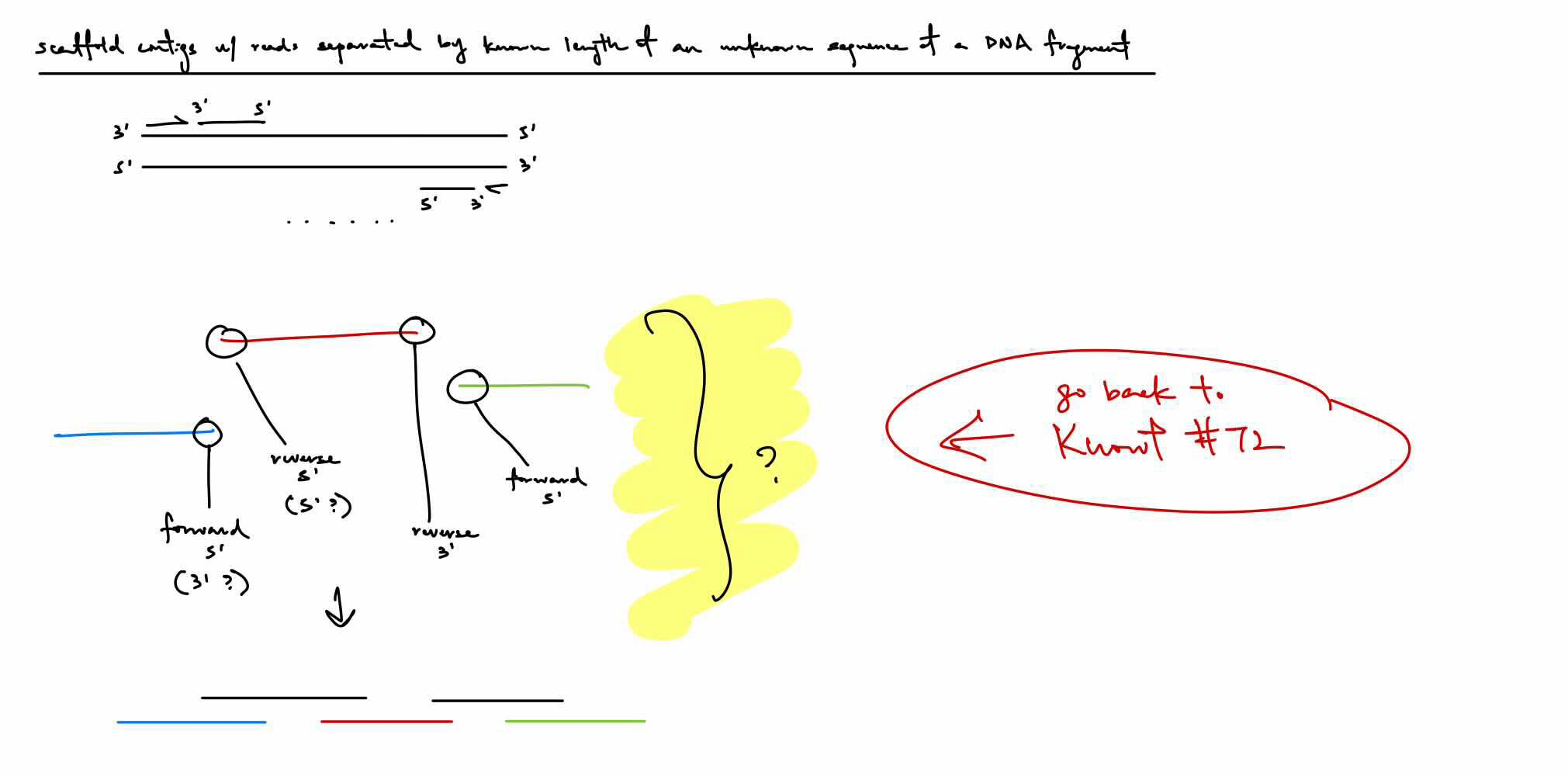
if have 3 contigs but don’t know the order, how do we determine the order (not the overlap)
we know that the contigs are from the same cluster with forward strand having the forward read in the 3' to 5' direction & with the reverse strand (5' to 3' relative to forward strand) having the reverse read in the 3' to 5' direction
if we have the forward contig 3' end & reverse contig 5' end, then that would determine the approximate ordering of the contigs (still considering the gap in between)
if forward and reverse contigs are inwards/towards one another, then follow that order
BUT: COULD BE THE 3’ OR 5’ END B/C IN THIS EX, THESE THREE CONTIGS MIGHT BE LOCATED ABOVE, BELOW, BESIDE, ETC. RELATIVE TO THE FORWARD OR REVERSE STRAND
^ just think this instead of the 2nd paragraph

Black strands are the connections of the 3’ and 5’ ends to determine the order of the contigs, knowing there’s an unknown amount of bp in between each contig
what is scaffolding with long reads sequencing
PacBio SMRT (single molecule real time sequencing)
single molecule is 50,000 bp
Oxford nanopore sequencing (minION)
single molecule is 100,000 bp
long read sequencing that uses these above technologies don’t need amplification b/c it is single molecule sequencing WHICH sequences the individual molecules instead of the amplified copies/clones