Protein Targeting: Lysosome, Nucleus, Mitochondria, & Perixome
1/30
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
31 Terms
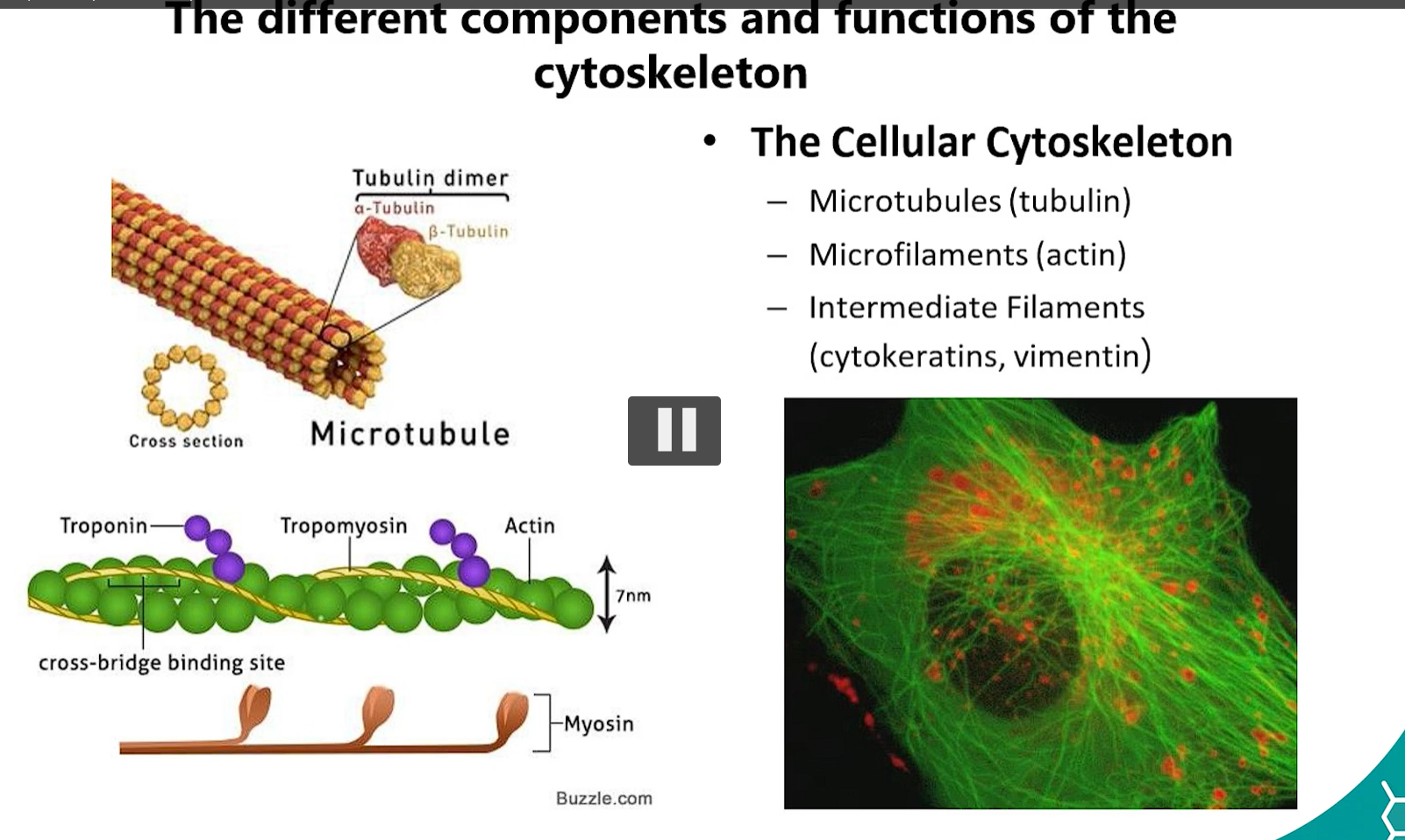
1. Branches of Protein Sorting & Targeting (cytosolic branch, endoplasmic reticulum ER)
Protein sorting begins after a protein is synthesized on a ribosome.
The pathway it takes depends on its "zip code" or signal sequence.
signal sequence= zip code= pathway it takes.
Cytoplasmic Branch: Proteins destined for the cytosol, nucleus, mitochondria, peroxisomes, or chloroplasts are synthesized on free ribosomes in the cytosol.
proteins are released directly into the cytosol and then:
Stay in Cytosol (no signal): If they have no signal, they function there.
Post-Translational Import (signal): If they contain the correct signal, they are imported into the target organelle after their synthesis is complete. This requires specialized translocation complexes (e.g., TOM/TIM for mitochondria, PEX for peroxisomes) and energy (ATP).
if there is no signal, the proteins in the cytosol stay in the cytosol
if there IS a SIGNAL, the proteins in the cytosol use translocation complexes (TOM/TIM for mitochondria, PEX for peroxisomes, and ATP) TO GO TO THE TARGET ORGANELLE (after synthesis is complete)
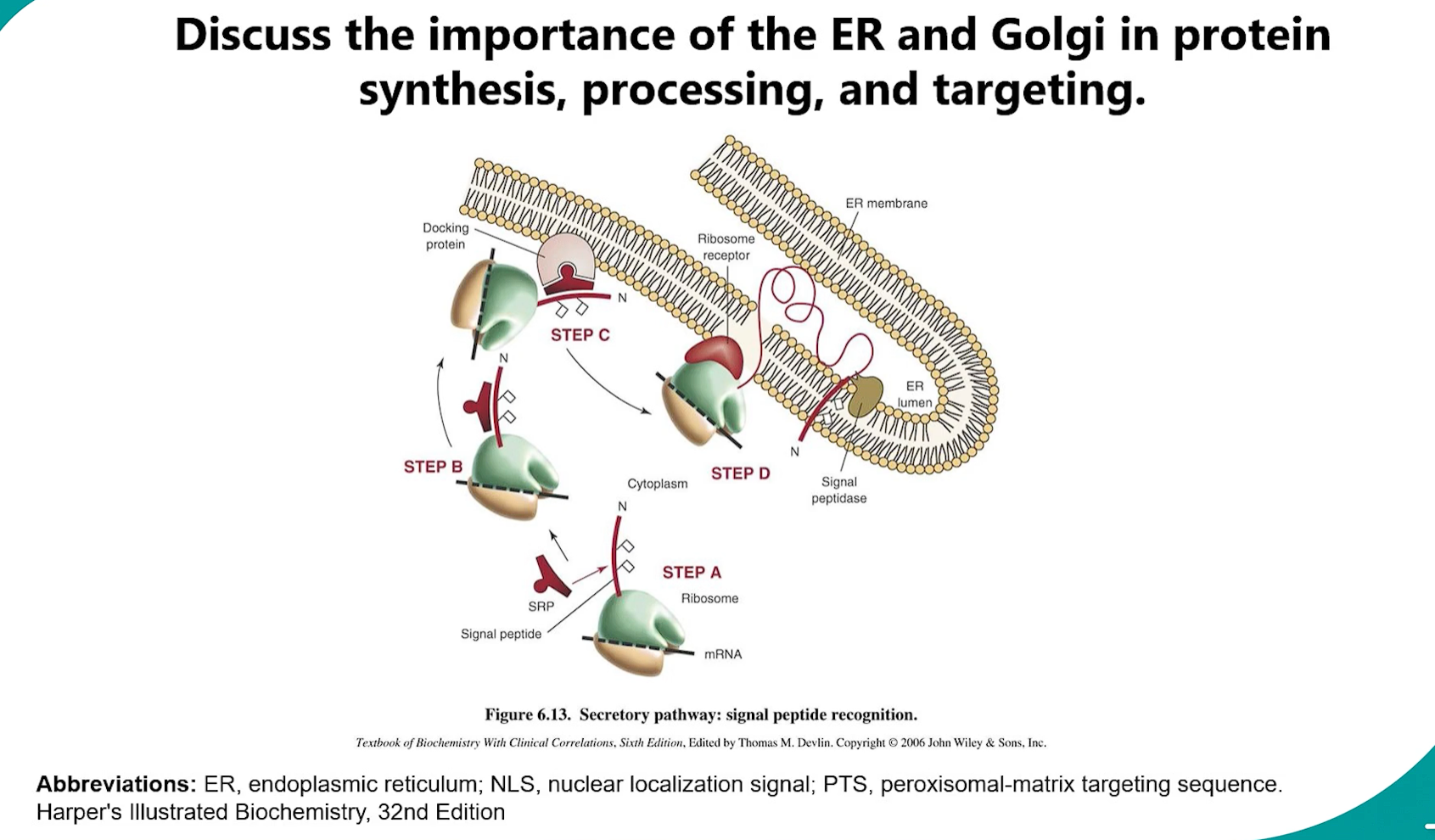
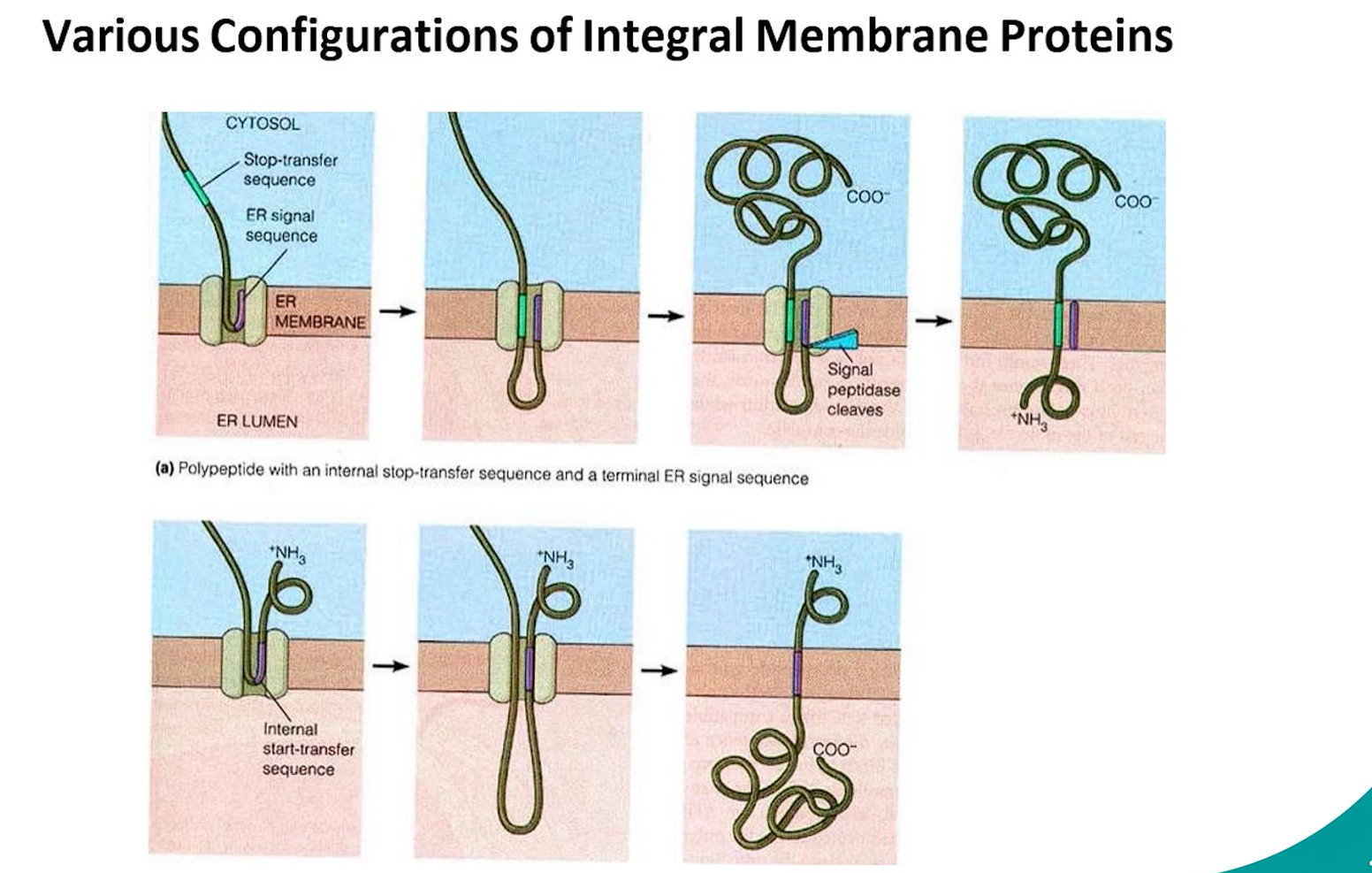
Endoplasmic Reticulum (ER) Branch: Proteins made on the ROUGH ER (RER)-bound ribosomes are destined for the ER, Golgi, Lysosomes, Plasma membrane, or secretion.
This is a co-translational import process:
a co-translational process is one that happens at the same time as translation. While the ribosome is still synthesizing the protein (the polypeptide chain is still growing), a specific cellular process begins.
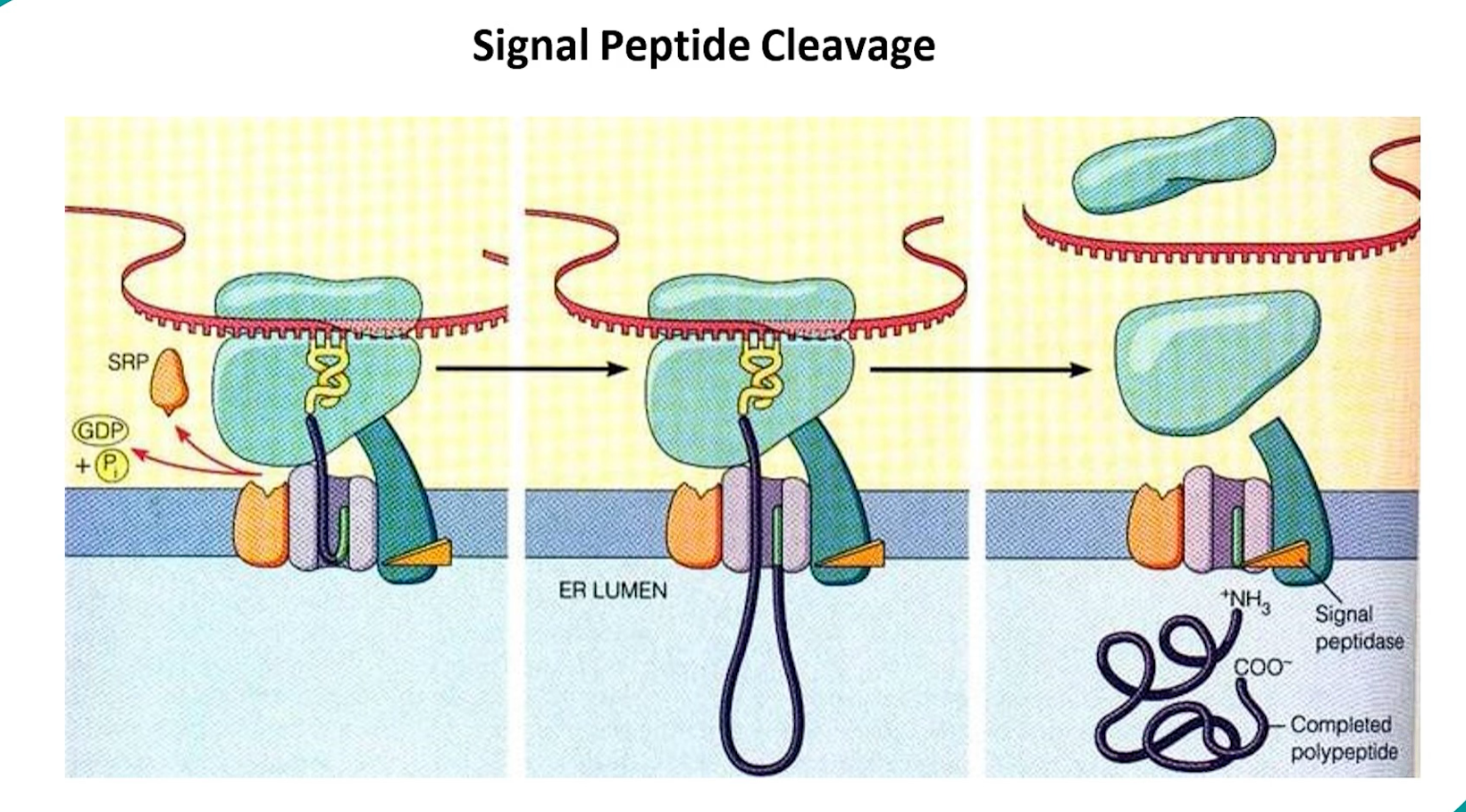
Signal Recognition: The signal sequence emerges from the ribosome.
A Signal Recognition Particle (SRP) binds to the signal sequence and pauses translation.
The SRP-ribosome complex docks onto an SRP receptor on the RER membrane.
The ribosome transfers to a translocon (Sec61 complex).
Translation resumes, and the growing polypeptide chain is threaded through the translocon into the ER lumen.
Entry into Secretory Pathway: Once in the ER, the protein enters the secretory pathway (ER → Golgi → various destinations).
signal recognition → SRP binding (translation pauses) → SRP-ribosome complex docks onto SRP receptors on RER membrane → ribosome transfers to a translocon complex→ translation continues into ER lumen → secretory pathway (ER → Golgi → various destinations).
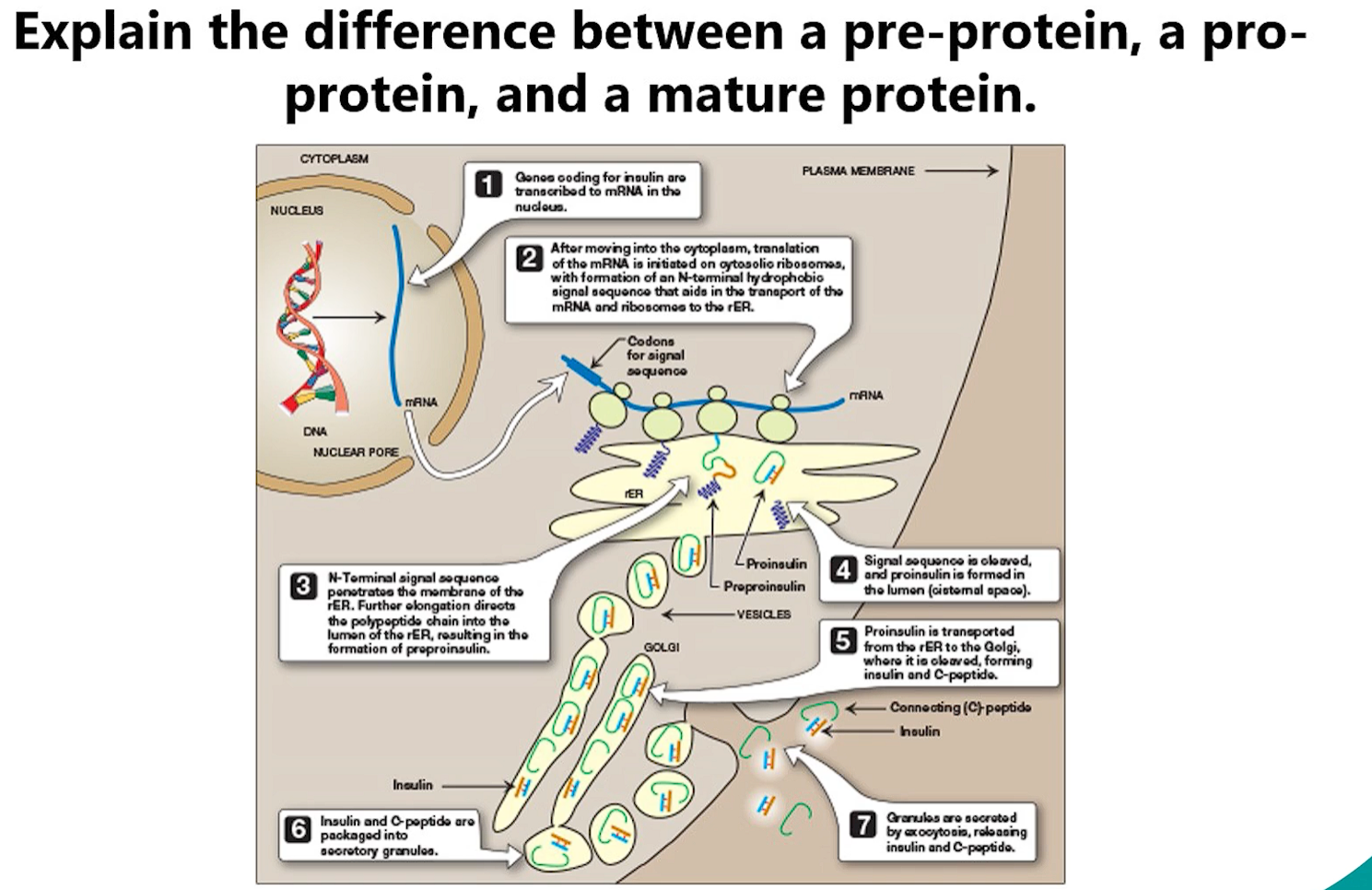
2. Pre-pro-protein vs. Pro-protein vs. Mature Protein
These terms describe stages in the maturation of many secreted proteins.
Pre-pro-protein: This is the initial, primary translation product. It contains two key parts:
"Pre-": An N-terminal signal peptide that targets it to the ER.
"Pro-": An internal propeptide that is often required for correct folding or to inhibit the protein's activity until it reaches its destination.
Example: Pre-pro-insulin.
Pro-protein: Once inside the ER, the signal peptide is cleaved off by signal peptidase. The remaining molecule is now a pro-protein. It is inactive and often folds into a stable, inactive conformation.
Example: Pro-insulin (after signal peptide removal).
Mature Protein: The propeptide is cleaved off later in the secretory pathway (usually in the trans-Golgi network or secretory vesicles) by specific enzymes called proprotein convertases. This cleavage activates the protein.
Example: Mature, active insulin (after propeptide removal).
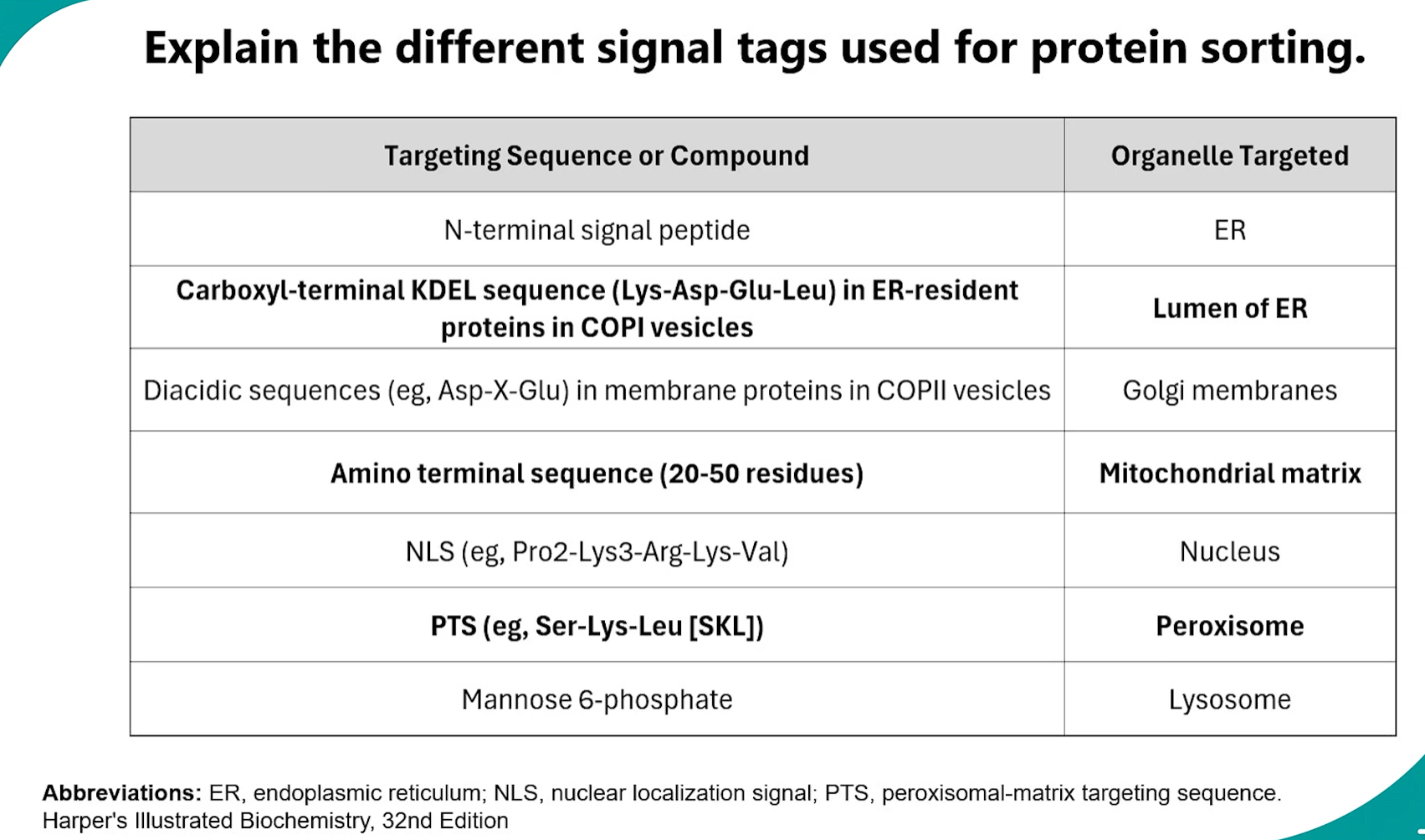
3. Signal Tags for Protein Sorting
Proteins contain specific short amino acid sequences that act as "sorting signals" to direct them to the correct organelle.
amino acid sequences= sorting signals = DIRECT THEM TO CORRECT ORGANELLE.
Signal | Sequence/Feature | Function |
|---|---|---|
ER Signal Sequence | N-terminal, 5-10 hydro-phobic amino acids | Targets ribosome to ER for co-translational translocation. Cleaved off after use. |
Nuclear Localization Signal (NLS) | Internal, rich in Lys (K) and Arg (R) (e.g., PKKKRKV) | Directs protein import into the nucleus through nuclear pores. |
Nuclear Export Signal (NES) | Rich in Leu (L) (e.g., LQLPPLERLTL) | Directs protein export out of the nucleus through nuclear pores. |
Mitochondrial Import Signal | Amphipathic α-helix at N-terminus | Directs protein into the mitochondrial matrix. Often cleaved off. |
Peroxisomal Targeting Signal (PTS1) | C-terminal tripeptide SKL or variant | Directs protein import into the peroxisome. |
Lysosomal Targeting Signal | Mannose-6-Phosphate (M6P) tag (added enzymatically) | Directs soluble enzymes to the lysosome. |
KDEL Sequence | C-terminal Lys-Asp-Glu-Leu (KDEL) | ER retention signal. Retrieves escaped ER proteins from the Golgi and returns them. |
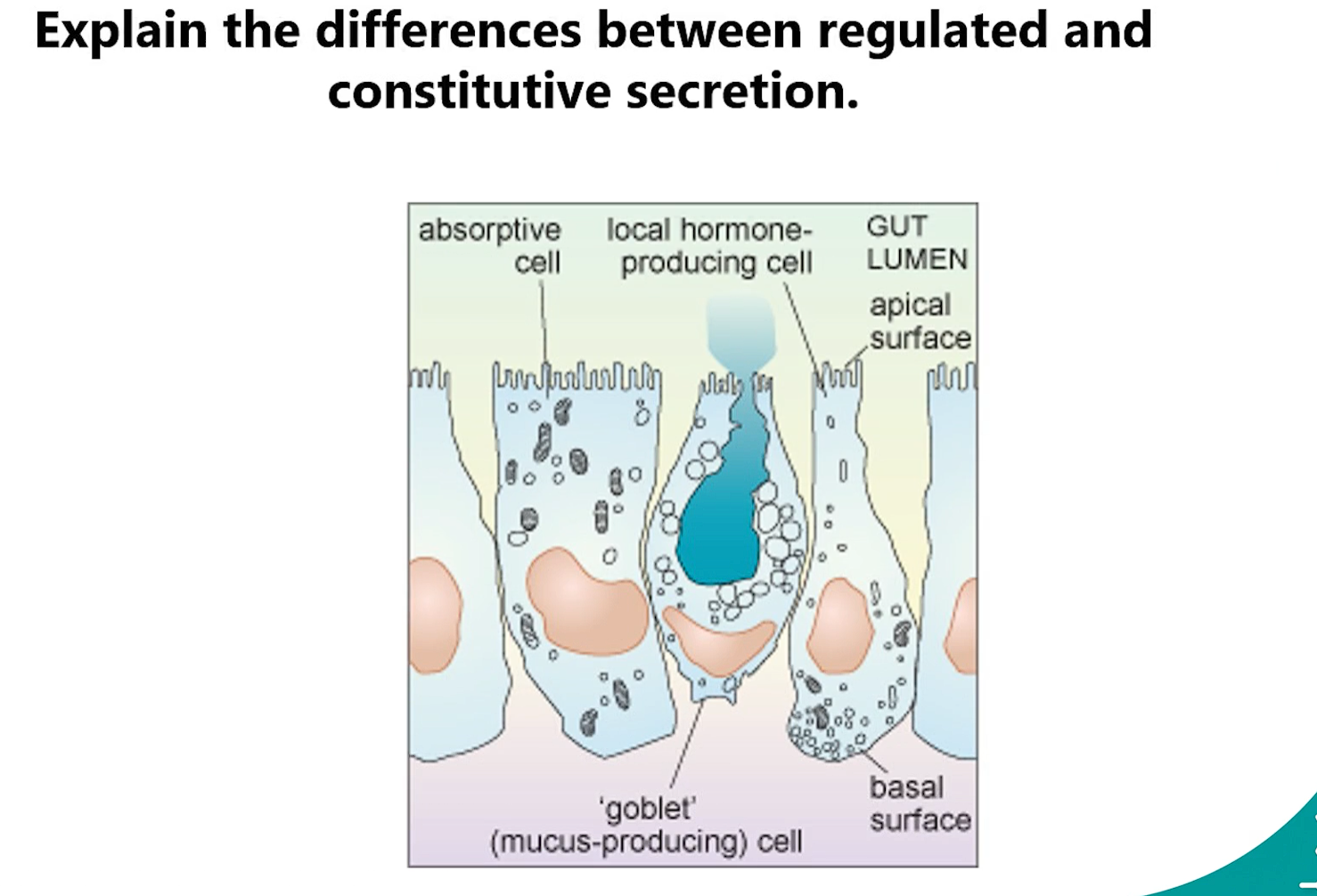
4. Regulated vs. Constitutive Secretion
Constitutive Secretion: A continuous, unstimulated process. Vesicles carrying cargo (e.g., plasma membrane proteins, extracellular matrix components) immediately bud from the trans-Golgi network and fuse with the plasma membrane. It happens in all cells.
Regulated Secretion: An on-demand, signal-triggered process. Secretory products (e.g., hormones, neurotransmitters, digestive enzymes) are stored in dense-core secretory vesicles near the plasma membrane. These vesicles only fuse and release their cargo in response to a specific extracellular signal (e.g., a hormone, calcium influx).
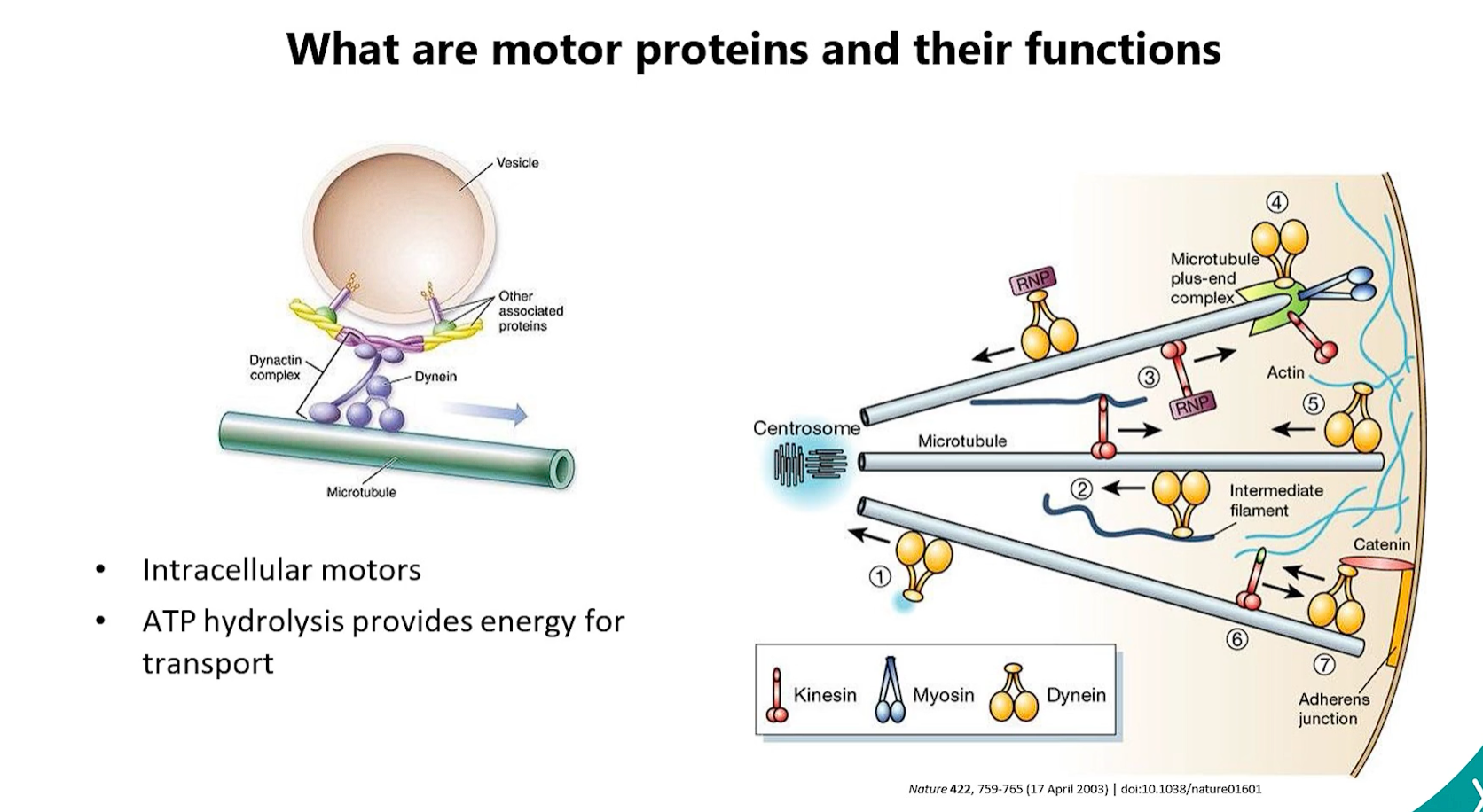
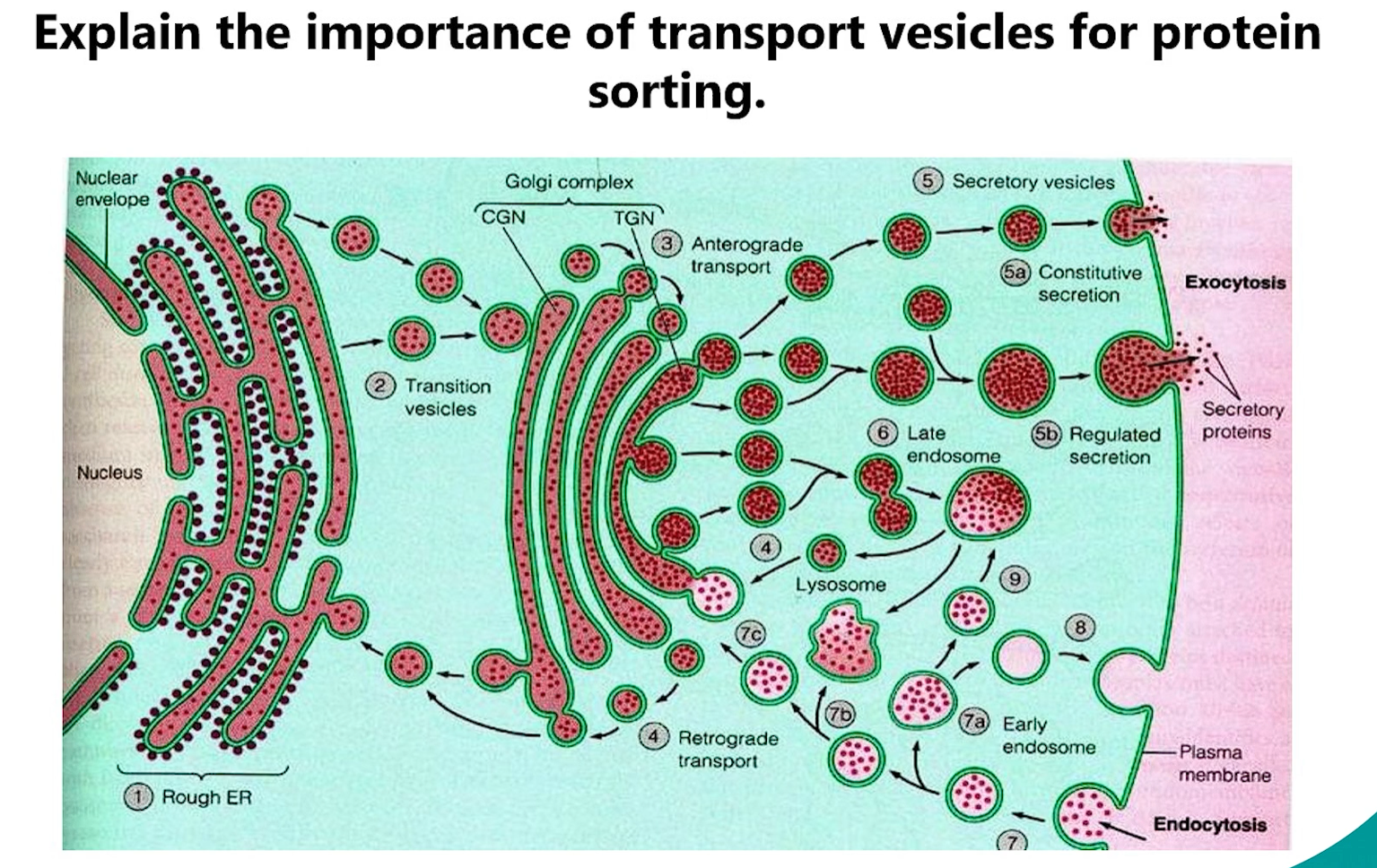
5. Importance of Transport Vesicles
transport vesicles = shipping containers of the cell
-selective cargo packaging
-directional transport
-maintenance of compartment identity
-driven by protein coats (COP I and COP II are examples, Clathrin)
II = forward, I = backward (easy)
Transport vesicles are the fundamental "shipping containers" of the cell. Their importance cannot be overstated:
Selective Cargo Packaging: They allow the cell to selectively package specific cargo (e.g., proteins with the same destination signal) together.
Directional Transport: They move cargo between membrane-bound compartments (e.g., ER to Golgi, Golgi to PM).
Maintenance of Compartment Identity: By budding from one compartment and fusing with another, they allow each organelle to maintain its unique protein and lipid composition despite constant traffic.
Driven by Protein Coats: Their formation is driven by coat proteins (e.g., COPII for ER→Golgi, COPI for Golgi→ER retrograde, Clathrin for TGN→Lysosomes/PM), which ensure specificity.
TGN → Lysosomes: This is the primary and most classic function of clathrin-coated vesicles originating from the trans-Golgi Network (TGN).
TGN → Plasma Membrane (PM): This is less common. While it can happen, the major pathway from the TGN to the PM is via clathrin-independent vesicles (e.g., for constitutive secretion).
6. Importance of ER & Golgi
ER
-synthesis
-folding
-quality control
-modification
golgi apparatus
-processing
-synthesis
-sorting hub
-packaging
Endoplasmic Reticulum (ER):
Synthesis: Site of synthesis for secretory/membrane proteins.
Folding (ER lumen has chaperones): Provides an environment for proper protein folding (lumen contains chaperones like BiP).
Quality Control: Misfolded proteins are detected and retro-translocated for degradation (ERAD).
Modification: Initial glycosylation (N-linked), disulfide bond formation, lipid assembly.
Golgi Apparatus:
Processing: Modifies N-linked glycans (trims mannoses, adds new sugars).
ER performs N-linked glycosylation, Golgi MODIFIES N-linked glycans by trimming mannoses and adding new sugars)
Synthesis: Synthesizes O-linked glycosylation and proteoglycans.
Sorting Hub: The trans-Golgi network (TGN) is the major sorting station, directing proteins to lysosomes, plasma membrane, or secretory vesicles.
Packaging: Packages sorted cargo into specific transport vesicles.
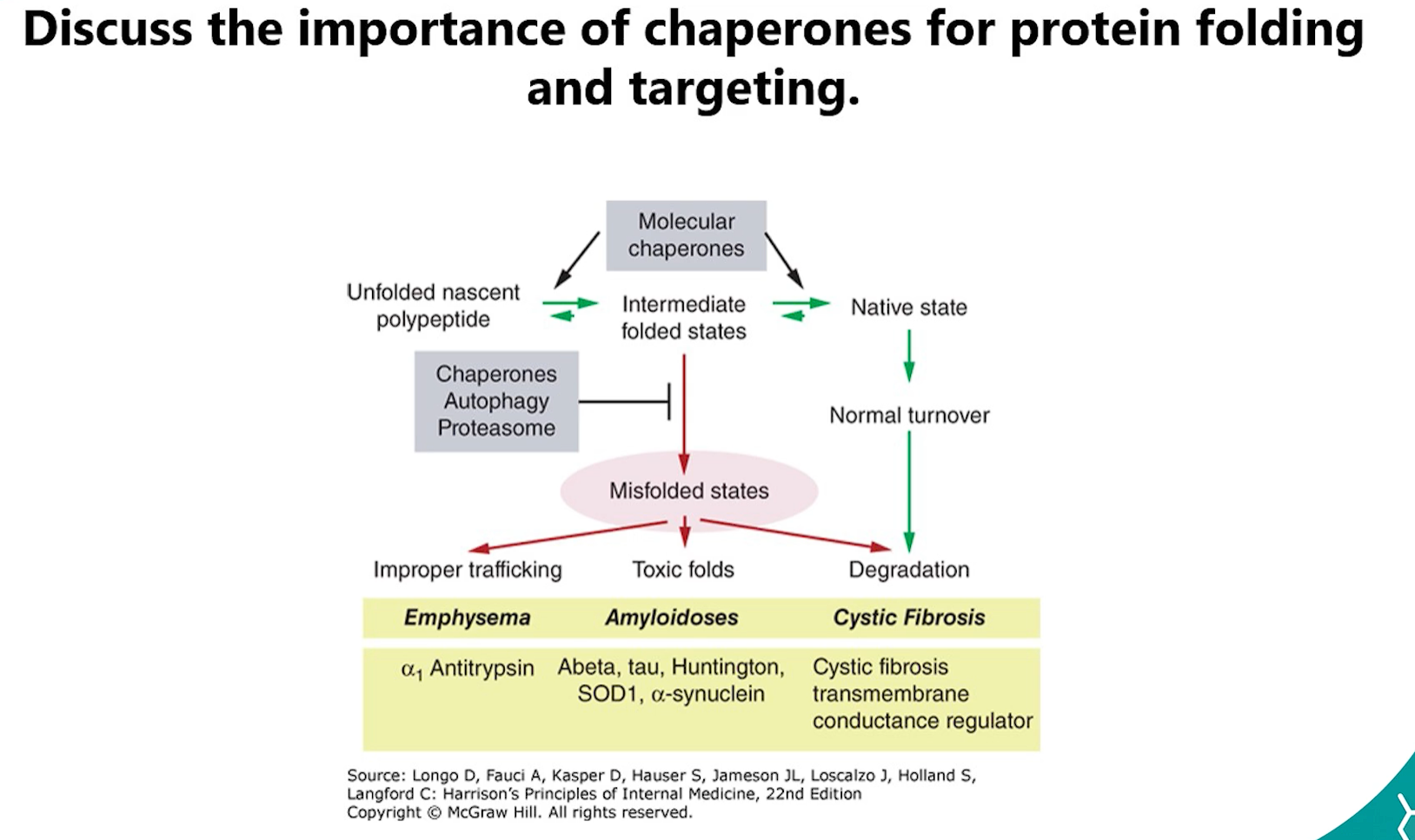
7. Importance of Chaperones (Hsp70, Hsp60, BiP)
Chaperones (e.g., Hsp70, Hsp60, BiP, Calnexin) are crucial for protein folding and targeting:
Prevent Misfolding: They bind to hydrophobic regions of unfolded polypeptides, preventing them from aggregating.
Promote Folding: They use ATP hydrolysis to facilitate the correct folding of proteins.
Quality Control: They identify terminally misfolded proteins and target them for degradation.
Mitochondrial Import: They keep cytosolic proteins unfolded so they can be threaded through the TOM/TIM complexes.
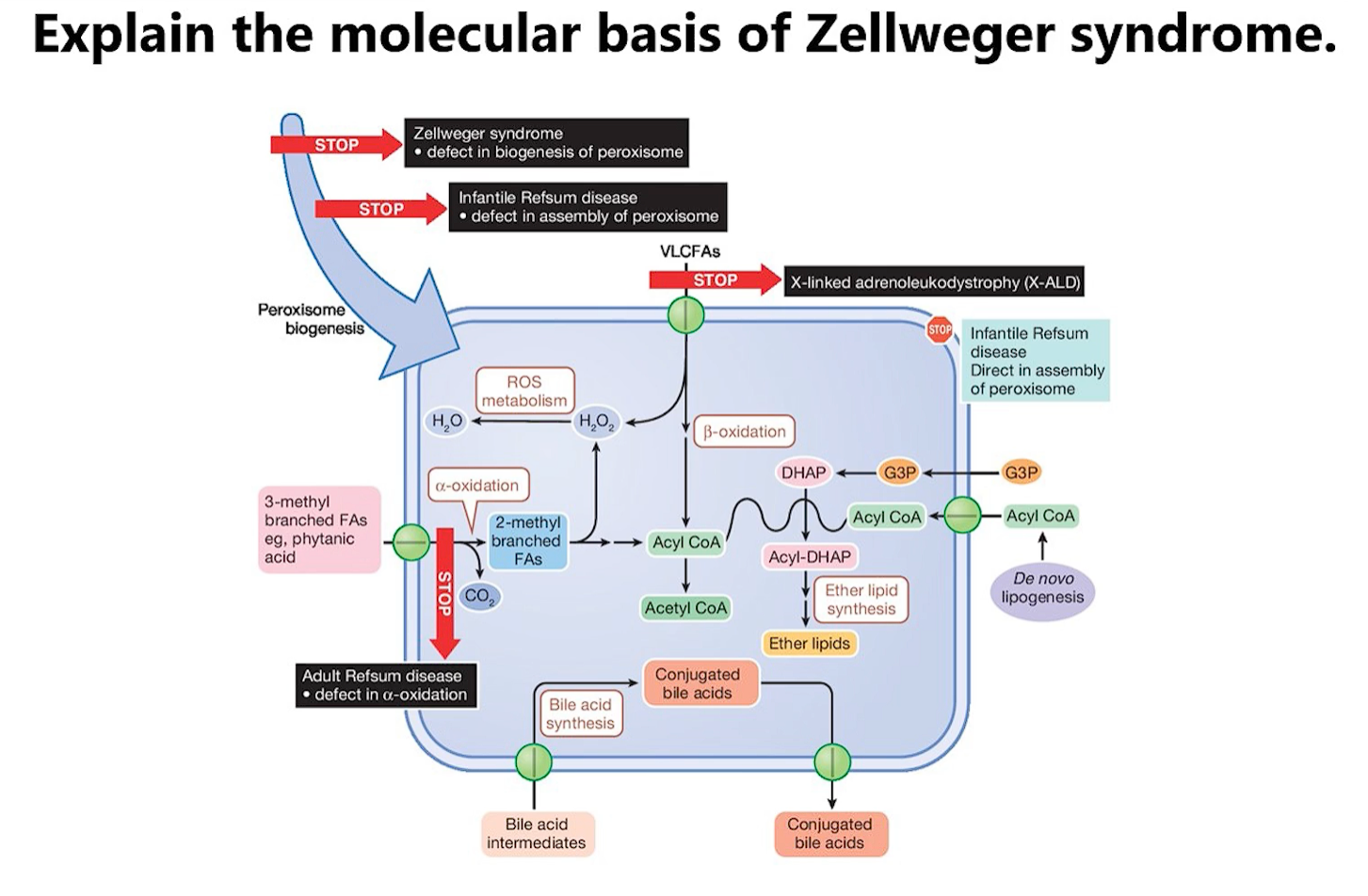
8. Molecular Basis of Zellweger Syndrome
Deep Dive into Zellweger Syndrome
Think of a cell as a large city. Peroxisomes are specialized recycling and power plants within that city, responsible for breaking down toxic waste and producing special materials.
In Zellweger Syndrome, the city has built the shells of these plants (the "ghosts"), but the machinery and workers that should be inside are missing because the import system is broken. This leads to a city-wide crisis.
1. The Core Problem: The Broken Import Machinery
The Genes (PEX Genes): These are like the "instruction manuals" for building the peroxisome import machinery. There are at least 13 different manuals (PEX genes), each for a different part of the machine.
The Machinery (Peroxins): The proteins built from these instructions are called peroxins. They form a complex system on the peroxisome membrane that recognizes proteins meant for the peroxisome, unfolds them, and pumps them inside.
The Defect: A mutation in any one of these PEX genes means a critical piece of the import machinery is missing or broken. The entire import line shuts down.
2. The Result: "Peroxisomal Ghosts"
Without the import machinery, the cell cannot bring any of its essential enzymes into the peroxisome.
The peroxisome membrane is formed, creating empty structures called "ghosts."
All the enzymes that are supposed to be working inside the peroxisome are left floating uselessly in the cytosol, where they are eventually degraded.
The metabolic functions of the peroxisome are completely lost.
3. The Consequences: A Metabolic Catastrophe
The peroxisome is involved in several crucial metabolic pathways. When they fail, the consequences are severe and multi-systemic.
Consequence 1: Accumulation of Very Long-Chain Fatty Acids (VLCFAs)
Normal Function: Peroxisomes are the primary site for breaking down (beta-oxidation) VLCFAs. These are fatty acids with long carbon chains that mitochondria cannot handle.
In Zellweger: VLCFAs are not broken down. They build up in the blood and tissues.
Toxic Effects: VLCFAs are incorporated into cell membranes (especially in the brain and nerves), disrupting membrane integrity and electrical signaling. This is a primary cause of severe neurological impairment, seizures, and vision/hearing loss.
Consequence 2: Accumulation of Branched-Chain Fatty Acids (e.g., Phytanic Acid)
Normal Function: Peroxisomes break down phytanic acid, a fatty acid found in dairy products, beef, and lamb.
In Zellweger: Phytanic acid accumulates.
Toxic Effects: High levels are toxic to neurons and contribute to skin abnormalities, neurological issues, and vision problems (retinitis pigmentosa).
Consequence 3: Deficiency of Plasmalogens
Normal Function: Peroxisomes are the exclusive site for the first steps of synthesizing plasmalogens. These are not waste products; they are essential, specialized phospholipids that are a major component of myelin (the insulating sheath around nerves) and cell membranes throughout the body, especially in the brain, heart, and immune cells.
In Zellweger: Plasmalogen synthesis is crippled. The body cannot produce enough of them.
Toxic Effects: The lack of plasmalogens has devastating effects:
Brain & Nerves: Myelin fails to form properly (dysmyelination), leading to profound neurological deficits, similar to diseases like Multiple Sclerosis but much more severe.
Bone Development: Contributes to the characteristic skeletal abnormalities seen in patients.
General Cell Function: Compromises the function of cells everywhere in the body.
Consequence 4: Accumulation of Oxalates and Bile Acid Precursors
Other toxic substances, like oxalic acid (which can lead to kidney stones) and immature bile acids (which cause liver damage), also build up, contributing to liver dysfunction and kidney problems.
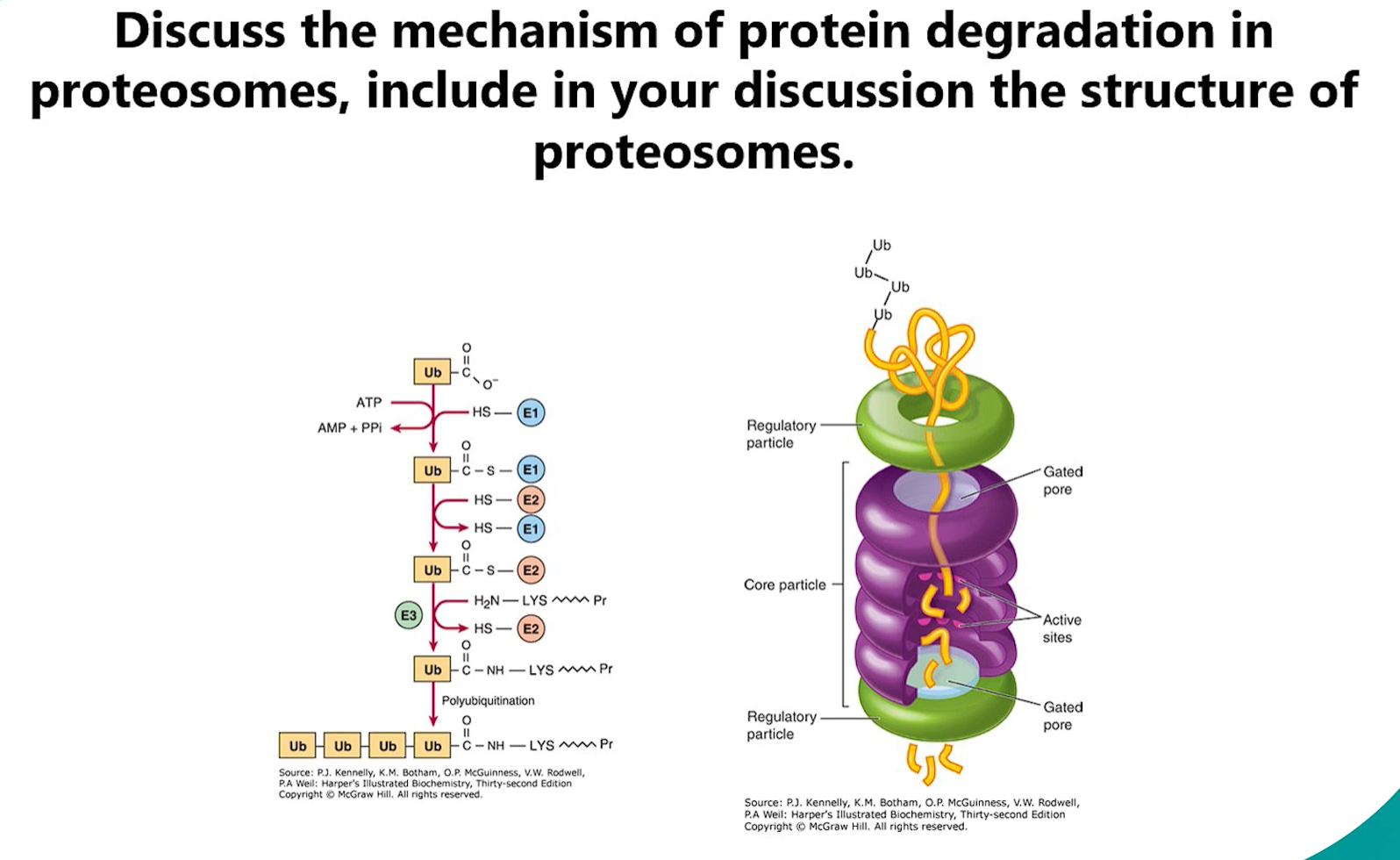
9. Protein Degradation by Proteasomes
The proteasome is the cell's primary machinery for targeted, efficient degradation of intracellular proteins. It is essential for maintaining cellular homeostasis by disposing of damaged, misfolded, or regulatory proteins.
1. The Ubiquitin Signal: "Marking for Death"
Before a protein is degraded by the proteasome, it must first be tagged by a ubiquitin signal. This is done through ubiquitination.
Process: A small protein called ubiquitin is covalently attached to a lysine residue on the target protein.
Enzymatic Cascade: The process involves three enzymes:
E1 (Ubiquitin-Activating Enzyme): Activates ubiquitin using ATP.
E2 (Ubiquitin-Conjugating Enzyme): Accepts the activated ubiquitin from E1.
E3 (Ubiquitin Ligase): Recognizes degron (a specific degradation signal) on the target protein and catalyzes the transfer of ubiquitin from E2 to the target. E3 ligases provide the specificity—there are hundreds, each recognizing a distinct set of target proteins.
Polyubiquitination: To be recognized by the proteasome, a chain of at least four ubiquitin molecules linked through lysine 48 (K48) must be attached to the target protein. This polyubiquitin chain is the definitive "degrade me" signal.
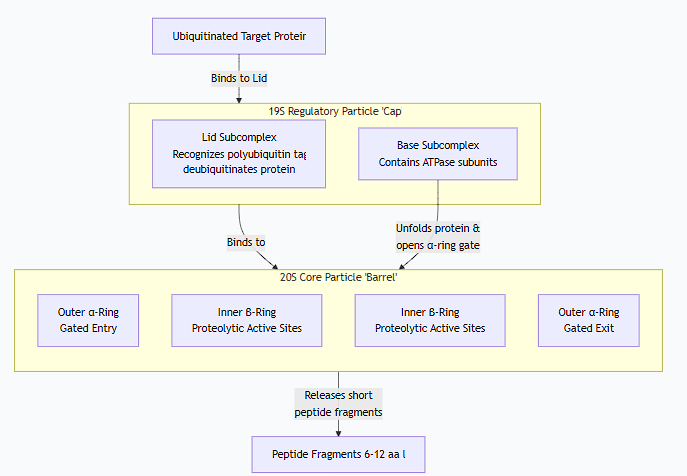
2. The Structure of the 26S Proteasome
The proteasome is a massive, multi-subunit complex composed of two main parts:
A. The 20S Core Particle (CP): The "Degradation Chamber"
Shape: Barrel-shaped, hollow cylinder.
Composition: Made of 28 protein subunits arranged in four stacked heptameric rings:
2 Outer α-Rings: Serve as a regulatory gate. They recognize the regulatory particle and control entry into the degradation chamber. The central pore is normally closed.
2 Inner β-Rings: Contain the proteolytic active sites. Each β-ring has three different catalytic subunits with distinct specificities:
Caspase-like activity (cleaves after acidic residues)
Trypsin-like activity (cleaves after basic residues)
Chymotrypsin-like activity (cleaves after hydrophobic residues)
This variety allows the proteasome to cleave any protein into short peptides.
B. The 19S Regulatory Particle (RP) or "Cap": The "Gatekeeper & Unfolder"
Function: Recognizes ubiquitinated proteins, prepares them for degradation, and regulates entry into the 20S core.
Composition: Made of at least 19 subunits, forming two subcomplexes:
Base: Contains ATPase subunits (Rpt proteins). These use the energy from ATP hydrolysis to:
Unfold the target protein.
Translocate the unfolded polypeptide into the 20S core.
Open the gate of the α-ring.
Lid: Contains subunits that:
Recognize the polyubiquitin tag.
Remove (deubiquitinate) the ubiquitin chain before degradation, allowing it to be recycled.
The complete complex, the 26S proteasome, consists of the 20S core particle capped at one or both ends by a 19S regulatory particle.
the mechanism of degradation (proteosome)
3. The Mechanism of Degradation
Recognition: The 19S cap recognizes and binds to the polyubiquitin tag on the target protein.
Deubiquitination: Enzymes in the 19S lid cleave off the ubiquitin chain, recycling it for future use.
Unfolding: The ATPase subunits in the 19S base use energy from ATP hydrolysis to unfold the target protein. This is essential because only unfolded proteins can fit through the narrow pore of the 20S core.
Translocation: The unfolded polypeptide is threaded into the central channel of the 20S core particle.
Degradation: Inside the sealed chamber of the 20S core, the polypeptide is cleaved into short peptides (typically 6-12 amino acids long) by the proteolytic active sites.
Release: The short peptide fragments are released into the cytosol.
Further Processing: These peptides are rapidly broken down into individual amino acids by other cellular peptidases, which are then recycled for new protein synthesis. In a crucial process for immune surveillance, some peptides are transported into the endoplasmic reticulum and presented on the cell surface as antigens by MHC class I molecules.
Biological Importance
The ubiquitin-proteasome system is critical for:
Protein Quality Control: Degrading misfolded, oxidized, or damaged proteins.
Regulation of Cell Processes: Controlling the levels of key regulatory proteins involved in the cell cycle (cyclins), apoptosis, and signal transduction.
Immune Response: Generating antigenic peptides for presentation to the immune system.
Stress Response: Rapidly removing proteins damaged by cellular stress.
Dysregulation of the proteasome is implicated in many diseases, including cancer, neurodegenerative diseases (like Alzheimer's and Parkinson's), and autoimmune disorders. This makes the proteasome a significant target for pharmaceutical drugs, such as Bortezomib, a proteasome inhibitor used to treat multiple myeloma.
Clinical Correlation (Cystic Fibrosis)
Cystic Fibrosis (CF) is a genetic, life-limiting disorder that primarily affects the lungs and digestive system. It is caused by a defect in a single gene that leads to the production of thick, sticky mucus throughout the body.
1. The Genetic Basis: Cause at the DNA Level
Inheritance: CF is an autosomal recessive disorder. This means:
A person must inherit two defective copies of the gene (one from each parent) to have the disease.
Parents who each carry one defective copy are called "carriers." They are healthy but can pass the gene to their children.
The CFTR Gene: The disease is caused by mutations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene, located on chromosome 7.
Over 2,000 different mutations have been identified, but the most common one is ΔF508 (a deletion of three nucleotides leading to the loss of a single amino acid, phenylalanine, at position 508).
Your 4-Step Process: Explained
CFTR Channel Opens: This is the key regulatory step. In response to specific signals (e.g., cyclic AMP), the CFTR chloride channel on the apical surface of the epithelial cell opens.
Cl⁻ Flows OUT into the Lumen: Driven by its electrochemical gradient, chloride ions (Cl⁻) exit the cell through the open CFTR channel. They enter the extracellular space, which is the thin layer of liquid (airway surface liquid, ASL) lining the airway lumen, right where the mucus sits.
Water and Na⁺ Follow: This is the crucial consequence.
Electroneutrality: The flow of negative charges (Cl⁻) out of the cell makes the lumen more negative. This creates an electrical gradient that "pulls" positively charged sodium ions (Na⁺) from the inside of the cell and the interstitial space into the lumen. Na⁺ often follows through tight junctions between cells (the paracellular pathway).
Osmosis: The net movement of salt (NaCl) into the lumen increases the solute concentration there. Water always moves to where the salt is to balance the osmolarity. So, water follows passively out of the cell and through the tight junctions into the lumen.
Mucus is Hydrated: The influx of water from step 3 is what directly hydrates the mucus, keeping it thin and fluid. This allows cilia to beat effectively and sweep the mucus layer, along with any trapped bacteria and particles, out of the lungs.
The Critical Contrast with Cystic Fibrosis (CF)
In Cystic Fibrosis, this entire process is broken at step 1:
Step 1 is broken: The CFTR channel is mutated, so it does not open or doesn't even reach the cell membrane.
Therefore, Step 2 doesn't happen: Cl⁻ cannot flow out.
Therefore, Step 3 is reversed: Without the Cl⁻ efflux to draw Na⁺ and water out, the system goes into reverse. The epithelial sodium channels (ENaC) hyper-absorb Na⁺ from the lumen into the cell. Water follows this salt movement back into the cell and tissue.
Therefore, Step 4 is reversed: The mucus layer becomes dehydrated, thick, and sticky, leading to the severe symptoms of CF.
So, your 4-step list is an excellent description of normal, healthy airway physiology

I-cell disease (COMPLETE deficienty Glc-NAc-1-phosphotransferase)

(clinical correlation) enzyme replacement therapy