Hematology - Hemolytic Anemia
1/42
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
43 Terms
hemolytic anemia
a state of accelerated erythrocyte destruction (loss of survival) characterized by premature removal of circulating red blood cells and increased bone marrow production of replacement cells
classification:
acute versus chronic
inherited versus acquired
intrinsic versus extrinsic
intravascular versus extravascular
fragmentation versus macrophage-mediated
anemia of increased destruction
hemolytic anemia -
decreased RBC survival
normocytic, normochromic anemia
polychromasia
reticulocytotic - increased in response to increased RBC destruction
haptoglobin - decreased
bilirubin - increased indirect (unconjugated)
increased LD
urine hemosiderin - increased
signs of hemolysis
hemoglobinuria
hemoglobinemia
→ hemoglobin in the urine
→ bilirubin is increased in plasma and haptoglobin is decreased
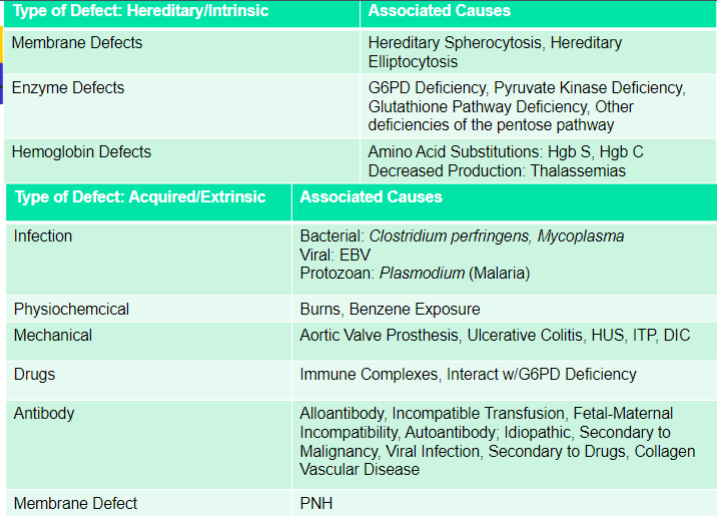
causes of accelerated RBC destruction
two main categories -
hereditary causes
intrinsic defects
acquired causes
primarily extrinsic defects
exception: paroxysmal nocturnal hemoglobinuria - intrinsic
diseases associated with accelerated RBC turnover
RBC life span - 120 days
three phases -
bone marrow production phase
circulating phase
removal phase
initial lab tests
CBC - Hgb, HCt, MCV, MCH, MCHC, RDW,
RBC morphology
reticulocyte count, polychromasia
serum haptoglobin
serum LD
serum bilirubin (direct and indirect)
direct antiglobulin test
automated hemogram
anemia - varies with the cause and rate of hemolysis
RBC indices - MCV clues:
slight macrocytosis <= 110 may be due to increased reticulocytosis
> 115 - think macrocytic anemia
< 70 with normal MCHC, think hemoglobinopathy i.e B-thalassemia
peripheral blood smear clues
reticulocytosis - causes polychromasia and macrocytosis
spherocytes - hereditary spherocytosis, autoimmune H.A, burns, ABO-HDN
target cells - hemoglobinopathies, jaundice, post-splenectomy
cell fragments - DIC, HUS, TTP, mechanical hemolysis
increased RBC production response
Increased renal erythropoietin (EPO) secretion due to anemia
marrow erythroid hyperplasia with increased storage Fe on biopsy
reticulocytosis - increased raw and corrected counts
circulating nRBCs if marrow stress is severe
chemistry tests
elevated LDH (LD) - directly released from RBCs
haptoglobin - decreased/depleted
elevated bilirubin
hemoglobinuria/hemoglobinemia
direct antiglobulin test
a positive DAT (firect anti-globulin test) indicates in-vivo coating of RBCs with immunoglobulins (IgG, IgM, + or C3)
suggests that the hemolysis is due to an immune mechanism
weak +DAT may or may not indicate immune hemolysis
DAT doesn’t separate autoimmune from alloimmune type hemolytic anemia
urine studies
hemoglobinuria gives pink/red to “smoky” or “cola-colored” urine
free urine Hgb can be measured
urine hemosiderin test - cytospin sediment stained with prussian blue, stains sloughed renal tubular cells containing Hgb breakdown Fe blue
develops late (days after the hemoglobinuria event)
RBC survival assay
Takes patient blood and mixes it with radioactive label
blood is given back to patient intravenously
measurements taken periodically to measure the circulating RBC
normal half life is 25-32 days
mild hemolysis - 20-25 days
moderate hemolysis - 15-20 days
severe hemolysis < 15 days
tempo and site of hemolysis
intravascular hemolysis - rapid tempo
free plasma hemoglobin, hemoglobinuria, unconjugated bilirubin, depleted haptoglobin
extravascular hemolysis - slower tempo, mainly spleen
conjugated bilirubin
hemolytic anemia - causes
intrinsic hemolysis
membrane abnormalities
metabolic abnormalities
hemoglobinopathies
extrinsic hemolysis
nonimmune
immune
hereditary shape abnormalities
all result from disturbances in either the make-up or function of the RBC membrane, resulting in abnormal shapes of the RBC and in decreased survival of erythrocytes
hereditary spherocytosis
hereditary elliptocytosis
hereditary pyropoikilocytosis
hereditary stomatocytosis/hydrocytosis
hereditary xerocytosis
acanthocytosis
structure of RBC membrane
Lipid bilayer
central cytoskeleton
key normal functions:
flexibility - to squeeze through tiny capillaries
maintenance of biconcave shape - for max gas exchange and other functions
categories of RBC membrane defects
lipid content - e.g Acanthocytosis
cytoskeleton - e.g Hereditary spherocytosis, hereditary ellipotcytosis, hereditary pyropoikilocytosis
permeability of the membrane - e.g hereditary stomatocytosis, hereditary xerocytosis
acanthocytosis
cause - decreased plasma and membrane lipid balance, causing irregularly formed RBCs
genetic cause - abetalipoproteinemia, McLeod syndrome
acquired cause - alcoholic cirrhosis (Burr cell anemia), myelodysplasia, hypothryroidism, malnutrition
additional tests - decreased triglycerides and cholesterol, sphingomyelin increased, peripheral blood smear - marked acanthocytosis, retic count - normal to increased
hereditary spherocytosis
most common membrane defect
autosomal dominant
cause - decreased or dysfunctional cytoskeleton protein - spectrin, ankyrin, protein 4.2 or Band 3
results - loss of biconcavity and deformability of RBCs and increased turnover of cells
splenic environment degenerates cells leading to early lysis
Lab evaluation:
Hgb & Hct: Normal to moderate decrease
MCHC: usually increased (36 or greater)
Peripheral smear: Spherocytes and anisocytosis
Reticulocyte count: Increased
Osmotic fragility: increased (more fragile)
Treatment: Splenectomy for severe cases
Decreases hemolysis, increases RBC survival
Howell-Jolly Bodies, Target Cells & Pappenheimer
bodies usually seen on peripheral blood smear
osmotic fragility test
test ability of RBCs to swell in a hypotonic solution
normal RBC can swell to 1.8 x resting volume before lysis will occur
spherocytes have less redundant membrane and lyse earlier
osmotic fragility
Increased Fragility:
Hereditary Spherocytosis- the classic example
Hereditary Stomatocytosis
Warm AIHA
Hereditary Pyropoikilocytosis
Decreased Fragility
Hereditary Xerocytosis
Thalassemia
Hereditary Elliptocytosis-variable
hereditary elliptocytosis
Elliptical RBCs
Cause: Defective spectrin
Severity varies: 90% of patients are
asymptomatic and do not experience
significant hemolysis
Labs:
H & H: normal – decreased
Peripheral smear: Elliptocytes
Retic count: elevated
Osmotic fragility: variable
hereditary pyropoikilocytosis
Autosomal recessive, rare
Subset of HE
Cause: Defective spectrin
causes variable RBC sizes and
shapes
Labs:
MCV: decreased
RBC morph shows budding,
fragmentation,
microspherocytes, elliptocytes
Osmotic fragility: increased
Positive heat sensitivity test:
RBCs fragment when warmed to
45 C
permeability problems
H. Stomatocytosis:
Cause: Membrane defect
allows water to enter the cell
MCV increased
MCHC decreased
Osmotic fragility: Increased
H. Xerocytosis:
Membrane defect
allows water to leave the cell
Target cell morph
MCV increased, MCHC increased
Osmotic fragility: Decreased
Rh-null disease
Lack of all Rh-Hr antigens on red cells
Defect in membrane with increased
permeability to K
Mild anemia with stomatocytes and
elliptocytes
clinical findings in hereditary membrane defects
highly variable, many have well=compensated hemolytic anemia
trends: the more severe the disease, the earlier in life it’s detected
high incidence of bilirubin gallstones
hereditary spherocytosis can be clinically relieved by splenectomy
hereditary RBC disorders due to deficiencies of the glycolytic pathway
glycolytic pathway - Embden-meyerhoff pathway
Embden-Meyerhoff pathway
RBCs have no mitochondria, therefore no oxidative metabolism
energy created via E-M pathway in which 1 molecule glucose generates a net 2 ATPS
multiple enzymes involved, among which there may be inherited deficiencies, leading to hemolytic anemia
as a group, these are called “hereditary non-spherocytic hemolytic anemias”
metabolic defect
G6PD deficiency
Hexose monophosphate
shunt
Most common RBC
enzyme defect, >50
variants
X-linked
Low glutathione due to
low NADPH
Oxidative hemolysis
Heinz bodies,
Spherocytic
Primaquine, fava beans
Pyruvate kinase
deficiency
E-M Glycolysis
Low RBC ATP level
Non-spherocytic
hereditary non-spherocytic hemolytic anemia - general characteristics
early onset (infancy), with jaundice, splenomegaly, pigment gallstones
no associated with drug ingestion
autosomal recessive inheritance
hexose monophosphate shunt deficiencies
10% of energy created here, but critically generates NADPH, a reducing agent essential to prevent oxidative damage to RBC membrane
key enzyme - GLucose-6-phosphate-dehydrogenase (G-6-PD)
G-6-PD deficiency
inheritance - X:linked
fully expressed in males, females are heterozygous and have 2 populations of RBCs (those deficient and those note)
cause:
decreased G6PD prevents synthesis of glutathione which is required to prevent hydrogen peroxide buildup in RBC during oxidant stress due to infections, chemicals, and food. Increased H2O2 levels irreversibly denature hemoglobin resulting in Heinze bodies and hemolysis.
G6PD deficiency - lab results
reticulocytes counts - increased during hemolytic event
supravital stain for Heinz bodies = +
fluorescent spot test - negative
quantitative G6PD assay - decreased
clinical findings
Key: Episodic hemolysis
Differential narrowed to G-6-PD deficiency
OR PNH, Malaria, or some unstable Hgb
What brings on an episode of
hemolysis?
Oxidative stress : certain drugs, infections,
foods (fava beans)
drugs/chemicals/foods associated with hemolytic events in G6PD deficiency
anti-malarial agent primaquine
sulfonamides
naphthalene - moth balls
fava beans
lab findings
Variable degree of anemia, reticulocytosis
RBC Morphology:
HEINZ BODIES- clumps of denatured Hgb
“BITE” cells- where spleen has removed a Heinz
body, leaving a dent or bite out of the RBC
Enzyme screens for G-6-PD, also for
Glutathione Reductase activity
PK deficiency - lab approach
Inheritance: Autosomal recessive
Cause:
Missing Pyruvate kinase: Needed to convert
Phosphoenolpyruvate to pyruvate
A decrease of PK in the EMP prevents synthesis of ATP
needed for cell membrane function
Lab Evalulation:
Specific enzyme screening for PK
Fluorescent spot test for PK: Decreased
PEP + ADP ---PK ----> Pyruvate + ATP
Pyruvate + NADH + H -LDH----> Lactate + NAD
Quantitative PK: Decreased
Osmotic fragility: Normal
Methemoglobin reductase deficiency
Glycolytic pathway enzyme needed to
reduce ferric (Fe3+) to ferrous (Fe2+) iron
in hemoglobin, allowing O2 binding
Autosomal recessive inheritance
Labs: Increased methemoglobin on
assay
Decreased NADH- methemoglobin
reductase activity on enzyme screen
Paroxysmal Nocturnal Hemoglobinuria
A rare Acquired Stem Cell disorder that results in RBC
membrane defect
Missing PIG-A gene
Deficiency of phosphatidyl inositol glycan
Membrane defect makes the RBC have increased
sensitivity to complement-mediated cell lysis
As a result, red blood cells hemolyze too early
The red cells leak hemoglobin into the blood, which can
pass into the urine
This can happen at any time, but is more likely to occur
during the night or early morning
RBC precursors lack CD 55 and CD59 (complement
inhibiting)
block complement from binding to RBCs
PNH clinical features
Occurs most often in adults
Irregular Episodes of acute intravascular
hemolysis
Worse during sleep when pH is lower,
enhancing complement binding
Hemoglobinuria
Moderate thrombocytopenia → platelets may be affected
Venous thrombosis, infections
PNH - lab findings
Peripheral blood:
Normocytic to macrocytic → increased prod of reticulocytes
NRBCs
Neutropenia & thrombocytopenia → WBC and platelets increased
Indices: normal
BM: Normoblastic
Retic count: increased 5% - 10%
Additional tests:
Immunophenotyping: Decreased CD 55 & 59
Sucrose Hemolysis test positive
Acid Hemolysis Test (HAM’S test)
Decreased haptoglobin
Urine hemosiderin nearly always positive