Biopharm Mod 4: Non-linear Pharmacokinetics
1/41
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
42 Terms
What’s another term that describes non-linear pharmacokinetics?
“Dose-Dependent & Saturable”
What’s the assumption we have for linear model drugs?
The pharmacokinetic parameters for a drug will
not change when different doses or multiple doses of a drug
are given.
What occurs to the parameters of non-linear drugs when we increase the dosage?
increased doses or chronic medication can
result in deviations in parameters
Why is non-linear pharmacokinetic also termed “ dose-dependent”?
increased doses or chronic medication can
result in deviations
Describe the concepts of SATURATION KINETICS
The phenomenon where an enzyme becomes fully occupied by its substrate, leading to a maximal rate of catalysis
describe the factors influencing saturation kinetics
Substrate concentration: As substrate concentration increases, more enzyme active sites are occupied, leading to increased enzyme activity. However, at high substrate concentrations, the enzyme becomes saturated, and further increases in substrate concentration do not significantly enhance enzyme activity.
Enzyme structure: The structure of the enzyme, particularly the active site, plays a crucial role in determining its saturation kinetics. Enzymes with a high affinity for their substrate tend to become saturated at lower substrate concentrations.
Temperature and pH: Temperature and pH can significantly impact enzyme activity and saturation kinetics. Optimal temperature and pH conditions can enhance enzyme activity, while suboptimal conditions can lead to reduced activity and altered saturation kinetics.
Describe The Role of the Michaelis Constant (Km) in Saturation Kinetics
The Michaelis constant (Km) is a fundamental parameter in enzyme kinetics that describes the binding affinity of an enzyme for its substrate. Km is defined as the substrate concentration at which the enzyme reaches half of its maximal velocity (Vmax/2). A low Km indicates high affinity, meaning the enzyme becomes saturated at lower substrate concentrations, while a high Km indicates low affinity, requiring higher substrate concentrations for saturation.
What are the two main characteristics we can observe in saturation kinetics?
Elimination of drug does not follow basic first-order kinetics
The elimination half-life changes as dose is increased
T/F In Saturation Kinetics, the elimination half-life increases with increased dose due to saturation of an enzyme system
T
T/F In Saturation Kinetics, The Css and AUC is proportional to the drug dose
F: The Css and AUC is not proportional to the drug dose
T/F In Saturation Kinetics, Half-Life is time-dependent
T
In Saturation Kinetics. The elimination half-life might increase due to “self”- induction of liver biotransformation enzymes
F: The elimination half-life might decrease due to “self”- induction of liver biotransformation enzymes, as is observed for carbamazepine.
T/F: F is constant under linear kinetics
T: constant over the FDA approved dose range for the drug
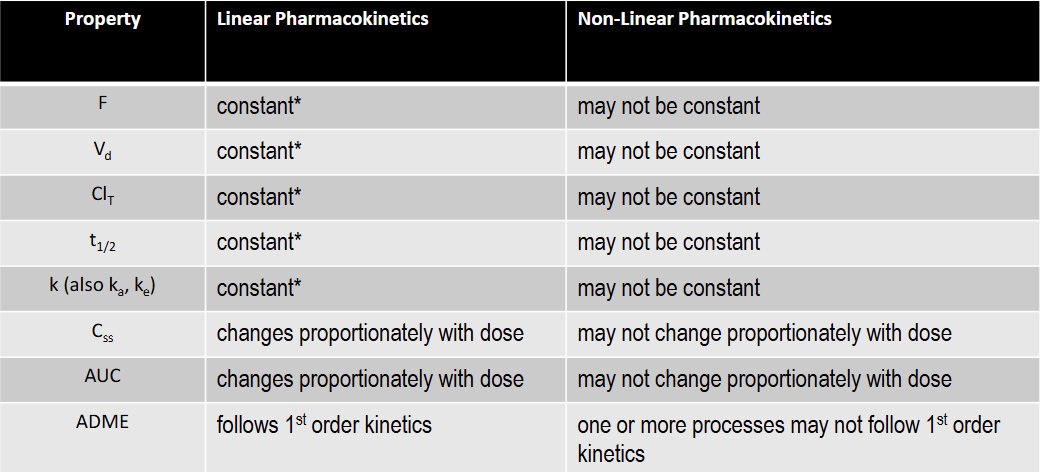
T/F: Vd is constant under linear kinetics
T: constant over the FDA approved dose range for the drug
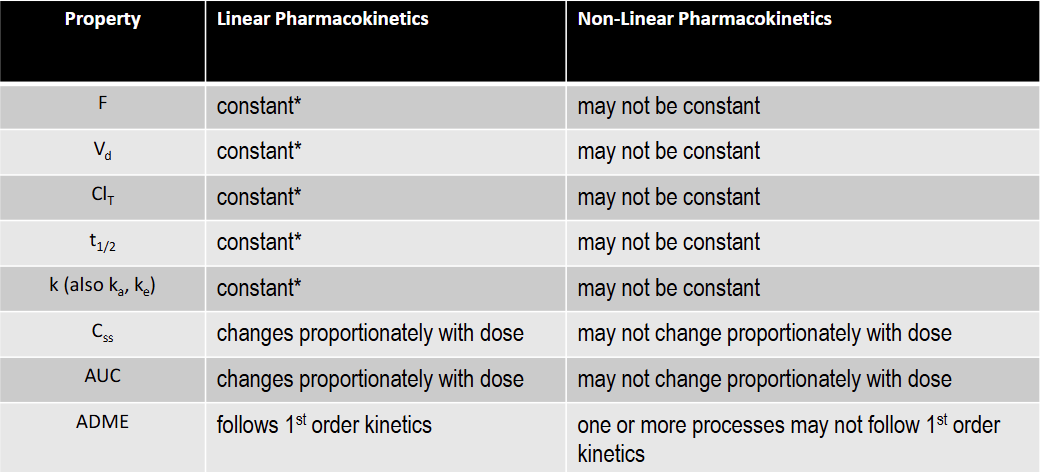
T/F: Clearance is constant under linear kinetics
T: constant over the FDA approved dose range for the drug
T/F: k is constant under linear kinetics
T: constant over the FDA approved dose range for the drug
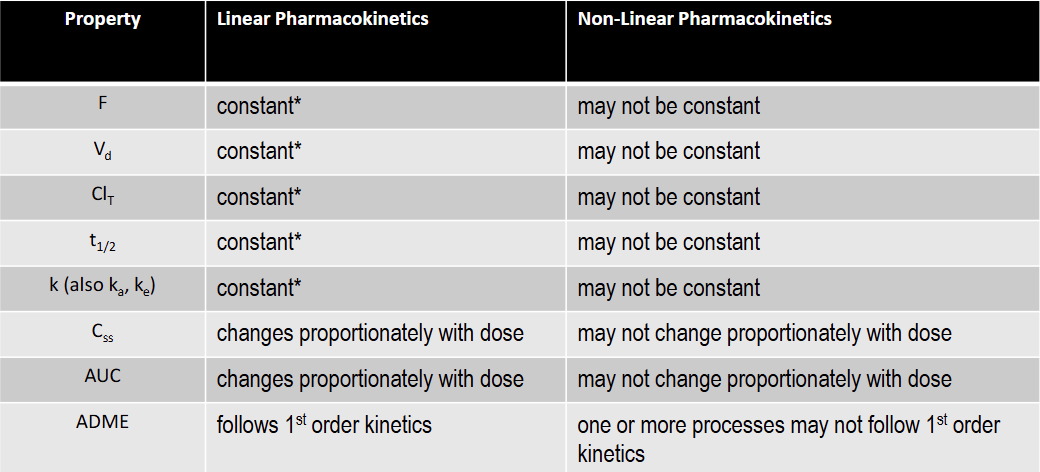
T/F: t1/2 is constant under linear kinetics
T: constant over the FDA approved dose range for the drug
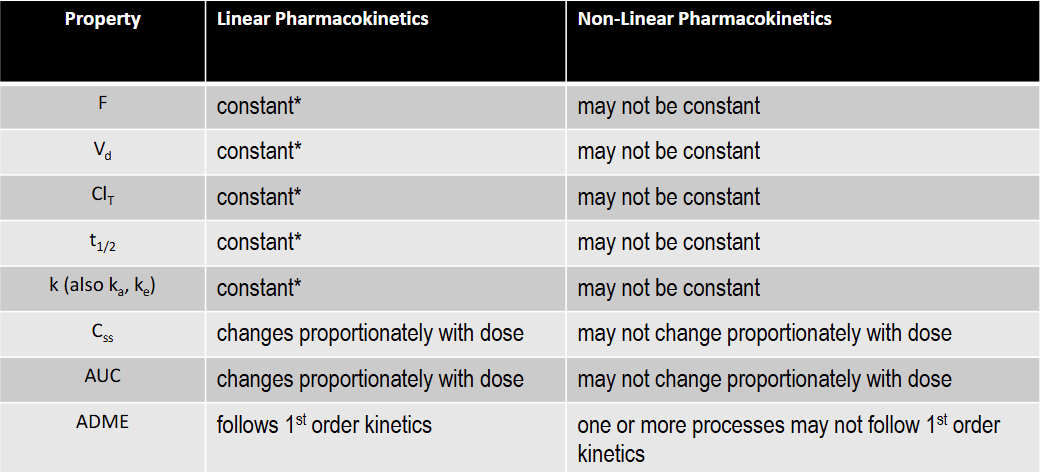
T/F: Css is constant under linear kinetics
F: changes proportionately with dose
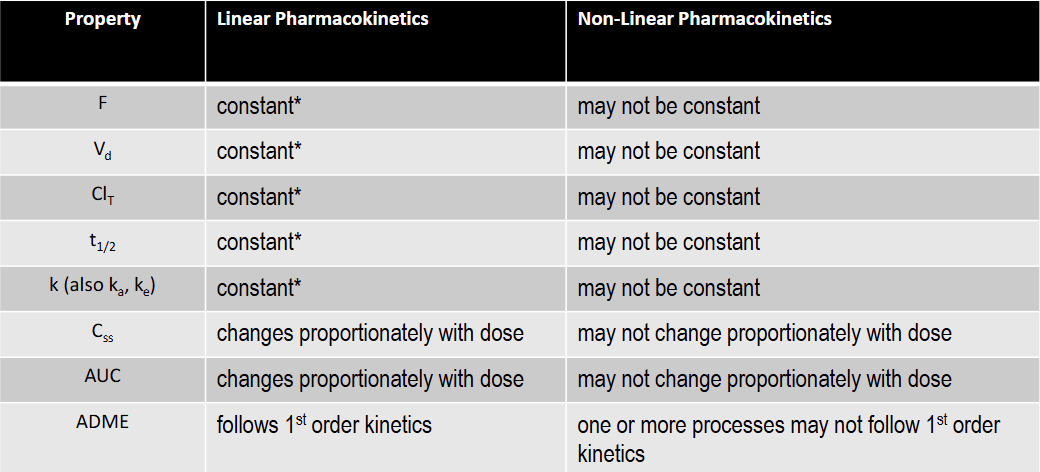
T/F: AUC is constant under linear kinetics
F: changes proportionately with dose
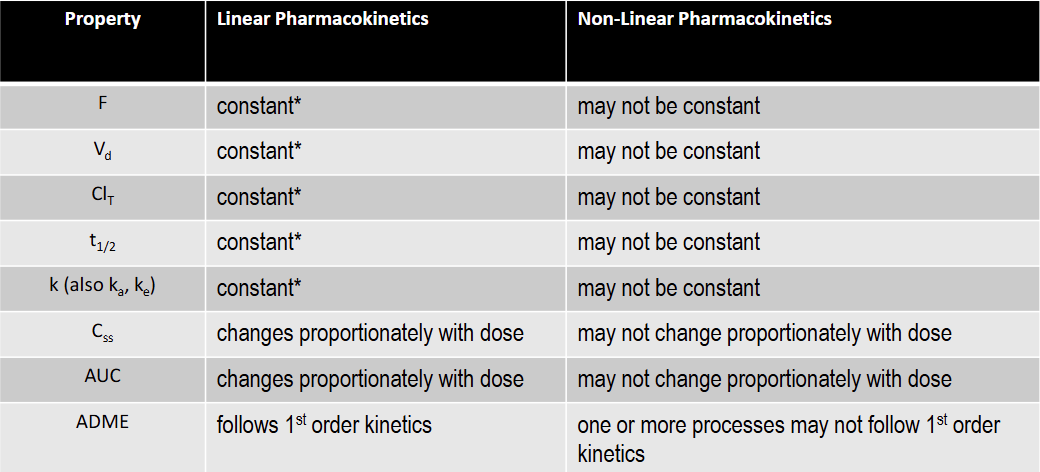
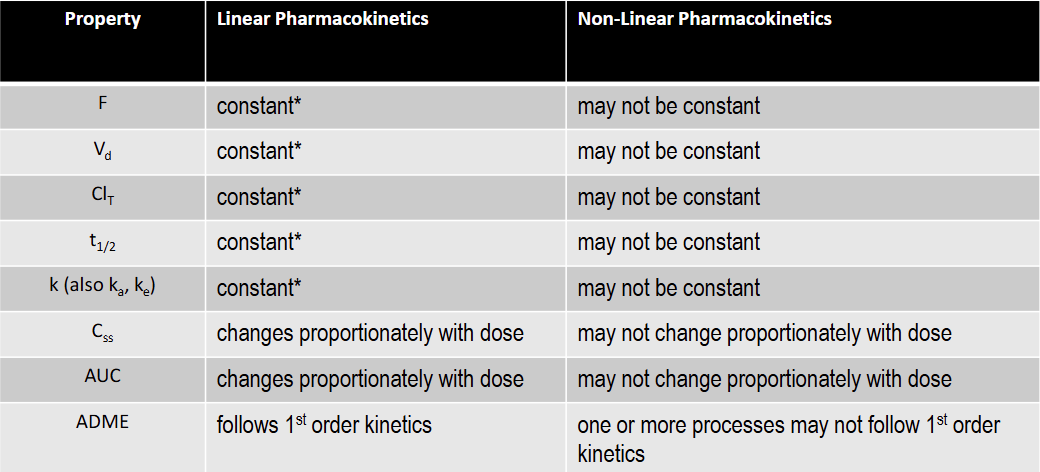
T/F: F is constant under non-linear kinetics
F
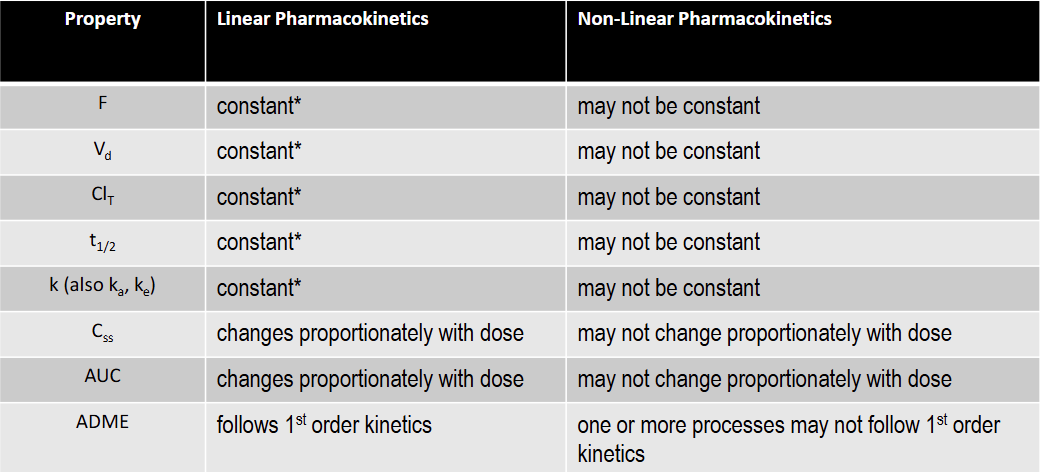
T/F: Vd is not constant under non-linear kinetics
T
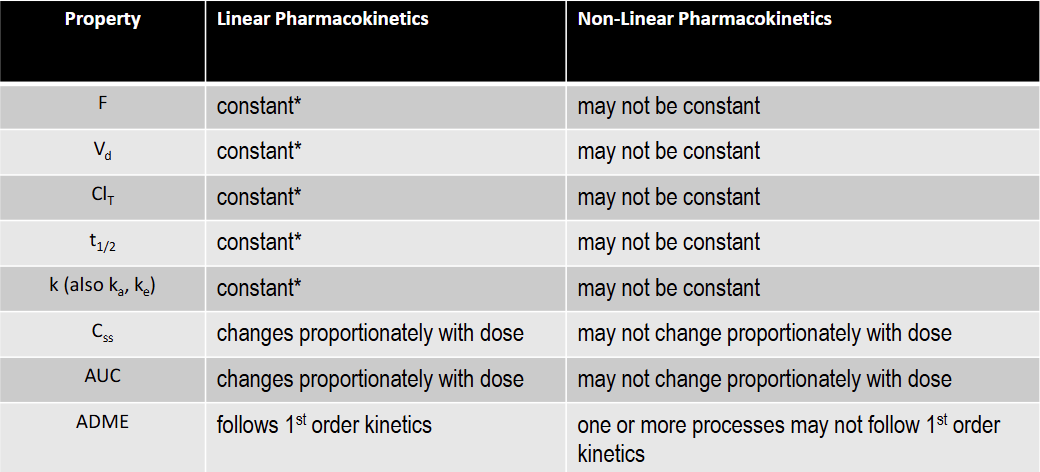
T/F: K is constant under non-linear kinetics
F
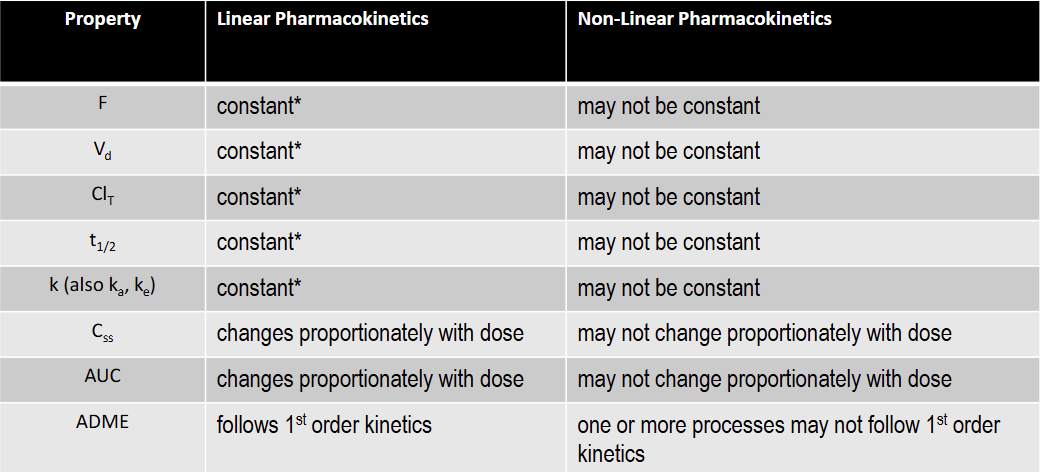
T/F: T1/2 is not constant under non-linear kinetics
T
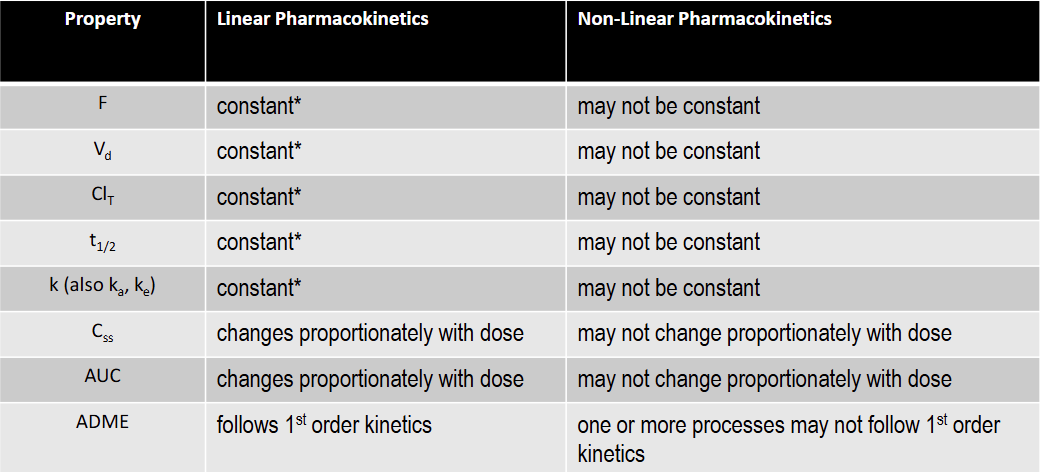
T/F: Total Clearance is constant under non-linear kinetics
F
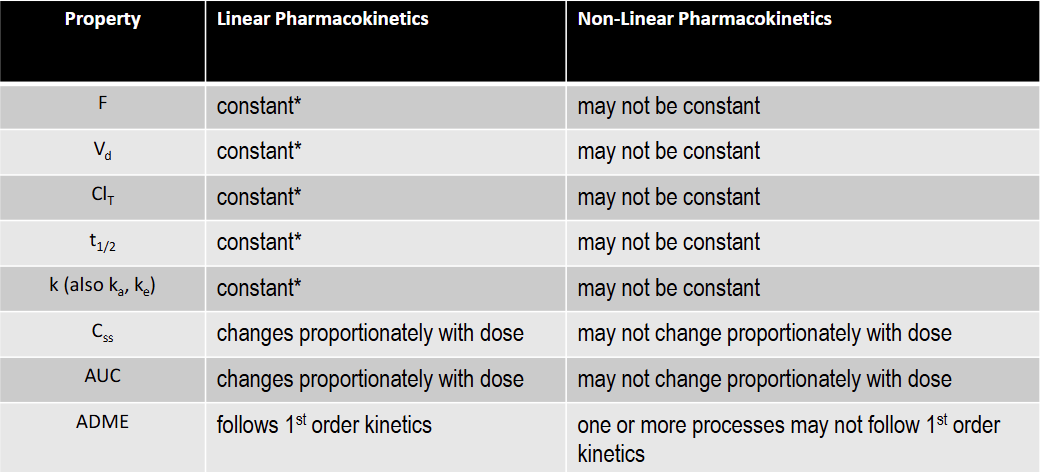
Why is non-linear evaluation important to drug therapy
Pharmacokinetic effects the ADME of the drug, and can effect it’s efficacy inpatients/ Css is extremely important in monitoring patient efficacy and can be analyzes using non-linear or linear kinetics
What happens when Cp is close to Km? Describe it’s effect on intrinsic clearance, hepatic, and total clearance for both a single and large dose.
non-linear kinetics
As ( Km + Cp ) incr = Cl instr decr - large dose scenario
As ( Km + Cp ) decr = Cl instr incr - single dose scenario
Clint Decr: Hepatic and Total clearance e decrease and are not constant
T/F Km describes elimination rate
F: Km describes concertation
In non-linear kinetics, R is assumed to be
Dose / Day; input rate is assumed to be the same value as elimination rate
A single oral dose (500 mg) of an antibiotic was given to a 48-year old
adult female patient weighing 50 kg. From the literature, the
pharmacokinetics of this drug fit a one-compartment open model. The
equation that best fit the pharmacokinetics of the drug in this patient was
Cp = 50 (e-0.23t – e-1.56t)
where Cp was in μg/mL and t was in hours
Instead of the 500 mg dose, if a dose of 750 mg was given, which of the
following would be TRUE?
I. The volume of distribution of the drug would increase.
II. The first order absorption rate constant (ka) would increase.
III. The peak plasma concentration of the drug would increase
III only; the rest of the parameters are constant under linear kinetics
Drug X is administered by IV infusion at a constant rate of 10 mg/hr. The infusion was started at 5:00 PM on Tuesday morning and discontinued at 11:00 AM the next day (Wednesday). The drug is eliminated from the body by first–order kinetics, the elimination half-life of this drug in the body is 3 hours and the volume of distribution is 10 liters.
ANSWER THE FOLLOWING THREE QUESTIONS:
If the infusion rate was 20 mg/hr instead of 10 mg/hr, which of the following would be TRUE?
A. The steady-state plasma drug concentration would increase.
B. The elimination half-life of the drug would increase.
C. The time to reach steady-state would decrease.
D. The volume of distribution of the drug would increase.
E. None of the above
A drug is given by intravenous infusion and has reached steady-state. And follows
1st order elimination kinetics. If the infusion rate is 200 mg/hr and the drug’s total body clearance (ClT) is 7.4 L/hr, then what is the steady concentration?
Provide the answer in units mcg/mL
What is the half-life (t1/2) of a Drug that has a clearance of 20 L/hr and a
volume of distribution (Vd) 100 liters?
If the elimination half-life of a drug is 12 hours and the volume of distribution is
140 liters, then what is the total clearance of the drug?
A patient receives 500 mg (IV bolus) of a drug with a half-life of 5 hours. How
much of the drug remains in the patient after 20 hours?
Drug concentration in the blood can
______ rapidly once an elimination process is
saturated
Increase
Name the metabolic processes that are likely to be the most saturated and their sites.
metabolism (biotransformation) and active
tubular secretion of drugs by the kidney are the
processes most usually can become saturated.
Describe Km
Michaelis constant, reflects the capacity of the
enzyme system , equal to the drug concentration
Describe Vmax
theoretical maximum rate of the process,
In an extreme case where the drug concentration, Cp, is small in relation to KM describe the pharmacokinetics that are likely to occur.
Saturation of the enzyme has not occurred
• the elimination proceeds at a log-linear fashion
• elimination of drug becomes a 1st-order process
rate decline of drug concentration/ change time = kCp
In an extreme case where the drug concentration, Cp, is large in relation to KM describe the pharmacokinetics that are likely to occur.
Saturation of the enzymes occurs
• the elimination proceeds at a fixed or constant rate equal to Vmax,
• the elimination of drug becomes a zero-order process.
rate decline of drug concentration/ change time = Vmax
Define Intrinsic Clearance
intrinsic ability of the hepatic enzymes to eliminate
drug when there are no limitations due to blood flow or protein binding
Css,min for phenytoin is 10 mg/L. Which patient would be
more likely to exhibit dose dependent pharmacokinetics of this anti-
seizure medication?
A. A patient with a Km = 30 mg/L and a Vmax = 1000 mg/day
B. A patient with a Km = 600 mg/L and a Vmax = 1000 mg/day
C. A patient with a constant half-life at 100 mg/day and 500 mg/day
D. A and C are most likely to exhibit dose dependent pharmacokinetics
E. A, B and C are all likely to exhibit dose dependent pharmacokinetics
A 10mgL (Css) is closer to 60 which is more likely to express non-liner kinetics. The other option are deemed as constant values