Tools of Protein Analysis & Proteonomics
1/16
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
17 Terms
learning objectives (you might have to go into the textbook for this one)
Define the following terms; Genome, Genomics, Transcriptome, Metabolome, Proteome, Proteomics.
Discuss methodological approaches to study expression and post-translational modifications in proteins.
Discuss methodologies used to detect protein-protein interactions.
Discuss experimental approaches to determine three-Dimensional (D) structures of proteins.
Discuss practical applications of proteomics to human health.
Define the following terms; Genome, Genomics, Transcriptome, Metabolome, Proteome, Proteomics.
These terms represent different levels of biological information, from the blueprint of life to its dynamic functional expression. Here are their definitions, organized from the fundamental blueprint to the active processes.
1. Genome
Definition: The complete set of genetic material (DNA, or RNA for some viruses) present in a cell or an organism. It includes all of the genes (coding regions) and the non-coding DNA.
Analogy: The entire instruction manual or the master blueprint for building and operating an organism.
Key Point: It is static—the sequence of nucleotides (A, T, C, G) is fixed for a given individual (barring mutations). The human genome is the sequence of over 3 billion base pairs.
2. Genomics
Definition: The scientific field that involves the mapping, sequencing, analysis, and functional interpretation of genomes. It is the study of the structure, function, and evolution of the entire genome.
Analogy: The science of reading and interpreting the entire instruction manual. It asks: What chapters (genes) are there? Where are they located? How do they differ between different editions (individuals or species)?
Example: Sequencing the human genome to find a gene associated with a disease is a genomics approach.
3. Transcriptome
Definition: The complete set of all RNA molecules (including messenger RNA (mRNA), ribosomal RNA (rRNA), transfer RNA (tRNA), and non-coding RNAs) expressed in a cell, tissue, or organism at a specific time and under specific conditions.
Analogy: The list of active chapters and paragraphs being copied from the manual at a given moment. It represents the genes that are actively being "read" or expressed.
Key Point: It is dynamic. The transcriptome changes rapidly in response to the environment, developmental stage, or disease state. It sits between the static genome and the functional proteome.
4. Proteome
Definition: The complete set of proteins expressed in a cell, tissue, or organism at a certain time and under specific conditions. This includes all the isoforms and modifications of proteins.
Analogy: The entire workforce of machines and workers that are actually built and operating in the factory at a given time, based on the active instructions.
Key Point: It is highly dynamic and complex. A single gene can give rise to multiple different proteins through alternative splicing and post-translational modifications (e.g., phosphorylation, glycosylation). The proteome is the functional effector of cellular processes.
5. Proteomics
Definition: The large-scale study of the proteome. It involves the identification, quantification, and characterization of the structure, function, and interactions of all proteins in a biological system.
Analogy: The science of cataloging and analyzing the entire workforce. It asks: What machines are present? How many are there? How are they modified? Who do they interact with?
Example: Using mass spectrometry to identify all proteins that are overexpressed in a cancer cell compared to a normal cell is a proteomics approach.
6. Metabolome
Definition: The complete set of small-molecule metabolites (such as sugars, lipids, amino acids, nucleotides, and other metabolic intermediates) found within a biological sample at a specific point in time.
Analogy: The complete inventory of raw materials, waste products, and fuel in the factory at a snapshot in time. It represents the end products of cellular processes and the building blocks for new ones.
Key Point: It is the most dynamic of the "omes," changing from second to second. It provides a direct, functional readout of cellular activity and physiological status.
Summary Flowchart
The following diagram illustrates the relationship between these "omes" and their corresponding fields of study:
In summary, this hierarchy moves from the foundational genetic code (Genome), to the expressed messages (Transcriptome), to the functional machines (Proteome), and finally to the chemical inputs and outputs (Metabolome). Genomics, Proteomics, and other "-omics" fields are the disciplines dedicated to studying these levels on a large, systematic scale.
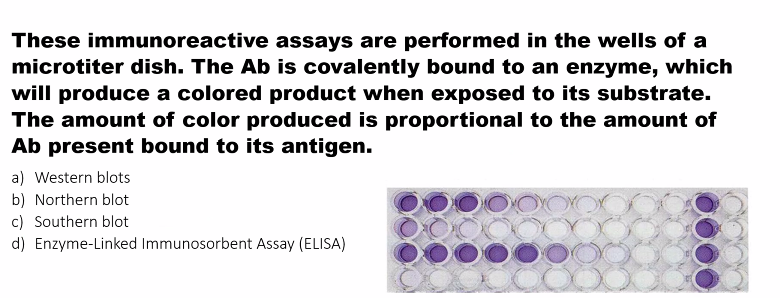
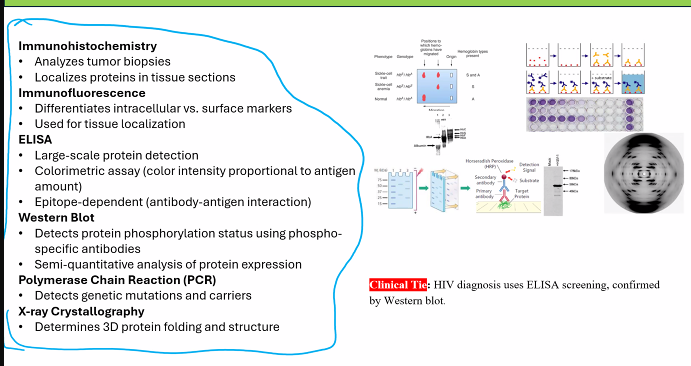
ELISA
• Mass spectroscopy
• immunohistochemistry
• X-ray crystallography
• SDS-PAGE
• Immuno-fluorescence
• Western Blot
• Northern Blot
• Southern Blot
• Polymerase Chain Reaction
This is an excellent review of key laboratory techniques. Based on the uses you've listed, we can determine the most appropriate technique for each scenario.
Here is a structured breakdown matching each diagnostic goal with the most suitable technique(s).
1. To analyze cancer tumor biopsies for assessing patient responsiveness to therapy
This often involves measuring specific protein biomarkers (like HER2 in breast cancer or PD-L1 in lung cancer) to see if a targeted therapy is suitable.
Primary Technique: Immunohistochemistry (IHC)
Why: IHC uses antibodies to detect specific proteins directly in a thin slice of the tumor tissue. It reveals not just the presence/amount of the protein, but also which cells within the complex tumor tissue are expressing it and the subcellular localization (membrane, cytoplasm, nucleus). This is critical for diagnosing targets like HER2 (a membrane receptor).
Immunohistochemistry (class)
• Analyzes tumor biopsies
• Localizes proteins in tissue sections
immunohistochemistry (IHC) is for BOTH cancer (biopsies) and proteins (localizes proteins in tissue sections)
A biopsy is a medical procedure where a small sample of tissue or cells is removed from the body for examination under a microscope
Supporting Technique: Immunofluorescence
Why: Similar to IHC but uses fluorescent tags. It can be more sensitive and allows for multiplexing (detecting multiple markers at once). Often used in research and increasingly in clinical diagnostics.
Immunofluorescence
• Differentiates intracellular vs. surface markers (can detect multiple markers at once)
• Used for tissue localization
2. For testing large-scale samples
This requires a technique that is automated, rapid, and cost-effective for processing hundreds or thousands of samples, like population screening.
Primary Technique: ELISA (Enzyme-Linked Immunosorbent Assay)
Why: ELISA is perfectly designed for high-throughput analysis. It can be performed in multi-well plates, is highly automatable, and provides a quantitative readout of a specific protein or antigen in a sample (e.g., serum, plasma, cell lysates).
Large-scale protein detection
• Colorimetric assay (color intensity proportional to antigen
amount)
• Epitope-dependent (antibody-antigen interaction
3. For assessing the phosphorylation status of proteins
This requires detecting a specific, small post-translational modification on a protein.
Primary Technique: Western Blot
Why: Western Blot is the gold standard for this. Researchers use phospho-specific antibodies that only bind the protein when it is phosphorylated at a specific amino acid. By running a parallel blot with a "total protein" antibody, you can determine the fraction of the protein that is phosphorylated. SDS-PAGE is the first step in a Western Blot.
Detects protein phosphorylation status using phospho-
specific antibodies
• Semi-quantitative analysis of protein expression
4. For differentiating between various genetic conditions and their carriers
This requires analyzing DNA to identify mutations, SNPs, or gene rearrangements.
PCR: Detects genetic mutations and carriers
Primary Technique: Polymerase Chain Reaction (PCR) and its variants (e.g., qPCR, RT-PCR, DNA sequencing).
Why: PCR is the fundamental tool for DNA analysis. It can amplify a specific gene or region of interest from a tiny sample, allowing for the detection of disease-causing mutations (e.g., in BRCA1 for breast cancer) and the identification of carriers (heterozygotes). Southern Blot was historically used for this but has largely been replaced by PCR-based methods due to their speed and sensitivity.
5. For differentiating intracellular vs. surface markers and among different tissues
This requires visualizing the location of a marker within the context of an intact cell or tissue.
For Different Tissues & Cellular Localization: Immunohistochemistry (IHC) or Immunofluorescence on tissue sections.
Why: As in point #1, these techniques provide spatial context, clearly showing if a marker is on the cell surface, in the cytoplasm, or in the nucleus, and which tissue types express it.
For Differentiating Surface vs. Intracellular in Cell Suspensions: Flow Cytometry (while not on your list, is the standard technique). If we must choose from the list, Immunofluorescence combined with microscopy, using cell permeabilization (to see intracellular markers) vs. non-permeabilization (to see only surface markers).
Summary Table of Technique Applications
Diagnostic Goal | Primary Technique(s) | Key Reason |
|---|---|---|
Analyze tumor biopsies | Immunohistochemistry (IHC) | Provides cellular and subcellular localization in tissue context. |
Test large-scale samples | ELISA (Enzyme-Linked Immunosorbent Assay) | High-throughput, automatable, and quantitative. |
Assess protein phosphorylation | Western Blot | Uses phospho-specific antibodies for modification-specific detection. |
Differentiate genetic conditions | Polymerase Chain Reaction (PCR) | Amplifies and detects specific DNA sequences/mutations. |
Differentiate marker localization | Immunohistochemistry (IHC) / Immunofluorescence | Visualizes protein location within cells and tissues. |
Clarification on Other Listed Techniques
Mass Spectrometry: Excellent for identifying unknown proteins, sequencing peptides, and comprehensively analyzing post-translational modifications (including phosphorylation), but it is not typically the first choice for rapid, routine clinical diagnostics of a known biomarker.
X-ray Crystallography: Used to determine the 3D atomic structure of a protein ("Protein Folding (3D)"), not for diagnostic testing of patient samples.
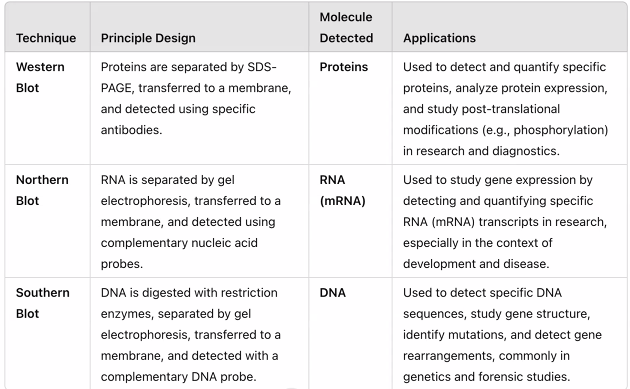
SDS-PAGE: Separates proteins by size but does not identify them. It is a preparatory step for Western Blot.
Northern Blot: Used to study RNA expression (mostly replaced by RT-qPCR and RNA-Seq).
Southern Blot: Used to study DNA structure (mostly replaced by PCR and sequencing).
Clinical Tie: HIV Diagnosis Algorithm
Your clinical example is a perfect real-world application of how these techniques are used in tandem, leveraging their respective strengths and weaknesses.
The standard diagnostic algorithm for HIV is a two-step process:
Screening: ELISA
Why? High Sensitivity. ELISA is designed to screen millions of samples cheaply and rapidly. Its extreme sensitivity ensures that it misses very few true positive cases (it has a very low false-negative rate).
The Catch: A very sensitive test can sometimes be less specific, leading to false positives (e.g., from cross-reactivity with other antibodies).
Confirmation: Western Blot
Why? High Specificity. The Western Blot is used to confirm a positive ELISA result. It doesn't just detect "anti-HIV antibodies"; it detects antibodies against specific HIV proteins (e.g., p24, gp41, gp120). A positive result requires a specific banding pattern.
This two-tiered approach provides a result that is both highly sensitive (thanks to the ELISA) and highly specific (thanks to the Western Blot).
Note: While this ELISA/Western Blot algorithm was the standard for decades, many modern testing guidelines now use a combination of 4th generation ELISA (which detects both HIV antibody and p24 antigen) and a differentiation immunoassay for confirmation, which is faster. However, the principle of a highly sensitive screening test followed by a highly specific confirmatory test remains the same, and the ELISA/Western Blot combo is a classic teaching example.
Your breakdown demonstrates a strong understanding of how the principles of each technique directly dictate its diagnostic application. Well done.
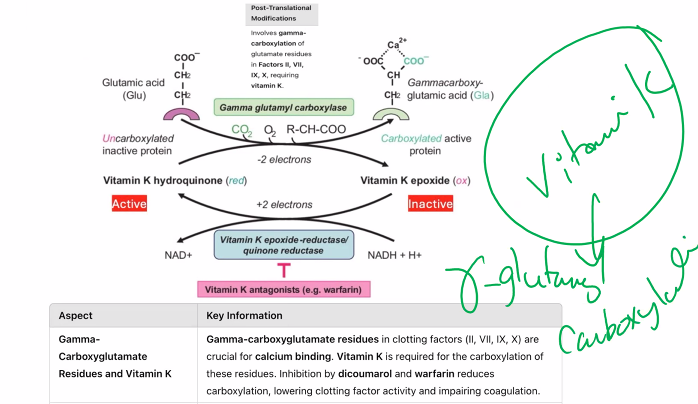
Discuss methodological approaches to study expression and post-translational modifications in proteins.
Studying protein expression and post-translational modifications (PTMs) is fundamental to understanding protein function, regulation, and their role in health and disease. The field dedicated to this is Proteomics.
Here is a discussion of the key methodological approaches, categorized by their primary goal.
Part 1: Methodological Approaches for Protein Expression
The goal here is to identify which proteins are present and in what quantity.
1. Electrophoretic Techniques
SDS-PAGE (Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis):
Principle: Separates proteins based primarily on molecular weight. SDS denatures proteins and gives them a uniform negative charge.
Use: A foundational method for visualizing the total protein complement (a "proteome snapshot") and preparing samples for further analysis (e.g., Western Blot). It provides low resolution and is not for precise identification.
2D-Gel Electrophoresis (2D-DIGE):
Principle: Separates proteins in two dimensions: first by isoelectric point (pI) and then by molecular weight.
Use: Allows for the resolution of thousands of protein spots on a single gel. By comparing gels from different samples (e.g., healthy vs. diseased), one can identify proteins that are upregulated, downregulated, or modified (a PTM often changes the pI of a protein, causing a spot to shift).
2. Immunoassays
Western Blot (Immunoblot):
Principle: Proteins separated by SDS-PAGE are transferred to a membrane. A specific primary antibody is used to detect a target protein, followed by a labeled secondary antibody for visualization.
Use: Excellent for targeted analysis of the expression level and approximate size of a specific, known protein. It is semi-quantitative.
Enzyme-Linked Immunosorbent Assay (ELISA):
Principle: Uses antibodies immobilized on a plate to capture a specific protein from a complex mixture. A detection antibody is then used for quantification, often via an enzymatic colorimetric reaction.
Use: Highly sensitive and quantitative method for measuring the concentration of a specific protein in a sample (e.g., in serum).
3. Mass Spectrometry (MS)-Based Proteomics
This is the cornerstone of modern, high-throughput proteomics.
Bottom-Up Proteomics (Shotgun Proteomics):
Principle: The entire protein mixture is digested with a protease (like trypsin) into peptides. These peptides are separated by Liquid Chromatography (LC) and introduced into a mass spectrometer.
The MS Workflow:
Ionization: Peptides are ionized (e.g., by Electrospray Ionization - ESI).
Mass Analysis 1: The mass-to-charge ratio (m/z) of all peptides is measured.
Fragmentation: Selected peptides are fragmented, often by collision with gas (Collision-Induced Dissociation - CID).
Mass Analysis 2: The m/z of the resulting fragment ions is measured.
Identification & Quantification:
Identification: The fragmentation pattern (MS/MS spectrum) acts as a "fingerprint" that is matched against theoretical spectra in a protein database.
Quantification: Methods like Label-Free Quantification (LFQ), TMT/iTRAQ (isobaric tags), or SILAC (stable isotope labeling with amino acids in cell culture) allow for precise comparison of protein abundance across multiple samples.
Part 2: Methodological Approaches for Post-Translational Modifications (PTMs)
PTM analysis is more challenging because they are often sub-stoichiometric (not on every copy of a protein) and labile. The general strategy is to enrich for PTM-containing peptides before MS analysis.
1. Enrichment Strategies (Pre-MS)
Phosphorylation (on Ser, Thr, Tyr):
Enrichment Method: Immobilized Metal Affinity Chromatography (IMAC) or Metal Oxide Affinity Chromatography (MOAC) using TiO₂. These resins have high affinity for the negatively charged phosphate group.
Acetylation (on Lys):
Enrichment Method: Immunoaffinity purification using pan-specific anti-acetyllysine antibodies.
Ubiquitination (on Lys):
Enrichment Method: Use of antibodies or ubiquitin-binding domains. A common trick is to express a tagged version of ubiquitin (e.g., His-tag or HA-tag) and use affinity chromatography against the tag.
Glycosylation (on Asn, Ser, Thr):
Enrichment Method: Lectin Affinity Chromatography (lectins are proteins that bind specific carbohydrate structures).
2. Analytical & Detection Methods
Mass Spectrometry (with enrichment): This is the primary tool. The workflow is similar to shotgun proteomics, but the sample is passed through an enrichment column first. The MS/MS spectrum can reveal the exact site of modification (e.g., a mass shift corresponding to a phosphate group (+80 Da) on a specific serine residue in the peptide sequence).
Western Blot with PTM-Specific Antibodies:
Principle: Uses antibodies that specifically recognize a protein only when it is modified (e.g., anti-phospho-ERK, anti-acetyl-p53).
Use: Simple, accessible method to monitor the modification status of a specific protein. Often used in signaling studies to see if a pathway is activated.
Edman Degradation & Amino Acid Analysis:
Principle: Classical chemical methods. Edman degradation can sometimes identify modified N-terminal, while amino acid analysis can detect unusual amino acids like phosphoamino acids.
Use: Largely superseded by MS but can be useful for specific applications.
Phos-tag™ SDS-PAGE:
Principle: A phosphate-binding molecule is incorporated into the polyacrylamide gel. This retards the migration of phosphorylated proteins, allowing for visualization of phosphorylation status directly from a gel.
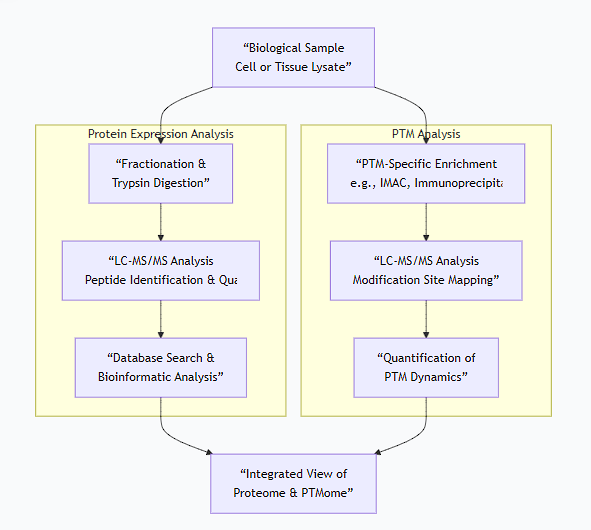
Integrated Workflow for Expression and PTM Analysis
A typical modern proteomics experiment integrates these methods, as illustrated below:
Summary Table of Key Methods
Goal | Method | Key Principle | Throughput & Application |
|---|---|---|---|
Protein Expression | Western Blot | Antibody-based detection of specific protein. | Low; targeted, semi-quantitative. |
Protein Expression | LC-MS/MS (Shotgun) | Mass spectrometry of all digested peptides. | High; global, discovery-level. |
Protein Quantification | ELISA | Antibody-based capture and colorimetric readout. | Low; targeted, highly quantitative. |
PTM Detection | PTM-specific Western | Antibody that recognizes the modification. | Low; targeted, status of known protein. |
PTM Discovery | Enrichment + LC-MS/MS | Isolate modified peptides then analyze by MS. | High; global, site-specific identification. |
In conclusion, the choice of method depends entirely on the question. Targeted questions about a known protein are efficiently answered with antibody-based methods (Western, ELISA). For discovery-level, global analysis of protein expression and PTMs, mass spectrometry-based proteomics, coupled with sophisticated enrichment strategies, is the powerful and indispensable approach.
Discuss methodologies used to detect protein-protein interactions.
The study of protein-protein interactions (PPIs) is crucial for understanding virtually all cellular processes, from signaling pathways to the formation of complex molecular machines. A wide array of methodologies has been developed to detect these interactions, each with its own strengths, weaknesses, and applications.
Here is a discussion of the key methodological approaches, categorized from in vitro (test tube) to in vivo (within living cells) techniques.
1. Affinity-Based Methods (Biochemical)
These methods rely on using a "bait" protein to physically pull down its interacting "prey" partners from a complex mixture.
Co-Immunoprecipitation (Co-IP)
Principle: An antibody specific to a bait protein is used to pull it out of a cell lysate, along with any proteins that are stably bound to it. The co-precipitated proteins are then identified, typically by Western blotting.
Strengths: Confirms interactions under near-physiological conditions; can detect endogenous complexes.
Weaknesses: Only identifies stable, strong interactions; high false-positive rate from non-specific binding; requires a specific, high-quality antibody.
Pull-Down Assays
Principle: A purified bait protein is tagged (e.g., with GST, His, or biotin) and immobilized on a solid support (e.g., glutathione beads for GST). The immobilized bait is then incubated with a cell lysate or purified proteins to "pull down" binding partners.
Strengths: Versatile; doesn't require a specific antibody for the bait; can use recombinant proteins.
Weaknesses: May miss interactions that require post-translational modifications or a cellular environment.
Cross-Linking
Principle: Chemical cross-linkers are used to covalently "freeze" transient or weak protein interactions in living cells or lysates before purification. The cross-linked complexes are then analyzed, often by mass spectrometry.
Strengths: Can capture transient and weak interactions that are lost in standard Co-IPs.
Weaknesses: Can create non-specific cross-links; complex data analysis.
2. Biophysical Methods
These methods measure the physical properties of interacting proteins, often providing quantitative data on binding strength (affinity).
Surface Plasmon Resonance (SPR)
Principle: The bait protein is immobilized on a sensor chip. A solution of the prey protein flows over it. Binding causes a change in the refractive index at the sensor surface, which is measured in real-time as "resonance units."
Strengths: Provides real-time kinetics (association and dissociation rates) and affinity (KD) without labels.
Weaknesses: Requires purified proteins; immobilization can affect protein activity.
Isothermal Titration Calorimetry (ITC)
Principle: Measures the heat released or absorbed when two proteins interact. By titrating one protein into another, it directly measures the binding affinity (KD), stoichiometry (N), and thermodynamics (enthalpy ΔH, entropy ΔS).
Strengths: Considered the "gold standard" for solution-based binding studies; provides a full thermodynamic profile.
Weaknesses: Requires large amounts of highly purified proteins; low-to-medium throughput.
Fluorescence Polarization/Anisotropy (FP)
Principle: A small fluorescently-labeled peptide or protein tumbles rapidly, resulting in low polarization. If it binds to a larger protein, its rotation slows, leading to an increase in polarization. This change is used to measure binding.
Strengths: Homogeneous (no separation needed); ideal for high-throughput screening of inhibitors.
Weaknesses: Typically limited to smaller ligands; requires labeling.
3. Genetic Methods (Yeast-Based)
These powerful in vivo systems use the modularity of transcription factors to detect interactions.
Yeast Two-Hybrid (Y2H)
Principle:
The bait protein is fused to the DNA-Binding Domain (DBD) of a transcription factor.
The prey protein is fused to the Activation Domain (AD).
If bait and prey interact, the DBD and AD are brought together, reconstituting the transcription factor and driving the expression of a reporter gene (e.g., for histidine or β-galactosidase).
Strengths: Can screen vast libraries (e.g., a whole genome) for interactors; performed in the native yeast nucleus.
Weaknesses: High false-positive rate; proteins may not fold or be modified correctly in yeast; cannot detect interactions that require more than two partners.
Related Systems:
Mammalian Two-Hybrid (M2H): Same principle but performed in mammalian cells, providing a more physiologically relevant environment.
Bacterial Two-Hybrid (B2H): Performed in E. coli, often faster and simpler.
4. Protein Complementation Assays (PCAs)
These methods split a reporter protein into two fragments that are fused to potential interacting partners.
Bimolecular Fluorescence Complementation (BiFC)
Principle: A fluorescent protein (e.g., YFP) is split into two non-fluorescent fragments. Each fragment is fused to a candidate protein. If the proteins interact, the fragments are brought together, reforming the fluorescent protein.
Strengths: Visualizes interactions in real-time within living cells; reveals subcellular location.
Weaknesses: Fluorescence complementation is often irreversible, "trapping" the complex.
Other PCAs:Use enzymes like Luciferase (Split-Luciferase) or β-lactamase, where complementation results in a luminescent or colorimetric signal.
5. Proximity-Dependent Labeling (Next-GenerationIn VivoMethods)
These are revolutionary techniques for mapping interactions in the native cellular environment.
BioID (Proximity-Dependent Biotin Identification)
Principle: A bait protein is fused to a promiscuous biotin ligase (e.g., BirA*). In living cells, this ligase biotinylates nearby proteins (~10 nm radius). The biotinylated "prey" proteins are then purified with streptavidin beads and identified by mass spectrometry.
Strengths: Captures weak, transient, and membrane interactions; "freezes" interactions in live cells; identifies the proximal proteome.
APEX (Ascorbate Peroxidase)
Principle: Similar to BioID, but uses an engineered peroxidase that, in the presence of H₂O₂, biotinylates nearby proteins in seconds. It offers superior temporal resolution and can also be used for electron microscopy.
Strengths: Extremely fast labeling (seconds); high spatial resolution.
Summary and Workflow
The choice of method depends on the biological question, as illustrated in the following workflow:
Comparison Table of Key Methods
Method | Context | Throughput | Key Strength | Key Weakness |
|---|---|---|---|---|
Co-IP | In vivo / Lysate | Low | Studies endogenous complexes | High false positives; requires antibody |
Yeast Two-Hybrid | In vivo (Yeast) | Very High | Genome-wide screening | Many false positives/negatives |
SPR | In vitro | Medium | Provides real-time kinetics | Requires purified proteins |
ITC | In vitro | Low | Provides thermodynamics | High protein consumption |
BiFC | In vivo | Medium | Visualizes subcellular location | Irreversible; delayed signal |
BioID/APEX | In vivo | High | Maps proximal interactions in live cells | Proximity, not direct binding |
In conclusion, a robust understanding of protein-protein interactions is best achieved not by a single method, but through a complementary approach using different techniques to validate and characterize the interaction from multiple angles.
Discuss experimental approaches to determine three-Dimensional (D) structures of proteins.
Determining the three-dimensional (3D) structure of a protein is fundamental to understanding its function at a molecular level. Several powerful experimental techniques are used, each with its own principles, requirements, and strengths.
Here is a discussion of the primary experimental approaches.
1. X-ray Crystallography
This is the most common method for determining high-resolution atomic structures of proteins.
Principle: A crystal of the purified protein is bombarded with a beam of X-rays. The regular, repeating lattice of the crystal causes the X-rays to diffract (scatter) in specific directions. The pattern of diffracted rays (spots on a detector) is used to calculate an electron density map. An atomic model of the protein is then built and refined to fit this map.
Workflow:
Protein Purification: Obtain a large quantity of highly pure, homogeneous protein.
Crystallization: The major bottleneck. The protein solution is slowly brought to supersaturation, allowing ordered crystals to form. This is a trial-and-error process.
Data Collection: The crystal is exposed to an intense X-ray beam (at a synchrotron facility) and the diffraction pattern is collected.
Phasing: Solving the "phase problem" to interpret the diffraction pattern into an electron density map. This often requires incorporating heavy atoms (e.g., selenium in Selenomethionine) into the crystal.
Model Building & Refinement: An atomic model is built into the electron density and computationally refined.
Strengths:
Provides very high resolution (often down to 1-2 Å), showing individual atoms.
Can handle very large proteins and complexes.
The "gold standard" for detailed mechanistic studies and drug design.
Weaknesses:
Requires protein crystallization, which is often difficult or impossible for many proteins (e.g., membrane proteins, flexible proteins).
Provides a static snapshot of the protein in the crystalline state, which may not represent its dynamic, solution-state conformation.
2. Nuclear Magnetic Resonance (NMR) Spectroscopy
This method is used to determine the 3D structures of proteins in solution.
Principle: In a strong magnetic field, the nuclei of certain atoms (e.g., ¹H, ¹³C, ¹⁵N) absorb and re-emit electromagnetic radiation. The frequency (chemical shift) of this resonance is exquisitely sensitive to the local chemical environment of each nucleus. Through-Nuclear Overhauser Effect (NOE) measurements, distances between nearby atoms (<5 Å) can be determined.
Workflow:
Isotope Labeling: Proteins are produced (in bacteria) using media containing ¹³C and ¹⁵N to make the carbon and nitrogen atoms NMR-active.
Data Collection: A series of complex multi-dimensional NMR experiments are performed.
Assignment: Each resonance in the spectrum is assigned to a specific atom in the protein sequence.
Structure Calculation: The experimental constraints (distances from NOEs, dihedral angles) are used in a computational process to calculate an ensemble of structures that satisfy all the data.
Strengths:
Studies proteins in a near-native, solution state.
Provides information on protein dynamics and flexibility on various timescales.
Does not require crystallization.
Weaknesses:
Limited to smaller proteins (<~25-30 kDa), though techniques for larger proteins are improving.
Lower throughput and requires significant expertise in data analysis.
Resolution is generally lower than high-quality crystal structures.
3. Cryo-Electron Microscopy (Cryo-EM)
Cryo-EM has undergone a "resolution revolution" and is now a dominant method in structural biology, especially for large complexes.
Principle: A solution of purified protein is rapidly frozen in a thin layer of vitreous (non-crystalline) ice, preserving its native state. An electron microscope is used to take thousands of 2D images of individual, randomly oriented protein particles. Computational algorithms then classify these images and reconstruct a 3D density map.
Workflow:
Sample Preparation: The protein complex is purified and applied to a grid, which is plunge-frozen in liquid ethane.
Data Collection: Automated cryo-electron microscopes collect millions of low-dose 2D micrographs to minimize radiation damage.
Image Processing: Particles are picked from the micrographs, classified into homogeneous groups, and used to reconstruct a 3D volume.
Model Building: An atomic model is built into the cryo-EM density map, similar to crystallography.
Strengths:
Can solve structures of very large and complex macromolecular machines (e.g., ribosomes, viruses, membrane proteins) that are difficult to crystallize.
Requires very small amounts of sample.
Can capture multiple functional states (conformational heterogeneity) of a complex from a single sample.
Weaknesses:
Traditionally had lower resolution than crystallography, but now often reaches near-atomic resolution (<3 Å).
Challenging for very small proteins (<~50 kDa), though techniques are improving.
Requires complex and expensive computational infrastructure and expertise.
4. Other and Complementary Techniques
Small-Angle X-ray Scattering (SAXS):
Principle: Analyses the scattering pattern of X-rays by proteins in solution.
Output: Provides low-resolution information about the overall shape, dimensions, and oligomeric state of a protein (e.g., is it elongated or spherical?). It is excellent for studying flexible systems.
Electron Crystallography:
Principle: Uses 2D crystals of proteins (often membrane proteins embedded in a lipid bilayer) and electron microscopy to determine a structure.
Mass Spectrometry (MS) coupled with cross-linking, HDX, or native MS:
These are not primary structure determination methods but provide powerful structural constraints (e.g., interaction interfaces, solvent accessibility, topology) that complement the other techniques.
Summary and Comparison
The following flowchart illustrates a typical decision process for choosing a structural biology technique:
Method | X-ray Crystallography | NMR Spectroscopy | Cryo-EM |
|---|---|---|---|
Sample State | Crystal | Solution | Vitreous Ice (Solution-like) |
Typical Size Range | Any size | < ~30 kDa | > ~50 kDa (ideal) |
Key Strength | High Resolution, Atomic Detail | Dynamics, Solution State | Large Complexes, Conformational Heterogeneity |
Key Limitation | Crystallization | Size Limit | Resolution (for some targets), Cost |
Information Gained | Static atomic coordinates | Ensemble of structures, dynamics, interactions | 3D Density Map, often at near-atomic resolution |
In conclusion, the choice of technique depends on the protein's properties (size, flexibility, ability to crystallize) and the biological question. Modern structural biology often uses an integrative approach, combining data from multiple methods to build a comprehensive and dynamic model of a protein's structure and function.
Discuss practical applications of proteomics to human health.
Proteomics, the large-scale study of proteins, has moved from being a basic research tool to a field with profound and direct applications in human health. Unlike genomics, which tells you what might happen, proteomics tells you what is actually happening in a cell or tissue at a given moment, providing a dynamic view of health and disease.
Here is a detailed discussion of the practical applications of proteomics to human health.
1. Biomarker Discovery: The Search for Molecular Signatures
This is one of the most prominent applications. The goal is to find specific proteins whose presence, absence, or modification is indicative of a disease state.
Early Detection and Diagnosis: Proteomics can identify protein "signatures" in easily accessible biofluids like blood (serum/plasma), urine, or cerebrospinal fluid.
Example: The PSA (Prostate-Specific Antigen) test is a classic, though imperfect, single-protein biomarker for prostate cancer. Modern proteomics aims to find panels of multiple proteins that together provide much higher accuracy for detecting cancers (e.g., ovarian, pancreatic) at early, more treatable stages.
Disease Subtyping: Not all cancers or neurodegenerative diseases are the same. Proteomics can identify distinct molecular subtypes that behave differently and require tailored therapies.
Example: Breast cancer is already subtyped using receptors (ER/PR/HER2), which are proteins. Proteomic profiling can reveal even finer subtypes and new therapeutic targets.
Monitoring Progression and Response to Therapy: By repeatedly analyzing a patient's proteome, clinicians can track whether a disease is advancing, stable, or responding to treatment.
Example: Monitoring changes in the serum proteome of a cancer patient to see if a tumor is developing resistance to a drug, allowing for a timely switch to another therapy.
2. The Quest for New Drug Targets
Proteomics helps identify proteins that are critically involved in disease pathways, making them ideal candidates for new drugs.
Identifying Dysregulated Pathways: By comparing the proteomes of diseased and healthy tissues, researchers can pinpoint which specific proteins and pathways are overactive or inactive.
Target Validation: Once a potential target is identified, proteomics can be used to validate its importance by showing that inhibiting it has the desired effect on the broader protein network.
Example: The discovery of the BCR-ABL fusion protein in chronic myeloid leukemia (CML) led to the development of the targeted therapy imatinib (Gleevec). Modern proteomics is used to find similar "driver" proteins in other cancers.
3. Personalized and Precision Medicine
Proteomics is key to moving from a one-size-fits-all model to treatments tailored to an individual's specific disease profile.
Stratifying Patients: Proteomic analysis of a patient's tumor biopsy can determine which specific proteins are present, helping to select the drug most likely to be effective.
Companion Diagnostics: These are tests developed alongside a drug to identify patients who will benefit from it. Many of these diagnostics detect specific proteins or their modifications (e.g., phosphorylated receptors).
Example: Patients with non-small cell lung cancer are tested for the presence of mutated EGFR protein. If the mutation is present, they receive EGFR inhibitor drugs like gefitinib, which are ineffective for patients without the mutation.
4. Understanding Disease Mechanisms (Mechanistic Proteomics)
To develop better treatments, we need to understand the underlying molecular mechanisms of disease.
Mapping Protein Interaction Networks (Interactomics): Identifying which proteins interact with each other in healthy vs. diseased states can reveal how a mutation disrupts a entire network, leading to pathology. This is crucial for complex diseases like Alzheimer's, where the interplay between proteins like amyloid-beta and tau is studied.
Post-Translational Modification (PTM) Analysis: Many diseases are driven not by changes in protein abundance, but in their modifications (e.g., phosphorylation, acetylation). Proteomics can globally profile PTMs to uncover aberrant signaling.
Example: Studying hyperphosphorylation of tau protein in Alzheimer's disease to understand neurofibrillary tangle formation.
5. Infectious Disease and Host-Pathogen Interactions
Proteomics provides insights into how pathogens cause disease and how our bodies respond.
Pathogen Virulence Factors: Identifying proteins that a bacterium or virus uses to infect host cells and evade the immune system.
Vaccine Development: By identifying surface proteins on a pathogen that are recognized by the immune system (antigens), proteomics can accelerate the design of new vaccines.
Host Response: Analyzing how the host's proteome changes in response to infection can reveal biomarkers of severity and potential host-directed therapies.
6. Toxicology and Drug Safety (Toxicoproteomics)
Proteomics is used to assess the safety of new drugs during development.
Identifying Toxicity Signatures: By analyzing the proteome of organs like the liver or kidney from animals treated with a drug candidate, researchers can identify protein patterns associated with toxicity. This allows for the early failure of unsafe compounds.
Mechanism of Toxicity: Understanding the protein pathways involved in drug-induced injury can help in designing safer molecules.
A Practical Workflow in Clinical Proteomics
The following chart illustrates a typical pipeline for applying proteomics to a human health problem, such as biomarker discovery:
Summary
Application Area | Key Question | Proteomics Contribution |
|---|---|---|
Biomarker Discovery | Is the disease present, and what is its status? | Identifies protein signatures for early detection, diagnosis, and monitoring. |
Drug Discovery | What should we target, and does the drug work? | Finds new drug targets and validates their mechanism of action. |
Precision Medicine | Which treatment is right for this specific patient? | Matches patients to therapies based on the protein profile of their disease. |
Disease Mechanisms | How does the disease work? | Maps dysfunctional protein pathways and interactions. |
Infectious Disease | How does the pathogen operate, and how do we respond? | Identifies virulence factors and vaccine targets; characterizes immune response. |
In conclusion, proteomics is transforming human health by providing a dynamic, functional readout of the body's state. It is driving a shift towards more precise, predictive, and personalized healthcare, enabling earlier disease detection, smarter drug development, and more effective treatments tailored to the individual patient.
B)
400 blood samples= liquid.
if it was 1 or 2 samples, he would go with Western Blot.

A)
Western blot is preferred.

B)

D) immunohistochemistry

E)


B)

D)