L3 Polar Organometallic Reagents
1/23
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
24 Terms
Common useful polar organometallics

How does C-M bond polarity change as you go from p block through TMs to s block
p-blocks are significantly covalent while s blocks are significantly ionic.
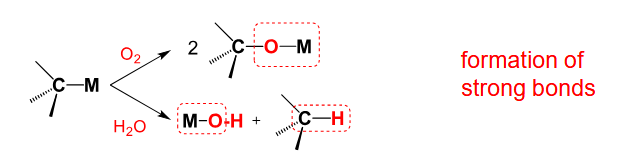
C-M reactivity with oxidation / hydrolysis
Generally the reaction will proceed due to the formation of strong bonds (M-O and/or C-H)
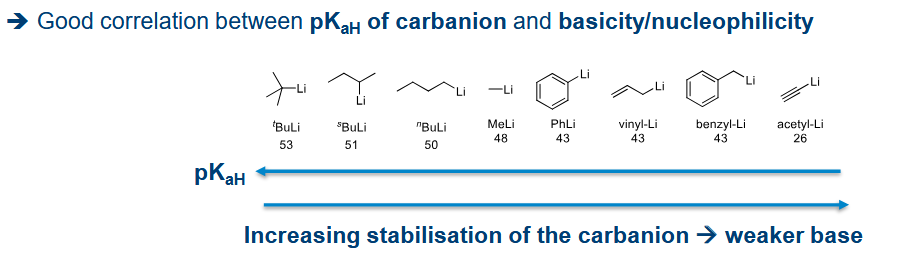
What determines the reactivity trend of R groups in R-M
More polar C-M bond = more reactive
More stabilised the partial negative charge on the carbon is by the R group (remember the C is more electrnegative than the metal)
Bearing in mind this stabilisation of the negative charge, what effects increase reactivity
sp3 > sp2 >> sp as the -ve charge is more stable closer to the nucleus hence less reactive.
More substituted carbons receive a greater +ve inductive effect, and of course allylic, aromatic stablisation also applies
Does the metal-carbon complex act as an acid or base. What numerical factor can help determine how reactive the complex is going to be
Base (carbon is a nucleophile here)
Organolithiums (R-Li) are electron deficient. How do they solve this.
Aggregation (both solids and in solution).
What is the degree of aggregation affected by
Bulkiness of the R group. e.g. nBuLi hexamer whereas tBuLi and sBuLi tetramers.
Solvents that donate electrons break down the aggregation - realistically this is only ethers and tertiary amines.
Why is lower aggregation desirable
It increases reactivity of the organolithium.
Grignard species structure
Less electron deficient than organolithiums. They form tetrahedral structures which are mono/dimeric
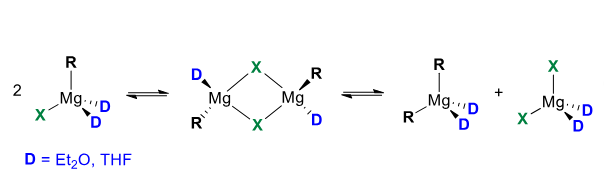
Schlenk equilibrium
Grignars exist in this equilibrium
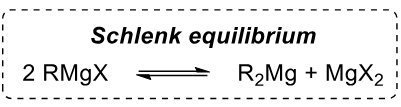
Which is more reactive out of R-Li and R-MgX
R-Li as it is more electron deficient and the bond is more polar
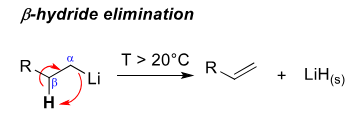
How does beta-hydride elimination occur for R-Li
At high temperatures the beta carbon is deprotonated to froma an alkene
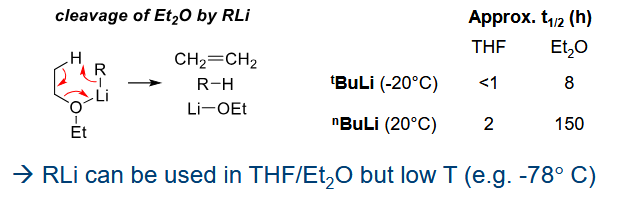
Why is R-Li kept in alkane solvents
It cleaves ether solvents if not at low temps.
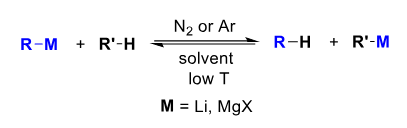
Direct Synthesis of R-Li and R-MgX
Start with a haloalkane under an inert atmosphere (of Ar for Li as N2 won’t workdue to LiN3)
For R-Li use an alkane solvent.
For R-MgX use an ether solvent or THF.
Solvent must be anhydrous and aprotic.
Mg may require induction e.g. using a high surface area powder

Synthesis via Metalation (metal-hydrogen exchange) - how can you shift the eq to the right
Proton exhange from one R group to another in exchange for a metal
You want R’ to have a lower pKa than R and R-H to be a gas
Synthesis via Metal-Halogen exchange - shifting equilibrium to right
Equilibrium shifted to the right when R’ > R stabilisation of carbocation. i.e. aryl or sp2
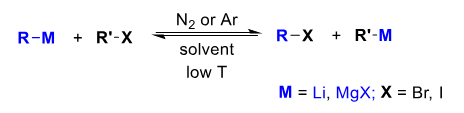
R-M often an alkyllithium.
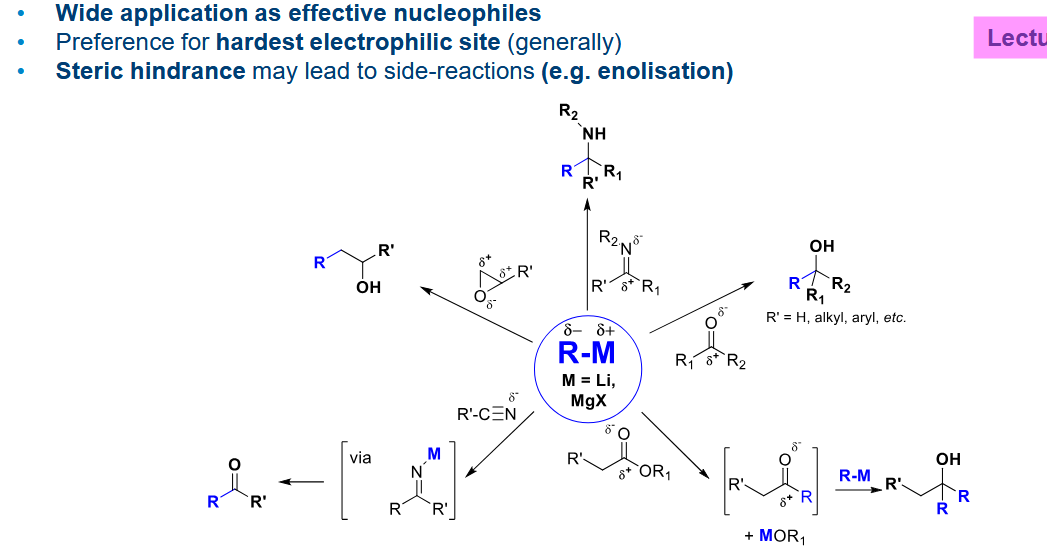
R-M overview
C-Cu(I) differences to s-block
Much more covalent than C-Li hence it is a softer nucleophile.
It will only react with a potent electrophile/ lewis acid
So how do we increase the reactivity, forming an organocuprate
Add a second R group to form a charge system.
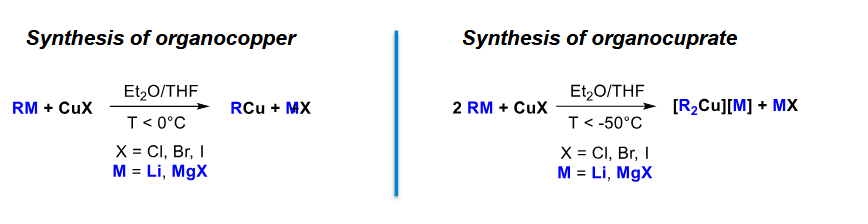
How do we form organocupper and organocuprate
Note the difference in T, otherwise the only difference is the equivalents
Reactivity of organocuprate [R2Cu(I)]- [M]+:

Organozinc synthesis and features
C-Zn more covalent than C-Li hence soft nucleophile.
Synthesised via direct synthesis from activated Zn or transmetalation. Activated Zn employs reduction with K or alloys to remain acitve at its surface.
It has good chemoselectivity and can tolerate many FGs
It is not that reactive much like R-Cu, so a Lewis acid can be used on the substrate (for more electrophilicity) or a Lewis base on C-Zn (for more nucleophilicity)
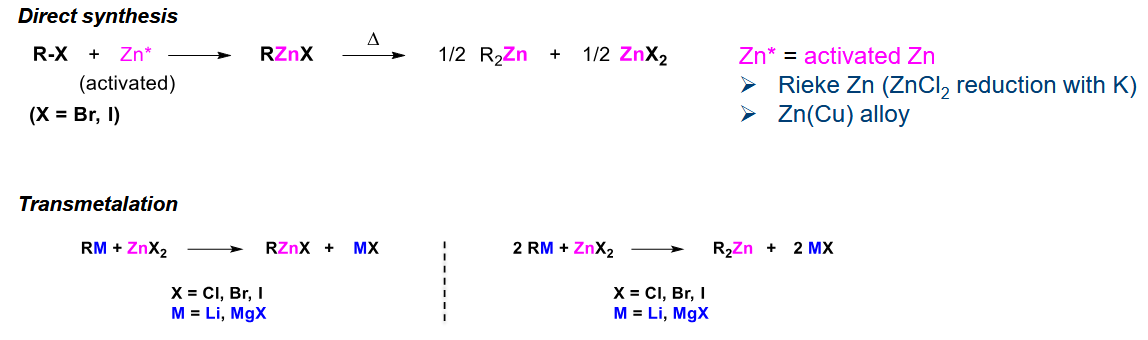
Organozinc reactions
Negishi cross-coupling (comes later)
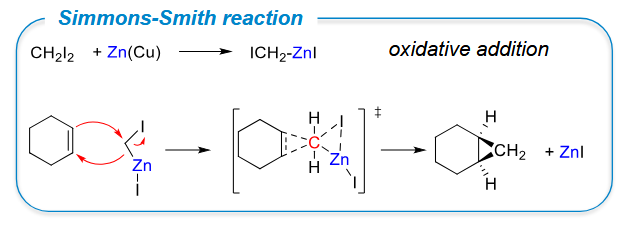
Simmons-Smith reaction - a concerted addition that conserves stereochemistry of reagent.