MBIO 2020 / Topic 6: Mutations
1/39
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
40 Terms
What is a mutation, & what causes it?
> Heritable change in DNA sequence.
> Due to a lesion in DNA that is not repaired prior to a replication round
What are the two types of mutations based on cause? Briefly describe each.
Spontaneous mutation
Errors caused by biological errors, e.g. DNA pol mistakes.
Induced mutations
Errors caused by external factors, e.g. UV.
How can mutant strains be derived in relation to the wild-type?
A mutant strain can be obtained from either (a) directly from the wild type or (b) from a parental mutant (a strain that was already mutated from the wild-type.
+ Depending on the mutation, a mutant may or may not differ in phenotype from its parent.
What are the two types of mutations based on survival? Briefly describe each.
Selectable mutations
> Confers a clear advantage on the mutant strain under certain environmental conditions, such that the progeny of the mutant cell are able to grow and replace the parent, e.g. antibiotic resistance of mutant over parent.
Non-selectable mutations
> Confers neither an advantage or disadvantage over their parent cells when grown in laboratory conditions.
What are the differences between selecting and screening to observe mutants in lab?
> Selection = Only mutants grow (e.g. antibiotic resistance).
> Screening = All grow; mutants identified by observable traits (e.g. colony color).
Why is selecting usually preferred over screening?
Selection is preferred over screening because selective conditions typically place severe restraints on the population that mutants are easily detectable.
What do auxotrophs require to survive, and how do you isolate against them?
> Auxotroph = Mutant needing extra nutrients than that of the wild type or parental strain (i.e. prototroph).
> Isolation requires selective medium with none of their extra requirement.
What are the two types of mutations based on change? Briefly describe each.
Point mutation (aka base-pair substitutions)
> Only 1 bp changes.
Frameshift mutations
> Insert/delete 1–2 bp = Shifts reading frame = Scrambling polypeptide sequence downstream of mutation.
Macrolesions
> May span 1 or more genes.
What are the five different point mutations? Briefly describe the impacts on the protein whenever applicable.
> Transition = Change within base category, e.g. adenine to guanine (purine to purine).
> Transversion = Change between base categories, e.g. adenine to cytosine (purine to pyrimidine).
> Silent = Different codon, but AA doesn’t change = Virtually no impact on final protein.
> Missense = Different codon + AA does change = Impact on final protein depends on location of changed AA & duration of effect on folding/activity.
> Nonsense = Different codon & AA changes into a stop codon = Incomplete polypeptide if mutation not at end of mRNA = Inactive protein or activity-lacking protein.
How can a point mutation in a coding region affect phenotype?
> Point mutation in a gene’s coding region may change the AA sequence = Alters protein structure/function = Affects phenotype.
> What is a reversion?
> What is a revertant strain?
> Reversion = Reversing the point mutation.
> Revertant strain = Strain regaining original phenotype due to a second mutation.
+ Spontaneous or induced.
What are the two types of revertants?
> Same-site revertant = Second mutation happens at the same site as the original
+ If it restores the original DNA sequence, it’s a true revertant.
> Second-site revertant = Second mutation is at a different site; acts as a suppressor mutation that compensates for the original defect
What are the three classes of suppressor mutations?
> Mutation in the same gene restoring original function.
> Mutation in a different gene that adjusts an interacting protein to function with the originally mutated one.
> Mutation in a different gene produces a new enzyme to assume role of originally nonfunctional, mutated one.
What are the four types of macrolesions?
> Large insertions or deletions, e.g. insert/delete a codon.
> Duplications = DNA segment repeated at different site on either strand.
> Translocations = DNA segment moved to different location on same strand.
> Inversions = DNA segment removed & inserted in opposite direction to preserve proper 3’ to 5’ reading for RNA pol.
What non-structural DNA regions can mutations affect, & how? HINT: There are 6 of them.
> Promoter (−35 & −10 boxes) mutations = RNA pol binding.
> Inverted repeats forming stemloops = Transcription termination.
> Shine-Dalgarno / RBS mutations = Translation initiation.
> tRNA secondary structure = Proper amino acid charging or pairing.
> Mutations in regulatory protein binding sites = Activator or repressor control.
> Mutations in regulatory non-coding RNAs = Gene silencing or activation.
What are the four types of spontaneous mutations?
> mutations @ apurinic/apyrimidinic sites → loss of base.
> deamination → loss of amino group from C/A/G but not T/U (no NH2)
> tautomeric shift → temporary change of base to a tautomeric form w/ TG and CA = new pairs
> insertions & deletions → DNA pol slips during replication esp w/ repetitive seq like AAAA
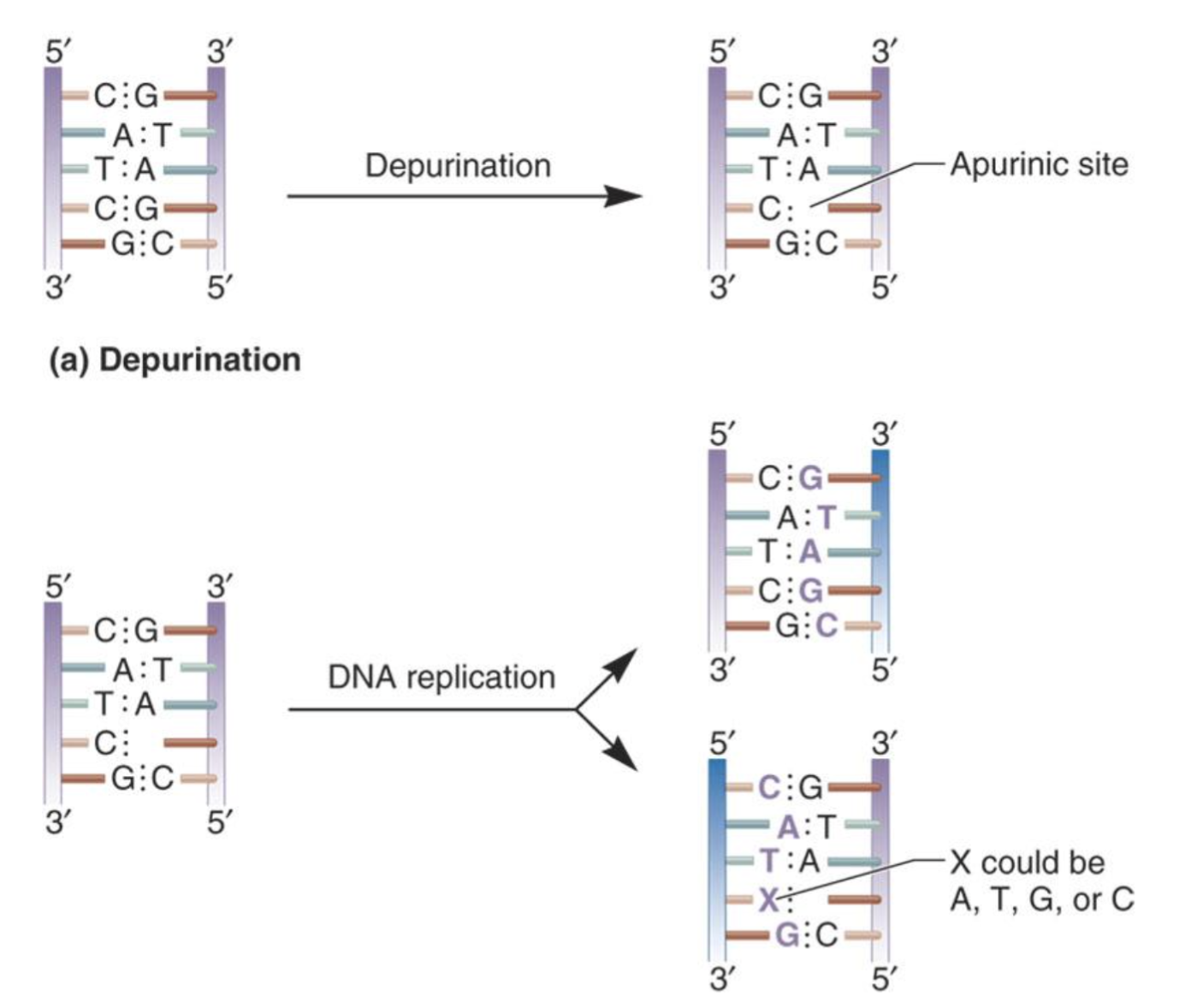
what’s the problem & solution of mutations @ apurinic/apyrimidinic sites? What happens if the solution isn’t done?
> Problem: Hydrolysis of covalent bond between base & deoxyribose removes base (apurinic/apyrimidinic site).
> Solution: DNA repair enzymes recognize & fix the site.
+ If not repaired, a random base is inserted during replication = May lead to a transition/transversion mutation.
+ In the image, whether a mutation occurs depends on which strand is used as the template. If the strand with the AP site is used, the mutation is likely.
what base changes happen during deamination and how do they affect base pairing?
> adenine:thymine → adenine becomes hypoxanthine → hypoxanthine looks most like guanine → guanine pairs with cytosine → hypoxanthine:cytosine
> guanine:cytosine → guanine becomes xanthine → xanthine still most looks like guanine → xanthine:cytosine
> cytosine:guanine → cytosine becomes uracil → uracil:adenine
what’s the problem & solution of deaminations?
> problem: becomes point mutation if not repaired before round of replication
> solution: specific repair mechanisms in place
which mismatches can occur due to tautomeric shifts in DNA bases?
> thymine (keto):adenine → thymine shifts to enol form → hydrogen bonding pattern fits better with guanine → thymine (enol):guanine
> cytosine (amino):guanine → cytosine shifts to imino form → hydrogen bonding pattern fits better with adenine → cytosine (imino):adenine
> adenine (amino):thymine → adenine shifts to imino form → hydrogen bonding pattern fits better with cytosine → adenine (imino):cytosine
> guanine (keto):cytosine → guanine shifts to enol form → hydrogen bonding pattern fits better with thymine → guanine (enol):thymine
how does a tautomeric shift in adenine lead to a point mutation over two rounds of replication?
> first round: T-A → A shifts to imino → mispairs with C → C-A
+ adenine still kept, not a point mutation technically.
> second round C-A → A shifts to amino, but C stays → C-G
+ neither T or A kept, now a point mutation = 1bp changed
what’s the problem & solution for taut shifts?
> problem: wrong bp from original
> solution: DNA pol proofreading activity
what’s the difference between insertion and deletion?
> a loop is formed in non-temp strand → addition of an extra base on non-temp unneeded
> a loop is formed in temp strand → one less base is not added into non-temp strand
what’s the problem & solution for insertions/deletions?
> problem: frameshift mutations if not in multiples of 3
> solution: DNA pol proofreading activity
what are the four types of induced mutations? describe each.
> mutations from nucleotide base analogs → fake bases looking like normal ones but can mispair with the wrong partner → causes transition mutations
> mutations from intercalating agents → flat molecules wedge between bps, pushing bps apart → distorts helix → leads to single base insertions or deletions → frameshift mutations
> alkylation of bases → add alkyl groups to bases → transition mutations
> deamination of bases
how do intercalating agents cause single base (a) insertions and (b) deletions?
> intercalator wedges between bps in non-template strand → creating extra space
insertions
> DNA pol thinks a base is missing when there really isn’t a missing base → single base insertion → frameshift mutation
deletions
> DNA pol thinks a base is present when there really wasn’t a base present → single base deletion → frameshift mutation
how does the Ames test detect if a chemical is mutagenic, and why are some revertants called spontaneous?
> goal: test if a chemical causes mutations by reverting bacteria with a point mutation back to wild type
> method: plate two plates, both with limited his:
(a) no mutagen = baseline for spontaneous revertants (control)
(b) with mutagen = tests for induced revertants
> result: if plate (b) has significantly more colonies, the chemical is a mutagen
+ the his is only there temporarily to let bacteria survive a few divisions. any reversion happens naturally (in plate a), without mutagen exposure—that’s why it’s called spontaneous
what are the two types of radiation mutagens? describe each.
non-ionizing radiation (UV)
> bases absorb UV
> results in pyrimidine pyrimidine dimer
> DNA pol cannot use strand bec of distorted track of DNA + dimers cannot undergo normal base pairing
ionizing radiation
> water and other substances to ionize
> radicals produced
> ds and ss breaks
> rearrangements and deletions bec of being “shot” through by radicals
why is leading strand synthesis more affected by pyrimidine dimers than lagging strand synthesis?
> leading strand is made continuously → a single dimer blocks DNA pol and halts synthesis
> lagging strand is made in short Okazaki fragments → if a dimer is present, pol can start the next fragment downstream → less affected
+ pyrimidines don’t have to be the same (e.g., T–C or C–C) to form dimers
+ dimers not passed to progeny & cause no change in nt sequence but can cause DNA damage → causes mutation
what are the two types of DNA repair systems?
> error-free repair systems → restore original bp seq
> error-prone repair systems → repair incorrectly and introduce mutations
what are the five error-free repair systems?
> alkyltransferase
> photoreactivation
> base excision repair (BER)
> nucleotide excision repair (NER)
> methyl-directed mismatch
how does the alkyltransferase repair?
> recognizes alkylated DNA
> catalyzes transfer of methyl/ethyl group from base to its cys side chain → restores base
> enzyme inactivated once alkyl transferred
how does the photoreactivation repair?
> photolyase binds to the dimer
> contains FADH₂, which stores light energy used to break the covalent bond
> activation triggered by 350–500 nm light (blue light)
> in absence of light, the nucleotide excision repair system steps in to remove the dimer
how does specific BER repair?
> specific DNA glycosylase identifies and cleaves glycosyl bond between damaged base and sugar, leaving an AP site
> AP endonuclease cuts the DNA backbone at the 5′ side of the AP site
> DNA pol removes the AP site and a few surrounding nucleotides
> DNA pol fills in the gap with the correct bases
> DNA ligase seals the nick in the backbone to complete the repair
how does general BER repair?
> AP site already present from spontaneous base loss or another process
> AP lyase cleaves the backbone at or near the AP site
> DNA polymerase fills the gap
> DNA ligase seals the nick to complete repair
what does NER repair?
> thymine dimers
> missing bases
> modified bases
how does NER repair?
> 2UvrA/1UvrB scan for distorted DNA
> UrvA detects kink and leaves; UrvB stays bound and unwinds region
> UvrC binds to UvrB and makes cuts on 5’ side and 3’ side of thymine dimer.
> UvrD helicase removes damaged oligonucleotide; UvrB/UvrC and UvrD releases; DNA naturally reseals
> DNA pol fills gap and DNA ligase seals nicks.
how does methyl-directed mismatch repair work?
> MutS dimer senses mismatch via kink in DNA and binds to mismatch.
> 2MutL binds MutS and recruits MutH.
> MutH nuclease finds the nearest methylated GATC site → nicks unmethylated/new strand
> UvrD helicase + exonucleases remove small segment containing from the nick through the mismatch
> DNA pol III fills the gap w/ correct bases
> DNA ligase seals the nick
> GATC sites get DAM’d
+ Hemimethylation tells cell which strand is the original and which one might be wrong.
how is the bacterial SOS response regulated from activation to shutdown?
> LexA at least partially represses SOS repair genes @ normal conditions to prevent unnecessary error-prone enzymes but allow DNA repair @ normal conditions
> When DNA damage is high, DNA pol stalls @ lesions while helicase keeps unwinding → ssDNA builds up
> RecA binds ssDNA → forms RecA* → stimulates LexA autoproteolysis
> LexA repression lifted → SOS genes and lexA regulon expressed + enzymes from other repair systems (@ high levels of these other enzymes in SOS) → accelerated repair
> Error-prone polymerases (e.g. DNA pol IV/V) come in to allow replication to continue but w/ increased mutation risk
> ssDNA damage repaired → ssDNA disappears → RecA inactivates + LexA is resynthesized → SOS genes’ repression resumes → SOS shuts off
what features of DNA pol IV/V allow replication to continue in high damage conditions? why is there increased mutation risk?
> synthesizes DNA even if template is trash (w/ lesions/errors)
> bypasses obstructions that make normal DNA pol quit → synthesizes DNA over bulky lesion and past it
> cannot proofread like normal DNA pol → likely to insert incorrect bases → increases mutation risk