Urea Cycle
1/32
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
33 Terms
Differentiate between a positive and negative nitrogen balance and what conditions could lead to this? What percentage of excreted nitrogen is in urea?
+: intake > out; growth and recovery from illness and malnutrition
-: intake < out; illness, malnutrition, burns
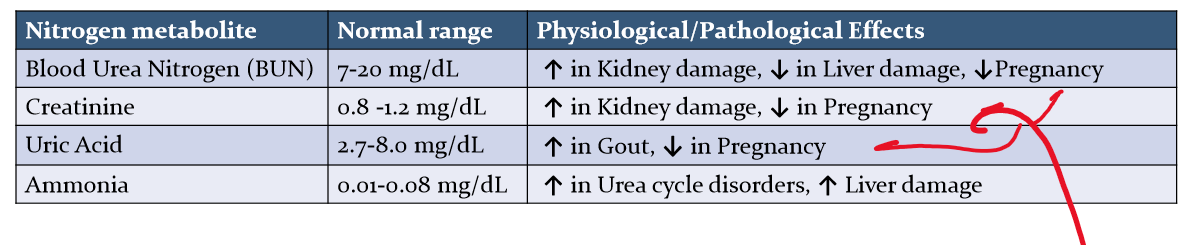
Why does BUN levels decrease when pregnant? What else decrease with BUN? What happens when there is kidney damage? gout? Liver Damage? and other urea cycle disorders?
During pregnancy, the glomerular filtration rate increases
what are the three major steps in amino acid degradation?
removal of nitrogen (transamination, deanimation, deamidation)
clearance of NH4+ (urea cycle, ammonia itself is toxic)
utilization of carbon backbone (Fed state: synthesis of glycogen and triglycerides; Fast state: energy production)
Where does Urea gets its nitrogen and carbon from?
Urea gets one nitrogen from aspartate and one from NH4+; it gets its carbon from CO2
Differentiate deamination and deamidation in the context of nitrogen removal from amino acids? (definition? Which AA? Enzyme used?) Why is deamidation important?
deamination: removal of amino group of some amino acids (glycine, threonine, histidine, serine, glutamate)
Uses glutamate dehydrogenase (reversible); glutamate —> alpha-ketoglutarate in liver, kidney; opposite way in other tissues
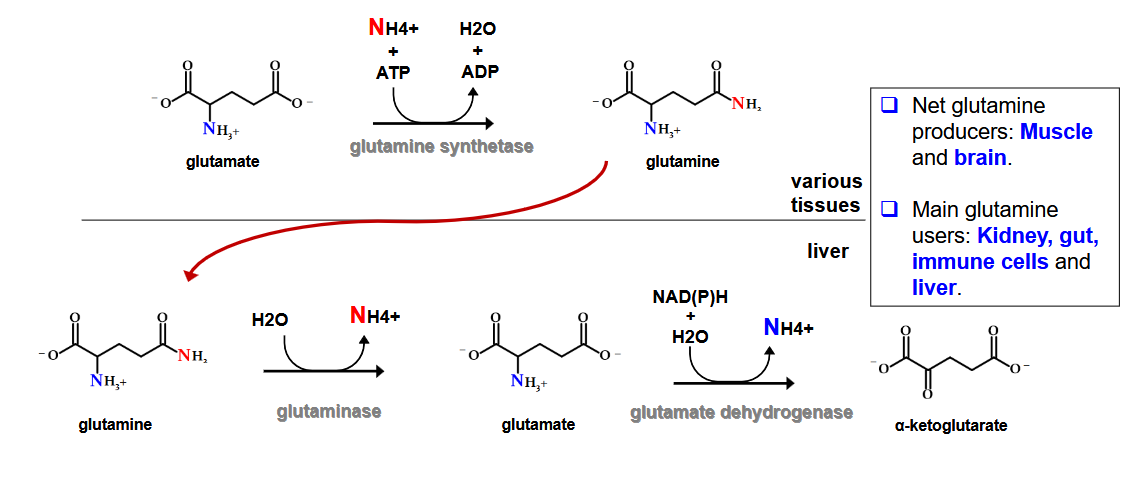
Deamidation: removal of amide group from glutamin and asparagine ; enzyme = glutaminase
most important in kidney
provides most of the NH4+ in urine
NH3 helps balance pH
describe transmination as it relates to nitrogen transfer. General Pathway?
main pathway of amino acid nitrogen removal
It uses transaminases (ALT, AST) and requires PLP (B6) as coenzyme.
general pathway: amino acid loses its NH4 to become a-keto acid; a-ketoglutarate gains NH4 to become glutamate.
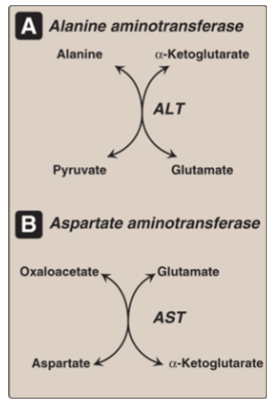
Draw out the reaction including AST and ALT; what is the difference and smilarity between them?
ALT > AST in specificity;
AST is used for diagnosing myocardial infarcation;
both: elevated levels = liver damage
What are the four central roles Glutamate have in nitrogen removal?
gets NH4 from other amino acids via transamination
provides NH4 to Urea cycle through deamination by glutamate dehydrogenase
it can transaminate OAA into aspartate. Aspartate can then provide an amino group to Urea
precursor of the allosteric activator of Urea
Where does the Urea cycle take place? How does nitrogen from other tissues get transported to this place? Where does urea go?
take place in liver
Amino acid nitrogen is carried to liver in the form of Alanine and Glutamine
Urea go to kidney to be excreted in urine. some urea also leave through intestines
describe how Glutamine transport Amino Acid Nitrogen to the liver? What is the net glutamine producer? main glutamine users?
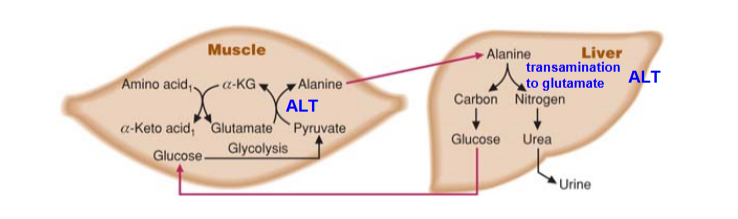
Describe the transport of Amino Acid Nitrogen to the liver via Alanine:
what is its two purpose?
Draw out how the muscle and liver is connected via this
What are the main sources of Alanine
GNG; removal of nitrogen for Urea Cycle
Sources:
Main: muscle (results of protein degradation/ transamination)
other = kidney, intestines (conversion of glutamine to alanine)
glutamine → (via glutaminase) glutamate → (via ALT) Alanine
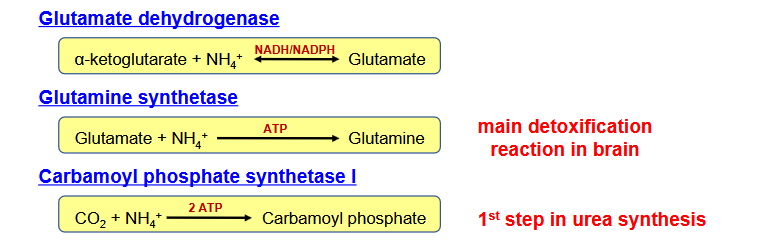
Why is ammonia dangerous? What are the two mechanism in which the body control this? What is the main detoxification reaction in the brain?
Ammonia is toxic
Two ways:
transamination reaction collect nitrogen on glutamate rather than release free ammonia
three eyzmes can fix free ammonia into organic molecules
glutamate dehydrogenase
glutamine synthetase
carbamoyl phosphate synthetase I
Draw out the Urea Cycle
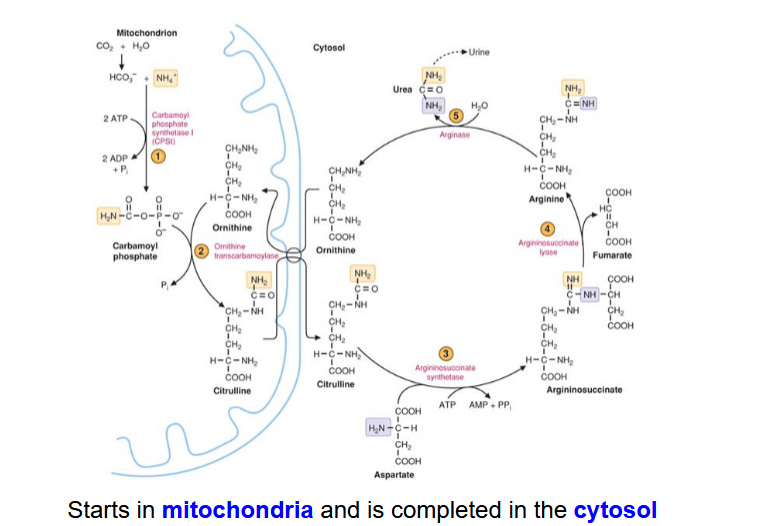
Describe the Carboamoyl Phosphate synthesis step in the Urea Cycle. How is it allosterically regulated
HCO3 + NH4+ → Carbamoyl Phosphate
catalyzed by carbamoyl phospahte synthetase I (CPSI)
Takes place in mitochondria
rate-limiting, committed step
requires 2 ATPS
allosterically regulated by N-acetylglutamate (NAG)
Describe the synthesis reaction of NAG
Glutamate + acetyl CoA —> NAG which activates CPSI
this reaction above is activated by Arginine
describe the reaction for citrulline synthesis
carbamoyl phosphate + ornithine —> Citruline
reaction is catalyzed by ornithine transcarbamoylase (OTC); takes place in mitochonria
What is ornithine translocase?
this enzyme helps transport ornithine from the mitochondria into cytosol
what causes type II hyperammononemia
deficiency of ornithine transcarbamoylase
what causes type I hyperammonemia?
deficiency of CPSI
what causes HHH syndrome?
deficiency in ornithine translocase
aka: hyperornithinemia-hyperammonemia- homocitrullinuria syndrome
describe the reaction for argininosuccinate synthesis
citrulline + aspartate —> arginosuccinate
catalyze by argininosuccinate synthtase;
requires 1 atp
the aspartate nitrogen will be incorporated into urae
location: cytosol
what is citrullinemia type I? What is citrullinemia type II?
I: deficiency in arginosuccinate synthetase
II: deficienct citrin (mitochonrial glutamate/aspartate antiporter)
describe the reaction for arginine synthesis
argininosuccinate —> fumerate and arginine
catalyzed by argininosuccinate lyase
cytosol
only way arginine is produced in the human body
what causes argininosuccinyl acidemia?
deficiency in argininosuccinate lyase
describe the reaction of cleavage of Arginine into Urea and Ornithine
what other pathways can arginine go into?
arginine —> urea + ornithine
catalyzed by arginase
cytosol
protein synthesis and nitric oxide synthesis
what causes arginiemia?
deficient in arginase
how many phosphate bonds are broken in the Urea cycle?
4
how does a high protein diet and excessive degradation affect the Urea cycle?
these causes increases in urea synthesis because an increase in arginine levels = increase in NAG = more urea produced because increase activity of CPSI
what are the symptoms of hyperamonemia? Compare and contrast primary an seconary hyperammonemias
hepatic encaphalopathy, cerebral edema, seizures, nausea, vomiting, lethargy, coma and death
primary: enzyme in urea cycle is defective
seconary: main cause is hepatic failure, can be caused by genetic defects not relate to urea cycle enzymes
what are the five primary hyperammonemias diseases? which ones are autosomal recessive?
type 1 (CPSI or NAG synthase deficiency_
type II (ornithine transcarbamoylase)
citrullinuria type I (argininosuccinate synthetase deficiency)
argininosuccinic aciemia (argininosuccinate lyase deficiency)
argininemia (arginase deficiency)
All are autosomal recessive except type II (x-linked)
What is the consequences of Type II hyperammonia in regards to carbamoyl phosphate
OTC is defective → increases in carbamoyl phosphate
accumulate leads to leakage out of mitochondria
accumulation of carbamoyl phosphate → increasess in Orate → pyrimidines and urine
what are the three treatments for reducing nitrogen load on urea cycle
low protein diet, avoid fasting (to avoi AA degradation)
scavengers drugs (conjugates amino acids and target them for urinary excretion)
benzoate: benzoate + glycine → hippuric acid → excretion
phenylbutyrate : phenylbutyrate + glutamine → phenylacetylglutamine → excretion
reducing ammonia from microflora: lactolose laxative decreases pH → gut ammonia = not reabsorbed;
also urease inhibitors can reduce nitrogen from this source as well
describe the amino acid supplementation to treat urea cycle deficiency
arginine: in all primary hyperammonemias, except for arginase deficiency, this is low; also promotess production of NAG → activates CPSI
citruline: in type I and II, low citruline; also captures aspartate so at least one nitrogen can be excreted
N-carbamoylglutamate: agonist for CPSI in case NAG synthase deficiency