BCHM112: Transition Metal Chemistry
1/72
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
73 Terms
define
transition elements
d-block elements
transition metals
(transition elements) elelements in the supposed transitional position between metallic elements (grp 1-2) and non-metallic elements (grp 13-18) on the PTable - a transition between EP periods → EN periods
(d-block elements) elements where electrons are filling d-orbitals (gr 1-2 = s-block, gr 13-18 = p-block)
(transition metals) d-block elements with partially full d-orbitals (d1-d9) of valence electrons - in one or more of their oxidation states
what characterises transition metals in the way they react with water (compared with s-block metals) to form aqueous solutions
(s-block metals) in aqueous solution react to give colourless solution, and an anhydrous salt (contains no water) after removal of solvant, which can go on to dissolve in water again to repeat the process (e.g. Na+(aq) + Cl-(aq) → NaCl(s))
(transition metals) react in solution to give a coloured solution, and removing the solvent gives a hydrated salt (associated with H2O) that is coloured (e.g. Co2+(aq) + 2Cl-(aq) → CoCl2 . 6H2O) and can be removed from the water with very high temperature boiling, it also is found to make the solution acidic (not the case in other metals)
this suggests water strongly associates with the transition metal (actually bonded H2O molecules of an exact number to the metal, exchanging slowly), compared to the s-block metal (hydrated by infinity H2O molecules exchanging rapidly with H bonds)
why are aqueous transition metal solutions acidic (pKA ~9) while s-block metal aqueous solutions are not?
the charge on the transition metal complex means that a proton can be donated (lower the overall charge) from an H2O ligand of the TM, to another H2O molecule, to form H3O+ (increases acidity)
can donate multiple protons if a multiply positive charge, and be even more acidic
does not happen in s-block metals as they are not specifically bonded to H2O, instead associated with infinity via H bonds
how are square brackets used to denote TM chemical and structural formulas?
(structural formulae) enclose the structure in square brackets with the charge outside the bracket to represent the complex, any counter ions are on the outside, ligands on the inside
(chemical formulae) enclose the specific things interacting with the mtal centre (ligands) in square brackets, with the counter ions outside the bracket, along with the charge of the complexI am a hard working and motivated university student, looking for further challenge, namely in giving back to my community.
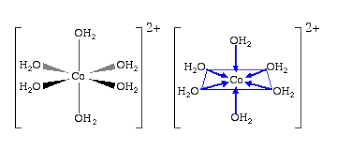
what does complex / coordination compound / TM complex mean
a TM cation centre, with coordination bonds to ligands surrounding it (molecules with atoms with lone pairs that can act as ‘donor atoms’ to form dative bonds to metal ion centres)
what is the typical covalent bond
what version of the covalent bond is formed with ligands to metal ion centres? what are these called?
what are the requirements of this bond
(typical covalent bond) each atom contributing 1 electron to form a 2-centre 2-electron bond (sharing these electrons)
(coordination / dative-covalent / donor-acceptor bond) the 2 electrons shared in the bond come from one atom’s lone pair, interacting with an ion’s charge, forming a 2-centre 2-electron bond
here the metal cation acts as an EP / electron acceptor / lewis acid
here the ligand acts as a Nu / electron donor / lewis base
(requires) a positive metal cation with an empty orbital (found in partially filled d-orbital), and an electron rich ligand (with a charge or with a lone pair available to donate)
this results in orbital overlap, forming a covalent bond with high electrostatic interaction too
what are the characteristics of a ligand
have ‘donor atoms’ most commonly from groups 15-16 (N, P, As, O) - have lone pairs, group 17 in anionic form only, and group 14 in carboanion form after reaction with a strong base
most ligands are either neutral (e.g. :NH3, H2O:) or anionic (e.g. :CN-, Cl-) to have this electron density (lone pair available) to bind with the metal catin
what is a ligand displacement reaction
what are the consequences on the colour of solution
this changes the ligands attached to a metal centre in a complex, a substitution reaction to replace these with another ligand
typically taking the TM aqueous solution, and replacing the H2Os associated with other ligands - these are in equilibrium but lying heavily to the right
this changes the colour depending on the complex formed / different TMs / different ligands
how are ligands classified based on denticity
(denticity) refers to number of donor atoms within the molecule that can coordinate to a metal ion - alike teeth (Dentate) biting onto the metal
(monodentate) can coordinate to a TM ion through 1 donor atom only (e.g. :NH3, Cl-)
(ambidentate) ligands with 2+ potential donor atoms (same or different, so can form same complex or different), can coordinate both at the same time (bidentate) or only 1 at a time (depends on relative reactivity to the TM), (e.g. :SCN:-, :O=N=O:) - so can chelate or not, and bridge complexes or not
(bidentate) can bind to a metal ion through 2 donor atoms (e.g. en = ethylene diamine, acac = acetyl acetonate ion) simultaneously, so can chelate
(polydentate) tri, tetra, penta dentate ligands (chelating ligands) as they form numbers of chelate rings when binding to a metal ion
what is a chelating ligand
what are the requirements for these to form
give an example
when bidentate / polydentate ligands attach to the same metal ion from both donor atoms, thus forming a ring
most commonly 5-6 membered rings (most stable)
only form if donor atoms are in correct position to bind, both able to be in the same position to interact simultaneously with the metal - so easier for flexible ligands (easily change donor atom position)
those who dont chelate, still act as ligands, just not to the same metal (may bridge compounds)
e.g. ethylene diamine (en), acetyl acetonate ion (acac-)
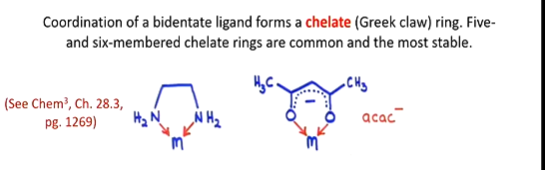
draw the structure of
en (ethylene diamine)
acac- (acetyl acetonate ion)
what does coordination number mean
factors that affect this
periodic table trends
what is the range of coordination numbers
most common coordination numbers
(coordination number) how many donor atoms can bind to a metal ion centre, of a complex
(factors)
size (atomic radii) of the ligand & metal ion (considering steric bulk around the centre)
larger radii metal centre will house more ligands, larger atomic radii ligands will fit less on metal centres
charge (if anionic will start to electrostatically repulse eachother, if multiple anions)
(period table trends) radii increases across and down the table, so consider these for ligands and metals
(range) 2-9
(most common) 4 & 6 (total donor atoms) - the rest being rare
how do the square bracket formulae notation of complexes, relate to the chemistry of these complexes
coordinated ligand groups (represented as being INSIDE the square brackets) are held more tightly in the complex (covalently bonded directly to the metal centre)
this means they are less chemically accessible / reactive with other molecules, compared to counterions outside of the square brackets (associated / electrostatic, but not covalently bonded, so more reactive to chemistry)
therefore when we dissolve these complexes in water, it solvates (seperates) those in the square brackets, and those outside (seperates the complex with the counter ions)
e.g. Co…..+, and Cl-
what determines the stereochemistry of complexes
metal ions can have multiple coordination numbers possible, each charactersed by a certain stereochemistry (3D shape)
the arrangement in practice is the one that reduces steric bulk / is the most stable / has the lowest energy - so minimises repulsions, maximises attraction - favoring a symmetric arrangement of ligands around the metal
this is considered when deciding between shape for coordination no with multiple possible shapes
what shape (& bond angle) arises from coordination number 2
linear
bond angles 180
e.g. [Ag(NH3)2]+
what shape (& bond angle) arises from coordination number 3
trigonal planar
bond angles 120
e.g. [Pt(PPh3)3] - large ligands so not many fit
what shape (& bond angle) arises from coordination number 4
(tetrahedral) bond angles 109 equally
e.g. [FeCl4]-
(square planar) all atoms in the same plane (due to 2 lone pairs) - more common in later TM
e.g. [Pd(CN)4]2-
what shape (& bond angle) arises from coordination number 5
(trigonal bypyramid) 3 ligands & metal in the plane (90 degrees), 2 ligands above & below the plane (120 degrees)
e.g. [CuCl5]3-
(square pyramid) 4 ligands & metal in the plane (90 degrees), 1 ligand above (90 degrees)
e.g. [Ni(CN)5]2-
differ little in energy so can be interconverted between easily , but steric constraints may favor a square pyramid - better at minimising repulsions
what shape (& bond angle) arises from coordination number 6
octahedral
bond angles 90 (between adjacents)
bond angles 180 (between opposites - above & below plane atoms)
4 ligands and metal in one plane, 2 ligands above & below the plane
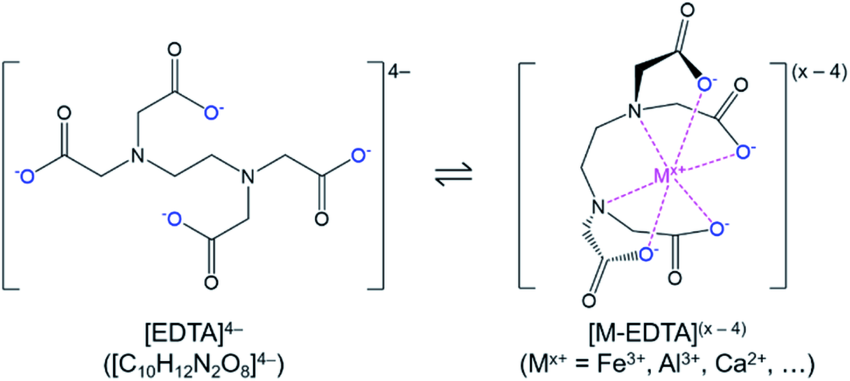
what is edta4-
why is it important
ethylene diamine tetracetic acid
a powerful chelating hexadentate ligand, when droptonated, with strong attractions to metal centres due to these negative charges (electrostatic attractions)
also powerful due to extreme stability (large stability constant eq lying strongly to right) as when one donor dissociates, the ligand wont fall off as 5 other is attached - unliikely for all to dissociate
also due to formation of multiple 5-membered chelate rings
(applications) strong binding to metals is important to treat metal poisioning, as it can remove excess metal ions from the body (e.g. mercury poisioning, lead poisioning, removing excess irons) - however can remove essential metals too (not specific)
what type of isomers can coordination complexes form
geometric (cis/trans & E/Z)
optical (enantiomers, diasteromers)
what two shape complexes can form Geometric Isomers (considering monodentate ligands)
how do these differ depending on type of groups coordinated
how do these differ between each other
(Square Planars)
(cis/trans) form if have 2 different donor groups
with 4 different donor groups, form 3 geometric isomers, as these each involve different groups near different groups
(Octahedrals)
(cis/trans) form if have 4 same, 2 different donors, based on if 2 same are opposite OR adjacent
(mer/fac) form if have 3 same & 3 same, (Mer = Meridian) 3 groups in the same plane, (Fac = Facial) 3 groups in the same triangular face (adjacent but not in the same plane)
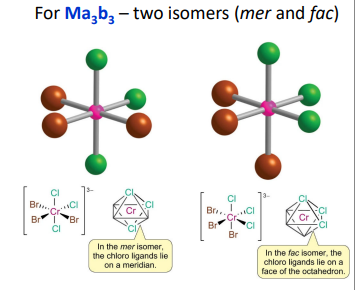
what Geometric Isomers can form for complexes with Polydentate Ligands?
(cis/trans) can usually only form cis isomers / adjacent positions on the coordination sphere, as trans are too far apart / get in the way of other ligands - considering the donor atoms on an individual Polydentate
(cis/trans for bidentates) in octahedral, bidentate ligands can form cis/trans isomers on complexes with 2 bidentates & 2 other groups (cis = bidentates beside eachother) (trans = bidentates opposite eachother)
(cis/trans for asymmetric bidentates) in square planar, can form cis/trans based on if like donors are adjacent (cis) or opposite (trans)
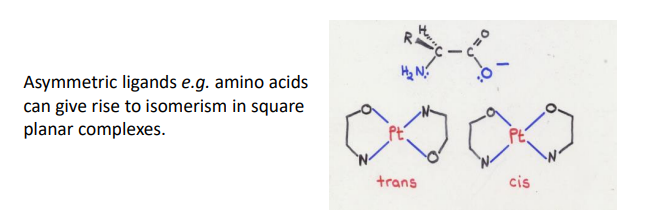
what are the requirements for optical isomers in complexes
what 2 shapes, with which type of ligands, can form them
(requirements)
for enantiomers, must form non-superimposable mirror images, so be chiral compounds, with no internal mirror plane (same molecule rotated)
(Octahedrals)
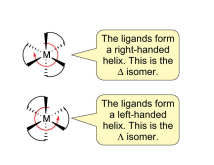
with 3 bidentate chelating rings, can form enantiomers, but only certain geometric isomers (cis CAN, trans CANNOT (has internal mirror plane)
denoted delta / lambda based on direction of helix / screw axis, based on starting from the donor pointing OUT of the plane
to stay on the same ligand and go towards the donor pointing BEHIND the plane, if it goes to the left (= Lambda), if it goes to the right (= Delta)
(diastereoisomers) stereoisomers that are not enantiomers, isomers on one point of chirality (stereocentre) rather than both
(Tetrahedrals)
with 4 different ligands, the metal centre becomes chiral, so can form enantiomers
(Ligands)
if chiral ligands are coordinated, enantiomers can formed based on the arrangement of these coordinated ligand stereocentres
thus can form enantiomers for square planars, which would usually not do enantiomers around the metal centre, as are all in the same plane
what are the 6 steps for naming complexes
(Order) name groups in the usual order (cation → anion for complex & counterion) & (number of ligands → name of ligands → metal ion → oxidation state)
(Naming Ligands) name negative ligands their anionic name but e→ o (e.g. Cl- → Chloride → Chlorido), name neutral ligands with usual name (EXCEPT H2O → Aqua, NH3 → Ammine)
(Listing Ligands) list alphabetically
(Number of Ligands) use usual prefixes for monodentate ligands (di- / tri - / tetra-), use new prefixes for polydentate ligands (bis - / tris - / tetrakis -) - and enclose in brackets
(Oxidation State) denote in Roman Numerals (e.g. I, II, III, IV)
(Anionic Complex?) if the whole complex is anionic, name the metal centre using Latin Metal name + Ate suffix (Cu → cuprate, Ni → nickelate, Ag → argentate, Fe → ferrate
dont number / denote the charge on the counterion (given by OS)
write the complex name as one word, any counter ion as a seperate word
how are complexes named in terms of chemical formula
square brackets to enclose the complex
metal written first
ligands written after (in alphabetical order)
charge of complex ion written outside the square brackets
counterions placed outside the square brackets
what theory explains why certain metal ions prefer to interact with certain ligands over others
explain this
Hard Soft Acid Base theory
(Hard) small highly charged / high charge density atoms, not polarisable (valence e held tightly, dont want to share), metal-ligand bonding has high ionic character (big charge difference, strong electrostatic)
e.g. Early TMs, ligands with higher EN (O, F-, Cl-)
(Soft) large atoms with low charge density, are polarisable (valence e held less tight so can have clouds distorted to create poles), metal-ligand bonding is more covalent character (sharing valence e)
e.g. Late TMs, ligands with lower EN (S, I-, P, C)
intermediates exist, in the middle TM section
(Acid) refers to metal ions, as these are lewis acids
(Base) refers to ligands, as these are lewis bases
THEREFORE
like attracts like, Hard ligands coordinate to Hard metal ions more readily, forming more stable complexes (high stability constant, strong bond) - and vice versa
will interact with opposites, but less coordinations will be formed, lower stability constant
helps determine ambidentate coordination - the donor atom of the ligand that will coordinate, depends on its similarity to the metal in terms of Hard / Soft characteristics
briefly describe the reaction mechanism of TM ligand substitution, for the 3 types
metal = EP, ligand donor = Nu, H2O = LG (for a complex in aqueous solution, surrounded by H2O ligands)
(Dissociative) a 2-step process alike Sn1, H2O departs in one step → intermediate with lower coordination number formed → Nu new ligand attacks and joins in another step
(Associative) a 2-step process alike Sn1, Nu new ligand attacks in one step → intermediate formed with both new and H2O ligand attached (increased coordination number) → in final step H2O LG leaves and substitution has occured
note - from intermediate can go in either direction
(Mid-Ground) alike Sn2 process, not really clear-cut steps, leaving & joining is simultaneous
how can we define different speeds of ligand exchange
how was this data found
how do these differ between TM, and between TM & S-block metals
what aspect of reaction does this determine
determines reaction rate
data found empircally - using a radiactive tracer to measure self exchange of water ligands (of metal in aqueous solution)
differs greatly between TM, tabulated data over 20 orders of magnitude (picosecond → years)
(s-block metals) groups 1& 2 are typically very fast
(d-block metals) are more variable, lower OS = faster, higher OS = slower, due to greater electrostatic attraction with higher charge (higher stability constant)
(fast exchange) reaction is chemically labile (half life < minute)
(slow exchange) reaction is chemically inert (half life > minute)
what are stability / formation constants for complexes
quantitative way of representing ligand exchange / reactivity of substitution reactions / stability of a complex (how likely ligand exchange)
uses K, equilibrium constant, a higher value meaning more favoring products - less stable, a lower value favoring reactants - more stable (measured by products / reactants, activities (= concentrations of solution)
but uses these cumulatively, to consider the differences in reactivity of the ligands binding successively (stepwise) - do all substituents come in one go? is it sequential? - each have their own equilibrium setup & stability constant
to find the overall stability constant therefore, sum the K for each step (= Bn, n being the number of ligands)

what are the trends seen in successive formation / stability constants
stepwise K values usually decrease as ligand coordination number increases, as to begin with there are many H2O to replace (a high probability of a ligand replacing one - readily react) but as ligands are replaced, the probability of replacing an existing ligand increases (reverse reaction, lowering K)
must also consider steric factors, K will decrease if crowding occurs (is increased as reaction proceeds, intereferring with subsequent substitutions - lowering rate of forwards reaction)
must consider coulombic factors, K will decrease if anion ligands are coordinated, as charged ligands repel, so gets less stable as number of anion ligand increases
what is the chelate effect
why does this occur (2 reasons)
how does size of chelate affect it
the enhanced stability of a complex containing chelate rings, compared to a complex with fewer / no chelates
(entropic reason) substituting with chelate increases disorder (forms more products as we displace existing ligands, and replace with fewer chelate ligands), this provides a large negative deltaS, which feeds into deltaG to be negative (thermodynamic driving force, spontaneous)
(statistical reason) chelates have 2 donor atoms, which increases their chance of coordinating as when one end attaches the other will readily bind, AND will be less likely to be removed as both donors must dissociate (if one dissociates the other will readily reassociate)
(more donors) increased stability with more donors
(bigger chelate) decreases stability as this effect occurs upto a point (5>6>7 members), due to furthering distance between donor atoms (less likely to coordinate together)
what coordination complex acts as an anti-cancer agent
how was this discovered and when
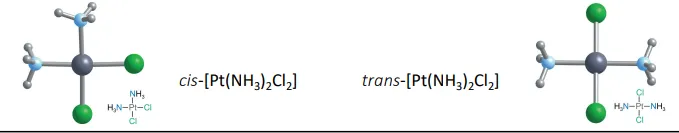
cis-platin, the square planar complex cis-[Pt(NH3)2Cl2]
discovered with Serendipity from Rosenberg (1964), who applied electricity current to EColi culture using Pt electrodes (thought to be inert) and NH4Cl medium → found cells didnt replicate, they instead grew to long filaments and didn’t reperoduce (prevented cell replication)
discovered to be the Pt complex causing this, rather than the electricity
thus found to be active against cancerous tumours in 1969 - a usually toxic metal, but helpful under small doses (high activity, specifity to target Purine DNA bases)
still today, only 3 on the market
oral administration (easy), administered with other drugs to target DNA replication & cell machinery, large doses spread out, may take months - years
![<ul><li><p>cis-platin, the square planar complex cis-[Pt(NH3)2Cl2]</p></li><li><p>discovered with Serendipity from Rosenberg (1964), who applied electricity current to EColi culture using Pt electrodes (thought to be inert) and NH4Cl medium → found cells didnt replicate, they instead grew to long filaments and didn’t reperoduce (prevented cell replication)</p></li><li><p>discovered to be the Pt complex causing this, rather than the electricity</p></li><li><p>thus found to be active against cancerous tumours in 1969 - a usually toxic metal, but helpful under small doses (high activity, specifity to target Purine DNA bases)</p></li><li><p>still today, only 3 on the market </p></li><li><p>oral administration (easy), administered with other drugs to target DNA replication & cell machinery, large doses spread out, may take months - years</p></li></ul><p></p>](https://knowt-user-attachments.s3.amazonaws.com/20be8f54-7d33-4bff-b7c6-cc1f109c3d26.png)
what are the chemical requirements for anti-cancer activity in cis-platin (and derived) drugs
active metal of Pt(II) - important as they have slow ligand exchange so are kinetically inert (~hours to react)
must be cis
must have X group of a good LG (e.g. Cl-, H2O)
must have 2 non LG (of R,R’NH - ammine group, with atleast 1 proton)
must be neutral (allowing passage across membrane)
must be water soluble (for use inside the cell)
= the gaps in these requirements are where things were changed to develop additional forms, to try minimise toxicity and side effects, and maximise efficiency
how does cis-platin interact with the body
easily passes into replicating cells only (readily allowing nutrients in for divison) be it cancer or not (cancer has uncontrolled cell replication, so targetting replications stops cancer growing - however stops other dividing regions like hair follicles hair / sperm production) - then passes through nucleus ion channels
the body detects it as foreign and wants to engulf and excrete, so is administered in large doses, and slow reaciton rate ensures it gets into the cancer part hopefully
hydrolysed in various steps, to form various intermediates prevalent at various times
(interaction w DNA) after first hydrolysis, it is charged so can interact electrostatically with DNA (draws closer), and can coordinate to the N in the 7th position of Guanine (N7G) base (has lone pair - attached at one point forming monofiunctional adduct)
can hydrolyse again, lose another Cl, and interact with 2 points of attachment (bifunctional), so it can stay long enough to do its job (before machinery removal)
(disrupt DNA) along the chain will have many Pt attached, chaning its strcuture and shape → unrecognisable to DNA rep machinery (enzymes & proteins) → rep prohibited → cell dies (gets too big to survive)
distinguish intra & inter strand bonding of cis-Platin to DNA
how does Cis-Pt incur DNA shape loss
(intra strand) two points of attachment with DNA in one strand
GpG is neighboruing Guanines
ApG is neighhbouring Guanine and Adenine
GpxpG is between non adjacent Guanines, of the same strand
(inter strand) more challenging bonding with seperate DNA strands Guanines, so happens less
(shape change) binding slightly unwinds double helix → backbone is kinked → H bonds weaken / disrupt (that hold DNA bases together) → DNA is no longer parallel, quite distorted
how may tumor cells become resistant to cis-Platin
not a case of genetic resistance, is patient specific where your body becomes smart to the administeration, prepares to remove the foregin agent, may be immediate in some people or developed over time
DNA is attacked all the time, a normal process of life, so the cell has ability to attack against it
(first level) reduce passage of drug into the cell (via reducing plasma membrane channels)
(second level) increase production (upregulation) of proteins that act to denature and remove Pt from the cytoplasm, so it can be immediately attacked, inactivated, and removed
(third level) even if it gets all the way into the nucleus, enzymes are upregulated here which act to remove it from DNA and make inactive
what are the 2 exceptions of usual electron configurations, for TM?
(Copper 0)
has more stability with 3d10 & 4s1 VS 4s2 3d9
note - at other O.S this d orbital is only partialyl filled, which is why it is still considered a TM
(Chromium 0)
more stability with 3d5 & 4s1 VS 4s2 3d4
what does TM electronic configuration determine
what determines TM electronic configuration stability
determines reactivity, stability, behaviour in reactions, speed of ligand exchange, and which ligands they readily react with - so provides good insight into properties of complexes
(Stability) determined by the net effect of nucleus-electron attractions, shielding from positive nuclus, inter-electron repulsion, and exchange forces
what is the core element & configuration for the first row of TM
what orbital electrons are ionised first to form different O.S?
[Ar] 4s2 3d______
[Ar] = 1s1 2s2 2p6 3s2 3p6
the 4s shell is filled first by valence e, however when ionised, e are lost from this shell FIRST (before 3d) - to form different O.S
what O.S are most common in TM
why are the others not as comon
+3 (Early TMs) & +2 (Late TMs) are most common
(Lower O.S) not stable in aqueous solution (goes negative), but can be possible if stabilised by ligands (e.g. CO)
(Higher O.S) quite rare, less stable
how do you determine TM electronic configuration, given its O.S
why are there different O.S for metal ions
(formula) group number - O.S = number of d electrons
e.g. Cobalt III is group 9…. 9 - 3 = 6d electrons
note - 4s electrons are removed first, so if not using this formula, consider removing 4s first, plus the appropriate number of 3d electrons
(why) to interact with ligands to form complexes, they must be at a state other than purely element 0
how does metal - ligand bonding vary on a spectrum?
what type of bonding is this (pi vs sigma)
how does this bonding occur
what type of bonding is more focused for this course
(spectrum) varies from ionic / electrostatic → covalent, depending on if the two are very different EN (electrons completely donated, attraction of + / -) VS similar EN (electrons shared from ligand donor)
many are somewhere in between, an admixture around the two
therefore TM complex bonding must consider a combination between covalent AND ionic
(type of bond) this is sigma, as the LP on ligand donor atoms is donated to form a bond
(how?) occurs via overlap of d orbitals, empty metal X full ligand, OR empty ligand X full metal
(for this course) ionic / electrostatic attraction bonding in focus
what are the 2 types of electrostatic attractions / ionic bonding common in TM complexes?
(charge - charge)
seen in cation metal X anion ligand
(charge - dipole)
sigma type bonding, LP on donor creates a dipole on that atom (not fully negative - neutral ligand - polarisation of the bond), which is able to interact with the positive metal cation
describe the Octahedral Crystal Field Theory
what is this a metaphor for
a free metal ion (in gaseous isolated state, no intearctions of anything, or influence of ligand binding) - therefore 5 d orbitals of each d shell, have equal energy (5-fold degenerate)
so inputting an electron (donating an electron AKA a ligand bonding) has equal chance to fill any orbital, first occupying singly, then doubly but at opposite spin (electron placement rules)
this theory looks at directing 6 point charges to the metal, along axes (x y z) in an octahedral plane, the 6 positions defining the octahedron verticies - from a long distance, closer and closer
as they get closer, they begin to interact with orbital electrons, differeing depending on orbital nature / shape (the 90% probability of an orbital electron being found here)
different orbital electrons will feel different repulsion (uneven energy disturbance), so all energies will increase (energy to input), but some more than others (those on axis directed at the incoming points)
therefore it will cost more energy to fill certain d orbitals with electrons, no longer 5-fold degenerate
crystal splitting is the difference in energy of the orbitals
= a metaphor for ligand bonding to a metal centre (where will electrons fill first donated from ligands?)
name and distinguish the 5 d orbitals
4 have the same shape, but different orientations (dx2-y2, dxy, dxz, dyz) - the final one is shaped differently (dz2) - all have 4 lobes pointed into different axis of the xyz coordinate system - are each imposed to create the overall d orbital
(dx2-y2) 2 lobes pointing to the x plane, 2 lobes pointing to the y plane
(dxy) rotated 45 degrees from previous, orbitals now pointing BETWEEN the xy planes
(dxz) rotated 45 degrees UP from the previous, now pointing in between x & z planes
(dyz) rotated 45 degrees from previous, now pointing between y & z planes
(dz2) 2 lobes pointing to the z plane, final 2 lobes in a circle around the middle of the plane
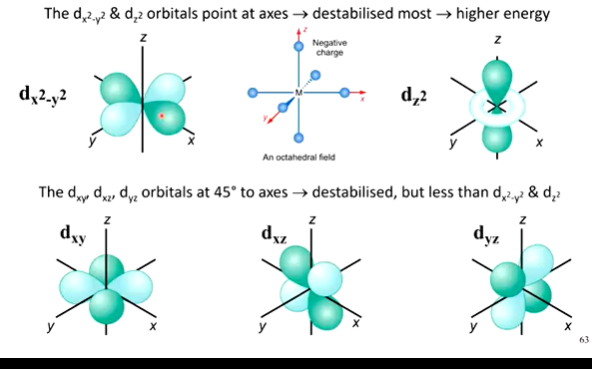
in Octahedral Crystal Field Theory, which d orbitals are higher energy
why?
what does an energy level diagram look like for this?
(dx2-y2 & dz2) higher energy (2-fold degenerate)- a
this is because they have lobes pointing directly at the axes, and in octahedral, 6 ligands come in from the 6 verticies of the octahedron - so are facing direct repulsion
therefore tehse orbitals are raised to higher energy, face more repulsion, so costs more energy to put electrons here
(dxy & dxz & dyz) lower energy (3-fold degenerate)
still are destabilised due to incoming negative charge, however are not pointing towards the axes so feel this less - are lowered in energy overall
therefore costs less energy to put electrons here
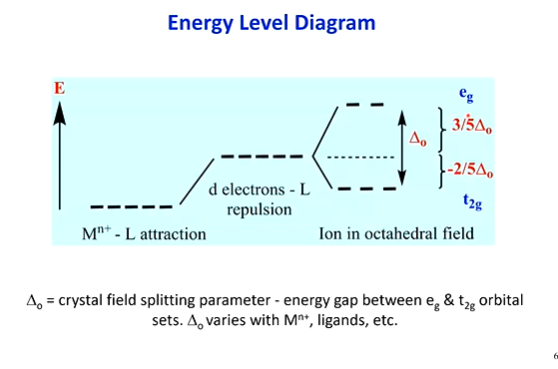
in an energy level diagram
what is deltaO
what is deltaT
what is eg
what is t2g
(deltaO)
total change in energy between lower and higher orbitals, in an Octahedral OR Square Planar EL diagram
(Octahedral) 3 orbitals lowered account for -2/5 deltaO, 2 orbitals heightened account for 3/5 deltaO
these cancel out to overall 0 energy
(deltaT)
total change in energy between higher and lower orbitals, in a Tetrahedral EL diagram
3 orbitals heightened account for 2/5 deltaT, 2 orbitals lowered account for -3/5deltaT
(eg & t2g)
for octahedral fields only, g = symmetry, so as e pass from one side of the complex through the other, they pass an inversion centre (internal mirror plane) - eg meaning doubly symmetric (for the higher 2 orbitals), t2g meaning triply symmetric (for the lower 3 orbitals)
tetrahedral fields have e & t2, but no g (no symmetric inversion), but opposite (Representing doubly vs triply)
how do added electrons fill TM d orbitals, considering Crystal Field Theory
which scenarios are high spin vs low spin configurations
not equal probability to fill from any of the 5 orbitals, first singly then doubly - as they are not 5-fold degenerate
depends on if deltaO / deltaT (energy cost of promoting electrons) VS pairing energy (similar between compounds - energy of unstability due to electron repulsion) is higher
(delta O/T < P) electrons will singly fill ALL orbitals first, then pair them, as it costs more energy to pair VS promote
(high spin) configuration with MAX number of unpaired electrons
(delta O/T > P) electrons will singly & doubly fill lower energy orbitals first, then singly & doubly fill higher energy orbitals, as it costs more energy to promote VS pair
(low spin) configuration with MAX number of paired electrons
this depends on the complex, as deltaO varies (size, ligands coordinated, ligand interacting, O.S of metal)
describe Tetrahedral Crystal Field theory
which orbitals are higher energy and why
(Theory)
similar to Octahedral, just considering 4 incoming point coordinates (4 ligands coordinating), from 4 opposite corners of a cube, with the d orbitals in xyz planes from the metal ion
this means that no d orbitals are pointing directly at incoming points, however some are pointed closer to their general direction (those between the axis) - therefore feel more disturbance / raised to higher energy
however lower overall energy & energy difference than octahedral (less splitting) due to only 4 ligands (less repulsion)
(Energy Level Diagram)
(dxy & dxz & dyz) 3-fold degenerate, higher in energy, as they point more in the general direction of the incoming charges (2/5 deltaT each) - t2 (triply), no g (not symmetric inversion) - destabilised
(dx2-y2 & dz2) 2-fold degenrate, lower in energy (-3/5 deltaT each) - e (Doubly), no g (not symmetric inversion) - stabilised
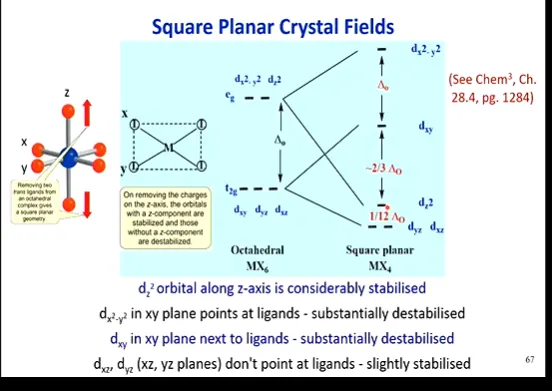
describe Square Planar Crystal Field Theory
describe the energy level diagram for this
(Theory)
4 ligand incoming point charges, from the verticies of an octahedron
however SP geometry removed 2 ligands off the z axis, so the 4 ligands bonded are in the same plane and move closer (favorable to stabilise the cation) - so greater repulsion is observed
this also means that lobes facing the z axis will be stabilised, feeling less repulsion from the incoming ligands - and those withouta z component will be destabilised
(Energy Level Diagram)
(dx2-y2) not degenerate, most destabilised (highest energy) as feels repulsion from incoming ligands AND closer 4 ligands of SP
accounts for deltaO
(dxy) not degenerate, quite destabilised (Second highest energy)
accounts for -2/3deltaO
(dz2) not degenerate, quite stabilised (Second lowest energy) as are pointing to ligand-less z axis
accounts for 1/12deltaO
(dyz & dxz) 2-fold degenerate, quite stabilised (lowest energy) as are pointing to ligand-less z axis
what is Crystal Field Stabilisation Energy (CFSE) and how is this calculated
what does this help determine
how much stabilisation is brought to the atom, by putting electrons into each orbital - so consider deltaO/T of stabilisation energy, between orbitals and the middle
(formula) no. of electrons X energies stabilised (+ pairing energy) = CFSE energies
e.g. 4 x (-2/5deltaO) (+ P) = CFSE
(purpose) helps determine how an electron configuration will be, if HS vs LS
considering Crystal Field Theory, which shape complexes are usually HS (deltaO/T < P) VS LS (delta O/T > P)
(Tetrahedral) HS - deltaT much lower than P
(Square Planar) LS - deltaSQ much higher than P
(Octahedral) HS OR LS - deltaO can vary
P stays the same between all shapes
what factors affect deltaO/T size?
(Consider ligand AND metal)
(Ligand)
type of ligand (weak field VS strong field - halides VS OH-/H2O VS amides / C-containing)
strong field interact more strongly with orbitals, creating a larger energy gap (increase deltaO) - the more covalent bonding the stronger field - the more ionic the weaker field
(Metal Ion)
charge / O.S, deltaO increases with increasing charge, as it wants to counter the charge by pulling ligands closer, creating greater electron X ligand repulsion (higher splitting energy)
element of the metal, deltaO increases down a group (d orbital size gets larger - closer association with ligands - greater repulsion - greater splitting)
what wavelengths of light is visible light?
how does light make colour?
visible light = 400-700 nm (below = ultraviolet, above = beyond red)
(colour) objects are coloured because they reflect / transmit visible light wavelengths corresponding to that colour (e.g. absorb all, except reflecting green) OR absorb the complementary / opposite colour (so appears the opposite colour as the other one is absorbed)
visible light is simply white light, all the colours mixed together, so when objects start absorbing different wavelengths, it is no longer white, colours are blocked out and therefore mean other colours are shown
so different colours relate to different wavelengths of light absorbed - just remeber the complementary colours (which absorbed, produces which observed)
how do electrons in TM complexes create colour, to make coloured solutions?
complexes can undergo electronic transitions (d electron excited to anothe d orbital) by absorbing light - whose wavelength / energy of the photon matches deltaO, allowing this photon to be absorbed to provide the energy to promote the electron to another d orbital
the wavelength of this photon absorbed, corresponds to which colour light is absorbed (if in the visible range) - so therefore which colour solution is produced (the complementary - nonabsorbed)
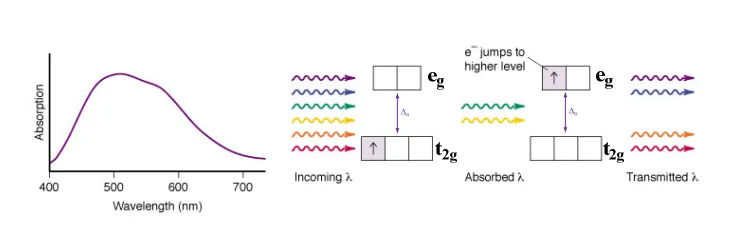
what do TM complex absorbance spectra look like - and why?
create absorbption peaks at certain wavelengths on the spectra, at the wavelengths absorbed most often by electrons
however we see a range of wavelengths absorbed (not just one peak matching deltaO), as bonds are vibrating (Decreasing & increasing in length), the complexes arent static, so at every different arrangement there are slightly different deltaO (ligands further vs closer, less vs more repulsion), so slightly differenet wavelengths of light are absorbed
to sharpen the spectra cool down the solution (lessens vibrations)
as we usualy have multiple d electrons, spectra usually have various wavelength peaks (visible d-d bands), depending on orbital splitting
helps distinguish how many d electrons in a complex , strength of deltaO (if wavelength peaks are far apart) - and therefore HS vs LS complexes
how can a solution’s absorbance be used to find concentration - and vice versa?
via Beer’s Law

name and define the 2 forms of magnetism
(dimagnetism)
a property of all elelements and compounds
caused by presence of electron pairs, in closed shells (e.g. pairs in a P shell orbital)
when a magnetic field is applied, this causes production of a small magnetic moment in the opposite direction to the field - substance is repelled by the field
degree of repulsion is proportional to the size of the field
(paramagnetism)
a property of TM complexes
caused by presence of unpaired electrons in orbitals - much greater in magnetude than dimagnetism
when a magnetic field is applied, the substance is attracted to the field - will produce a magentic moment in the direction towards the field
the size of attraction, is proportional to the number of unpaired electrons in the complex
in a compound with both, it will do paramagnetism
therefore can use to distinguish HS vs LS complexes (HS = greater attraction)
what are magnetic moments
how are these calculated for TM complexes
what do these tell us
(magentic moment)
measure of the strength of the magnetism of an electron, a consequence of the spin quantum number, causing it to possess a local permanent magnetic moment (acts as a tiny magnet)
this results in it trying to align up with the direction of a field
this course considers Spin-Only magnetic moments only (beyond this course to consider orbital motion of electrons - have an affect on magnetic moment)
(calculating)
see attached formula - for calculating expected magnetic moment (for a complex with known number of unpaired electrons)
can also work backwards to find number of unpaired electrons
expected moment will differ from tabulated data (as we consider Spin-Only) - but roughly the same
(purpose)
allows us to distinguish HS & LS complexes, as it tells us the number of unpaired electrons, which we can input into the Energy Level Diagram to determine the configuration
therefore tells us if deltaO > OR < P, AND helps us create Splitting diagrams
helps us distinguish shapes of complexes, depending on HS vs LS & no. of unpaired electrons => due to the usual configurations of shapes
what is the idea between electrochemical cells
what are they also known as
who created them
the idea of carrying out redox reactions, but with spatial seperation of the pairs, being linked by only electrical connection, to allow the passage of electrons via the current
this is what occurs in a battery - a spontaneous redox reaction generating a potential difference → generating an electric current in an external circuit
AKA Galvanic cell AKA Voltaic cell
created by Galvani & Volta
what two components of reaction are needed for Galvanic / Electrochemical cells
what else is required for these cells to enable reaction
(redox pairs)
are redox reactions, so require a reductant (is oxidised - increases O.S - releases electrons) & require an oxidant (is reduced - decreases O.S - accepts electrons)
reduction occurs at the cathode (Red Cat) & oxidation occurs at the anode (An Ox) - both required for reaction
(external circuit)
this wiring must connect electrodes, to enable flow of electrons, to enable transfer of electrons between redox pair (enable reaction to occur)
(salt bridge)
a tube connecting the two half-cells, with inert ions (often immobilised in gel / aqueous solution) that can pass through
these inert ions offset the accumulation of charge on the half cells, as electrons are lost / gained and anions / cations are produced - as these inert ions move towards the respective side
these move into the half cells, but are inert so dont react
this bridge must prevent mixing of the redox pairs themselves (otherwise direct reaction will occur and no current will generate)
e.g. KCl, NH4NO3, KNO3
how do electrochemical cells generate an electrical current
how is this measured, and what symbol is related - and its unit
these reactions involve electron transfer (oxidation releases electrons → reduction gains these electrons), which generates a current and electrical work is performed and can be utilised (chemical energy → electric work)
the Cathode is the positive electrode - electrons flow towards it while ions are los & the Anode is the negative electrode - electrons flow away from it as ions are gained
the spontaneous flow of electrons occur due to their electric potential difference (imagine a waterfall - the larger the difference the more spontaneous), flowing from high → low potential energy
this creates electric potential, the potential of energy dervied from the process that can be used to do work
the difference in potetial energy between cells (Cell potential / reduction potential => the likelihood to react spontaneously), is denoted E (in volts), for standard state (1M conc, 298K T) = E0
describe the Daniel Cell example
a model electrochemical cell of the typical process (spontaneous redox)
Zn(s) + Cu2+(aq) → Zn2+(aq) + Cu(s)
Zn solid is oxidised (electrons lost) as this anode dissolves (in ZnSO4 solution) to create ions, and electrons flow through the wire
Cu2+ ions (in CuSO4 solution) are reduced (electrons gained via the flow from Zn) as ions are lost as they precipitate onto the cathode (Cu(s))
so mass of Anode would decrease, mass of Cathode would increase
E0 = 1.10 volts
what is the conventional notation for electrochemical cells
(Anode) written on left, with a -
(Cathode) written on right, with a +
(Order for Half Cell species) write in order they appear in reaction
(Phases) denote each, then use a single vertical line between phase changes of the same species
(Salt Bridge) denote with a double vertical line seperating the 2 redox pairs
(Inert Electrode / Catalyst) seperated with another single vertical line, on respective Cathode / Anode side
what are the 3 methods of electrodes used for Electrochemical cells
describe each
(Active Electrodes)
where the electrode material itself enters the reaction, so the metal solid is immersed in the corresponding metal ion solution
(e.g. metal solid → metal ion / metal ion → metal solid)
e.g. Zn(aq) Zn(s), Cu(aq) Cu(s)
(Gas Electrodes)
redox process involves a gas, so reactant gas is bubbled over the surface of an inert metal electrode (e.g. Pt), in simultaneous contact with the gas & the electrolyte solution (typically acidic), with this electrode surface acting to catalyse the redox process
e.g. H3O+(aq) H2(g), Cl2(g) Cl-(aq)
(Inert Electrodes)
both redox forms are aqueous in solution, while an inert metal surface (e.g. Pt) is in contact with a ‘mixed’ solution of redox pairs
e.g. Fe3+ Fe2+, MnO4- Mn2+
what 3 factors influence E (reduction potential / cell potential)
nature of half-cells
concentrations / activities of electrolyte species present
temperature of the cell
considering these allows us to calculate E0 (Standard reduction potential)
how do the nature of half cells influencce E (cell potential / reduction potential)
how are these standardised
how is this helpful
(influence)
different redox pairs react in different ways, changing E
(standardisation)
more efficient than ranking all electrodes in all possible cells
done using the H electrode (H2 gas, H3O+ solution, Pt inert electrode half cell - can be either Anodic / Cathodic (depending on pair)) = E0 = 0
all other redox half-cells were reacted against this, providing their standard E values (E0 electrode), for REDUCTION potentials - simply reverse to find oxidation potentials
high positives = high tendency to be reduced (act as cathode), low negatives = low tendency to be reduced = tendency to be oxidised (act as anode) => so whichever is higher, will proceed as cathode in the reaction
changing reaction stoichometries does NOT change E0
(helpful)
this allows us to find E0cell = E0cathode - E0anode
what is the ‘diagonal rule’ for redox electrochemical cells?
why is this helpful?
any species on the LEFT of a given half-cell reaction, will react spontaneously with a species appearing on the RIGHT of any half-cell located below it in the tabulated data of Reduction Potentials
therefore the higher RedPot half-cell will be reduced (occur forwards), and the lower RedPot half-cell will be oxidised (occur backwards)
what sign E0 cell value describes spontaneous reactions
positive E0 cell values denote spontaneous reactions
E cell values of 0 mean it is at equilibrium
negative E0 cell values denote non-spontaneous reactions, and require energy input
what rules must you remember for balancing redox equations
(half-cells)
balance non O / H species
balance O by adding H2O to the other side
balance H by adding H+ to the other side
balance charge by adding electrons
(full cells)
multiply stoichometry of half reactions so that electron numbers match
what is the Nernst Equation
what are the components that make it up
what does it allow us to do
(Equation)
Ecell = E0cell - RT/nF lnQ
Ecell is reduction potential (E) at non-standard conditions
E0cell is redpot (E) at standard conditions
R is the gas constant, T is temperature (in K)
n is number of electrons transferred between electrodes, F is the Faraday Constant (for scaling up moles of electrons)
Q is the reaction quiotent (products / reactants conc)
(Purpose)
it allows us to calculate reduction potential (E) at non-standard conditions for any given cell (any set of concentrations, any T)
higher vs lower than E0cell, means it is more or less spontaneous under the new conditions
it also allows us to calculate deltaG0 (deltaG0 = -RT lnK = -nFE0)
it also allows us to calculate K (E0cell = RT/nF lnK)

how does deltaG relate to the work done by a system
how does the sign of deltaG relate to the sign of E and the spontaneity of an electrochemical cell reaction?
(deltaG and work)
the total work a system can do is -deltaG0 (so -deltaG0 = nFE0cell)
(signs of E & deltaG & spontaneity therefore)
-deltaG denotes a spontaneous reaction (opposite to S)
in the above equation to retain -deltaG, (F positive, n negative - by definition) so E0cell must also be positive
therefore a positive E0cell denotes a spontaneous reaction