Red Blood Cells Clinical Biomedicine
1/57
Earn XP
Description and Tags
Review of Red blood cells, haemoglobin and anemia
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
58 Terms
What is the size of RBCs
–Mean diameter 7μ,
–mean volume 78 – 101fl
What percentage of rbc is Hb
25% by volume, but 33% by weight
What is useful about the biconcave shape of red blood cells
Allows pliability as some rbc is bigger then capillaries allows it to squeeze, also increases surface area for gaseous exchange
How far will a rbc travel and how long is it’s lifespan
300miles roughly 170,000 circuits through the heart and they last about 120 days in a healthy individual
What are the % breakdown of a rbc membrane
40% lipids, 52% protein and 8% carb
Roughly what is the red cell cytoskeleton
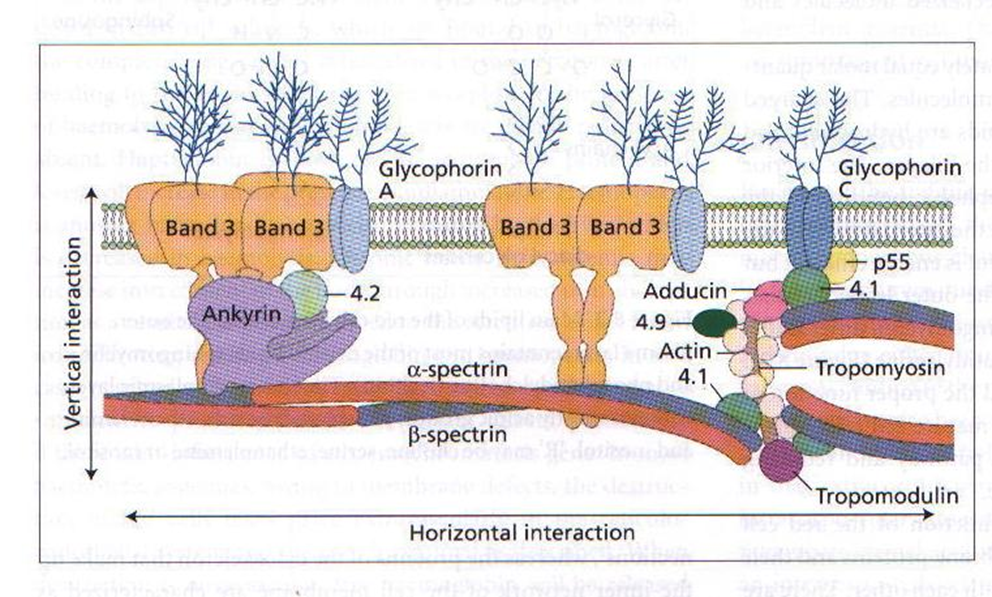
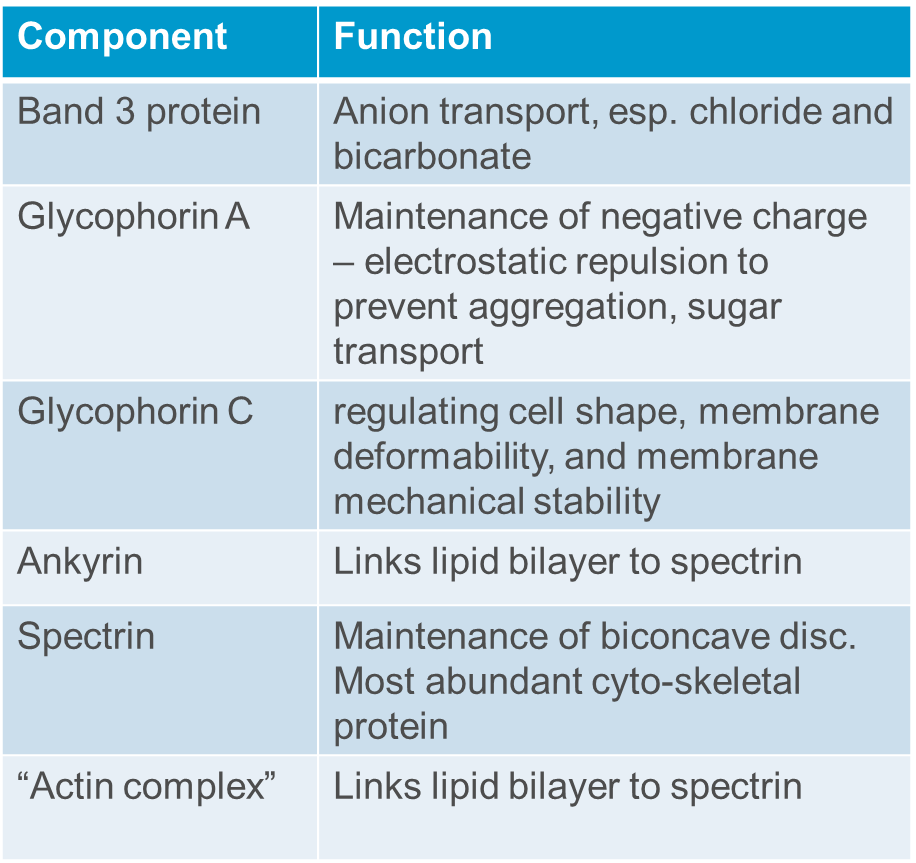
Describe the metabolic activity of a red blood cell
Anaerobic glycolysis to avoid oxidation of iron which creates an inoperative oxygen carrier. The ATP produced maintain membrane deformability and ion/water exchange. Glycolysis results in production of 2,3 DPG
In a healthy individual where are rbc made and how many are destroyed and replaced daily
In an adult, is usually confined to marrow spaces of sternum, pelvis and long bones
Approx 1011 RBCs are destroyed and replaced per day (nearly 1 million every second)
RBC production can be uprated by 10X
1mm3 blood normally contains 3.5 - 5.5 x106 RBCs
What are two types of immature rbcs
Nucleated RBCs or normoblasts
Reticulocytes, which aren’t nucleated but do have some organelles still present usually 1-2 days into ciruclation
What drives RBC production
EPO(erythropoietin), a hormone produced primarily by the kidneys in response to low oxygen levels.
How are RBC nuclei removed
They are extruded and phagocytosed by macrophages,
Which receptor allows a developing rbc get iron
CD71
What is haemolysis
A shortening of the RBC lifespan = haemolysis. (May be compensated by increased production, so not necessarily anaemic – “compensated haemolysis”)
Whats the Spleen job
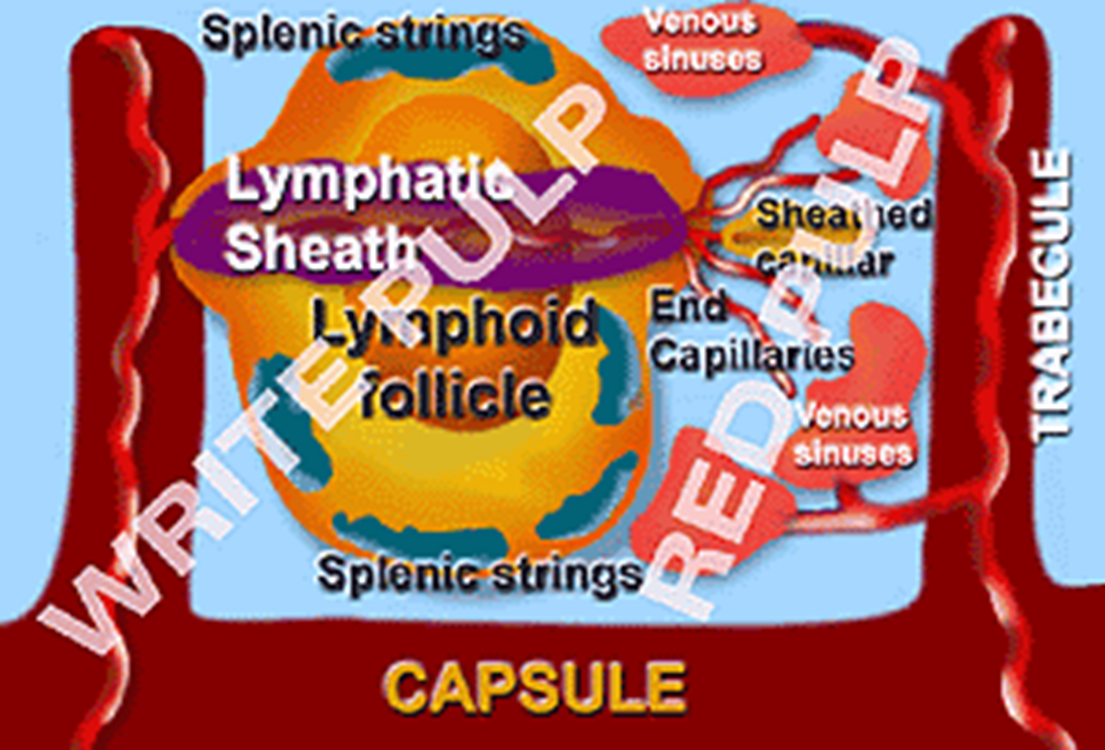
Discriminating filter that pluck out granular debris and defective membrane, and destroy senescent cells. Macrophages line the cords which filter into the splenic sinuses.
What is removed by macrophages in the spleen AND what feature makes this process more effective
nuclear material (Howell-Jolly bodies)
cytoplasmic organelles,
Siderotic (= iron containing) granules (Pappenheimer bodies),
oxidised Hb (methaemoglobin, i.e. Heinz bodies)
Reticulocytes are sticky which possibly slow the passage to allow greater time for the macrophages to work
How is oxygen transported from the lungs to respiring tissues
O2 is reversibly bound to a haem group (= an Fe atom in a porphyrin ring) – a physical, not chemical interaction
Fe atom remains in reduced form. (Fe3+ = non-functional methaemoglobin, MetHb).
How is CO2 transported
RBCs associated with 95% of total CO2 transport
25% bind directly to Hb while 70% is converted to bicarbonate ions (HCO3-) via carbonic anhydrase. The remaining 5% is dissolved in plasma.
How does Hb act as a buffer
Hydrogen ions left from dissociation of carbonic acid to bicarbonate bind to globin chains of Hb, preventing acidosis
What is the difference between oxygenation and oxidation
Oxygenation is the PHSYICAL binding of oxygen to haemoglobin whereas oxidation is the CHEMICAL binding which is incompatible for oxygen transport
What is the structure of Haemoglobin
Tetrameric molecule about 68kDa, 4 globin chains of two types in each Hb molecules.
3 different variations of Hb
Adult Hb (HbA) = α2ß2 Hb A2 = α2δ2 Fetal Hb (HbF) = α2γ2
Each globin chain surrounds a Haem group which comprises of 1 atom Fe & porphyrin ring C34H32FeN4O4 so 4 haem groups in each Hb molecule
Describe oxygen binding with reference to the oxygen dissociation curve
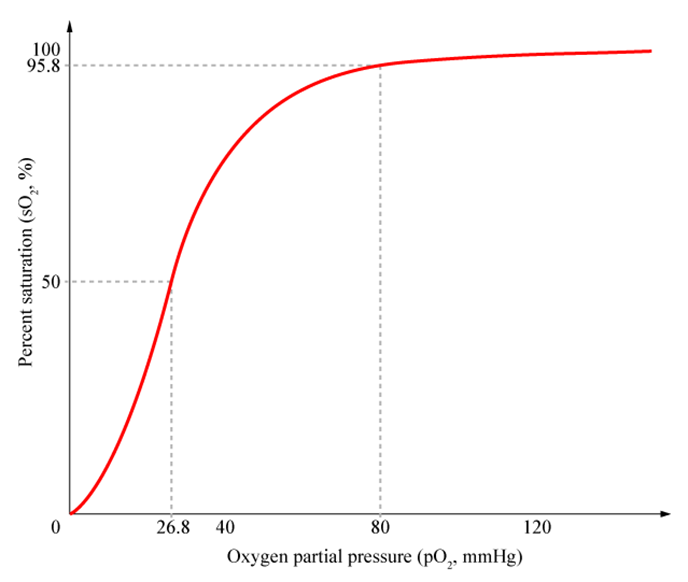
Binding of first oxygen is relatively difficult but after binding there’s a conformational change making the 2nd and 3rd binding a lot easier.
Relatively high affinity of fully oxygenated Hb prevents arterial deoxygenation but after first oxygen molecule is given up the next two are more easily released
What factors affect oxygen affinity
Confirmational change due to binding.
2,3 diphosphoglycerate or 2,3DPG promoting deoxygenation
Acidosis induces Bohr effect
Fetal Hb has increased O2 affinity due to less acitve binding to 2,3 DPG therefore reducing deoxygenation
Potential advantages of artificial RBCs
Storage temperature and shelf-life
Immediate, universal administration
No lag in effectiveness as with natural blood (due to 2,3-DPG and nitric oxide depletion during storage)
No risk of disease transmission
Not dependant on donors
Avoid religious / cultural issues, e.g. Jehovah’s Witnesses
Why can’t Hb be transfusedin solution
Free globin chains are toxic to kidneys, scavenge nitrous oxide leading to vasoconstriction and hypertension, short half life
What are some examples of artificial red blood cells
Re-manufactured” Hb (human or animal) – polymerised bovine globin chains, e.g. Hemopure (licensed in S. Africa in 2001).
Conjugated Hb (e.g. to polyethylene glycol) (Hemospan)
Artificial / bioengineered cells (e.g. Yeasts, E.coli, lipososmes - “neohemocytes”) containing Hb
Blood “pharming” – in vitro differentiation and growth of stem cells into RBCs. First clinical trial Nov 2022 (UK)
What is Anaemia
Reduced Hb in blood
What are the symptoms of anaemia and when does it typically display symptoms
In general appear when [Hb] falls below 90 – 100 g/l
• Slow-onset anaemia - much better tolerated than rapid
(as low as 60 g/l in an otherwise well, young patient)
• Shortness of breath
• Weakness
• Pallor
• Lethargy
• Palpitations
• Headaches
• Heart failure and confusion (older patients)
The clinical signs are pallor of mucous membranes and nails bed, depending on the anaemia, concave nails, jaundice, leg ulcers, bone deformities and recurrent infection could occur
How is anaemia classified
2 Parameters: Size (Microcytic, normocytic and macrocytic) and Hb concentration (Hypochromic, normochromic, hyperchromic)
What are the regular parameters for MCHC, MCH and MCV
mcv - 80-101fl
MCHC 300-350 g/l
MCV 27 - 34pg
What is normocytic, normochromic anaemia
MCV normal, MCHC normal, MCH normal but RBC count is reduced. This could be due to an acute bleed, marrow failure, haemolysis or renal failure
What is microcytic, hypochromic anaemia
RBCs reduced in volume and have less Hb. MCV, MCH and MCHC all reduced
Iron deficiency, thalassemia, anaemia of chronic disorder
Macrocytic, normochromic anaemia
RBCs increased in volume Hb at normal conc, RBC count low
MCV increased
MCHC is normal
MCH increased
B12 or folate deficiency
What levels or blood loss cause issues
1L-1.5L ok if lying or sitting but standing may be hard
1.5L - 2L loss, variable loss of consciousness, SOB and sweating
>2L severe shock possibly irreversible and potentially death
Does anaemia set in straight away after an acute bleed
No, as blood cell and fluid are both lost. It may take 2-3 days post-bleed before it becomes anaemia. Reticulocytes takes 3-5 days to respond
What does iron deficiency anaemia look like

Small, pale Red blood cells with some being miss-shapen
How do you treat IDA
Oral administration e.g. ferrous sulphate tablets or parenteral (injected)
Should rise about 10g/l per week and blood transfusion in severe cases
How does B12 or folate deficiency display in blood cells
Macrocytic, normochromic with oval macrocytes and hype segmented neutrophils, where the nucleus is split into clusters
What causes hereditary elliptocytosis
(Spectrin, Glycophorin C or protein 4.1 disorder)
Incidence 1in 3 - 4,000
A splenectomy may be required
This is normocytic and normochromic
What is hereditary spherocytosis
Incidence 1 in 2,000 in North Europeans (but much less common elsewhere)
Spectrin, Ankyrin, band 3 or protein 4.1 disorder
Many are clinically silent
Considerable genetic
heterogeneity
Increased osmotic fragility
Splenectomy may be required
How can a patient get acquired impairment of erythropoiesis
Bone marrow infiltration - Replacement of erythropoietic tissue by tumour usually from prostate, breast, leukemia or fibrotic tissues
Transient failure from parvovirus or drugs like cytotoxic drugs
How does anaemia due to bone marrow infiltration present
Tear-drop poikilocytes
If NRBCs and immature WBCs are also present, is suggestive of marrow infiltration leading to extra-medullary haemopoesis
What is extra-medullary haemopoesis
Usually associated with severe anaemia due to bone marrow infiltration or fibrosis
haemopoesis outside the bone marrow.
Indicated by:
–Tear-drop poikilocytes
–NRBCs
–Immature WBCs
What are two inherited impairment of erythropoiesis disorders
Fanconi Anaemia. 1 in 160,000 but strong racial association. Progressive BM failure causing death from haemorrhage or infection. Increased risk of solid tumours
Diamond Blackfan anaemia 7 in 1,000,000. Presents in early infancy decrease in erythroid precursors
What are some causes of AIHA
Causes of AIHA:
50% are idiopathic
Lymphoproliferative disorders
Mycoplasma and EBV infection
Drug-induced, e.g. Penicillin
Other autoimmune diseases
What is microangiopathic anaemia
RBC fragmentation within the circulation commonly associated with mechanical heart valves. DIC, HUS, TTP
You can see RBC fragments (schistocytes)
What does a pyruvate kinase deficiency cause
Haemolysis through failure of glycolytic pathway and inadequate ATP you can see Burr Cells produced by cellular dehydration
What does a G6PD deficiency mean
Hb oxidises to MetHb
Challenge by oxidising agent (fava beans, some malaria medication) results in significant haemolysis
Spleen then removes chunks of cells creating keratocytes or bite cells
What are the 2 categories of haemoglobinopathy
Structural variations (>800), where Hb is made in essentially normal amounts but its structure is abnormal
The thalassemia syndromes, in which there is a variable loss of ability to produce a particular type of globin chain.
What are some structural variations of Hb
1.HbS (Sickle cell) α2ß2 6 Glu → Val
2.HbD Punjab α2ß2 121 Glu→Gln
3.HbE α2ß2 26 Glu→Lys
4.HbC α2ß2 6 Glu → Lys
Alpha
HbG Philadelphia α268Asp→Lysß2
Hb Reading α1 or α2 48Leu→Pro ß2

What are the clinical significance of heterozygote and homozygote expression of Beta chain variation
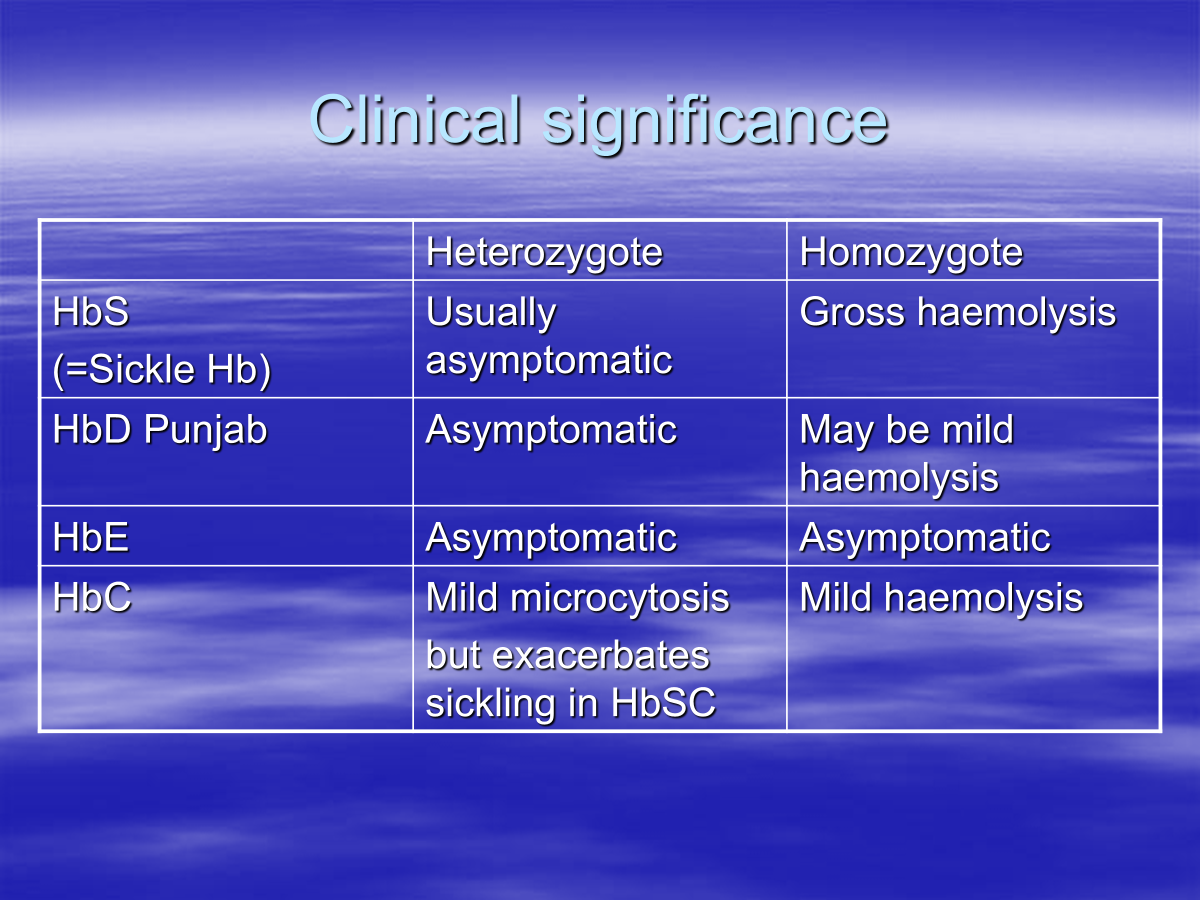
What is sickle cell disease
HbS is insoluble in deoxy state and forms tactoids distorting RBCs
This can trigger violent haemolytic episodes which causes clotting
How is the frequency of HbS gene maintained in malarial areas
In a balanced polymorphism:
–Heterozygote is more fit than either of the homozygotes
–Hb AA → P. Falciparum (malaria).
–Hb SS → life-limiting haematological disorder
Benefit to heterozygotes is at the expense of homozygotes
–HbS heterozygote has some resistance to Plasmodium falciparum whilst not usually sickling
What are some causes of unstable Hbs
1) abnormality of heme pocket, so heme is not firmly bound, and water can enter > metHb
2) Interference in binding of α & β chains
3) Interference with α chain structure
Most cases are new mutations with normal parents
What does unstable Hbs cause
Oxidation of heme precipitates and damages the cell membrane the precipitates are Heinz bodies and these are removed by macrophages
What is Thalassemia
A group of inherited RBC disorders characterised by reduced globin chain synthesis (α or β)
Generally prevalent in populations that evolved in warm, humid areas where malaria was endemic, but now affects all races. (Thalassemias provide varying resistance to malaria)

What is α-thalassemia
α-thalassemia – impaired ability to synthesise
α globin chains
→ excess β chains → β tetramers = Hb H
(in fetus, excess γ chains, = Hb Barts )
β tetramers (HbH bodies) aggregate, can be stained by e.g. brilliant cresyl blue
usually a mild, microcytic hypochromic anaemia.

What is β -thalassemia
β -thalassemia – impaired ability to synthesise
β globin chains.
Mutations in HBB gene on chromosome 11.
Many mutations identified
βo mutations allow no β chain production, β+ allow some
Excess α-chains bind to RBC membrane, damaging it → ineffective erythropoesis & reduced RBC survival
Reduction in available β-chains causes an increase in production of γ and δ chains, so get increased production of HbF (α2γ2) and HbA2 (α2δ2)
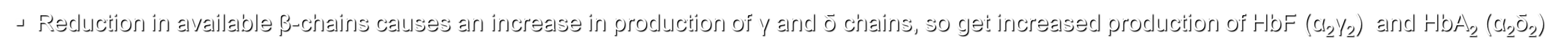
What is Casgevy
CasgevyTM is a gene therapy in which the regulator gene that normally stops HbF production at birth (BCL11A) is knocked out using CRISPR from the patient’s (harvested) stem cells.
The modified stem cells are re-infused and re-colonise in the (prepared) bone marrow, producing HbF-containing RBCs. (As HbF has γ, not β chains it provides viable RBC function and lifespan).
Approved by UK Medicines Regulator. Cost US$2million per patient.
