org chem reactions
1/47
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced |
|---|
No study sessions yet.
48 Terms
reactions for alkanes
combustion
free radical substitution
combustion
complete combustion occurs in excess oxygen and heat.
alkanes: CxHy + (x+y/4) O2 → xCO2 + y/2 H2O
alcohols: CxHyOz + (x+y/4-z/2)O2 → x(CO2) + (y/2)H2O
incomplete combustion in limited oxygen. produces C (soot), CO and CO2
alkanes vs alcohols
alcohol produces less energy: lower standard enthalpy of combustion
therefore gasoline more reliable
free radical substitution reagents and conditions
alkane + halogen → halogenoalkane
UV light/high temperature 250-400C
free radical substitution mechanism
initiation: homolytic fission of Cl-Cl bond → forms chlorine radicals
propagation: radicals consumed + regenerated
Cl• consumed as it removes H atom from molecule to produce H3C•, which reacts with another Cl2 molecule to produce CH3Cl and regenerate another Cl• and so on.
CH3Cl can undergo further substitution in the presence of excess Cl2 → to avoid, limit concentration of Cl
unable control which product forms, will always produce a mixture
termination: 2 radicals consumed (chain termination steps)
as reaction progresses, conc of molecules decrease, conc of radicals increase, probability of radical consuming itself increases
reactions for alkenes
reduction
electrophilic addition
addition of halogen
addition of hydrogen halide
addition of inter-halogen compounds
addition of water (hydration)
reduction of alkenes reagents and conditions
alkene + H2 (g) → alkane
Ni catalyst, heat 150C OR Pt/Pd catalyst, room temp
electrophilic addition mechanism
pi electron cloud polarises molecule
d+ electrophile attacks pi electrons in C=C, formation of sigma bond and carbocation intermediate
electron-rich d- donates electron pair to electron-deficient carbocation, forms sigma bond to form stable addition product
explanation for positive inductive effect (electron donating inductive effect)
“tertiary carbocation has more alkyl groups than primary carbocation, exerts stronger positive inductive effect, disperses charge to larger extent so more stable, formed more readily.”
no. of R groups/size of R groups increases, stronger electron donating inductive effect
no. of R groups is more significant
helps disperse positive charge to a larger extent
tertiary carbocation more stable, formed more readily
electrophilic addition of halogen to alkene reagents and conditions
alkene + halogen → dihalogenoalkane
CCl4 solvent, room temp
used to distinguish alkanes and alkenes. alkene decolourises red-brown bromine.
electrophilic addition of hydrogen halide to alkene reagents and conditions
alkene + hydrogen halide → halogenoalkane
dry HBr (g) , room temp
electrophilic addition of inter-halogen compounds to alkene reagents and conditions
alkene + BrCl → dihalogenoalkane
liquid BrCl, rtp
electrophilic addition of water to alkene (hydration) reagents and conditions
alkene + H2O → alcohol
lab: cold conc H2SO4 catalyst, followed by boiling with H2O
industrial: H2O (g), heat 300C, pressure 60-70atm, conc H3PO4
reactions for arenes (aromatic compounds)
electrophilic substitution: addition of nitrogen
reduction of nitrobenzene
nitration of benzene (electrophilic substitution) reagents and conditions
benzene + conc HNO3 → nitrobenzene
conc H2SO4 catalyst, 50C
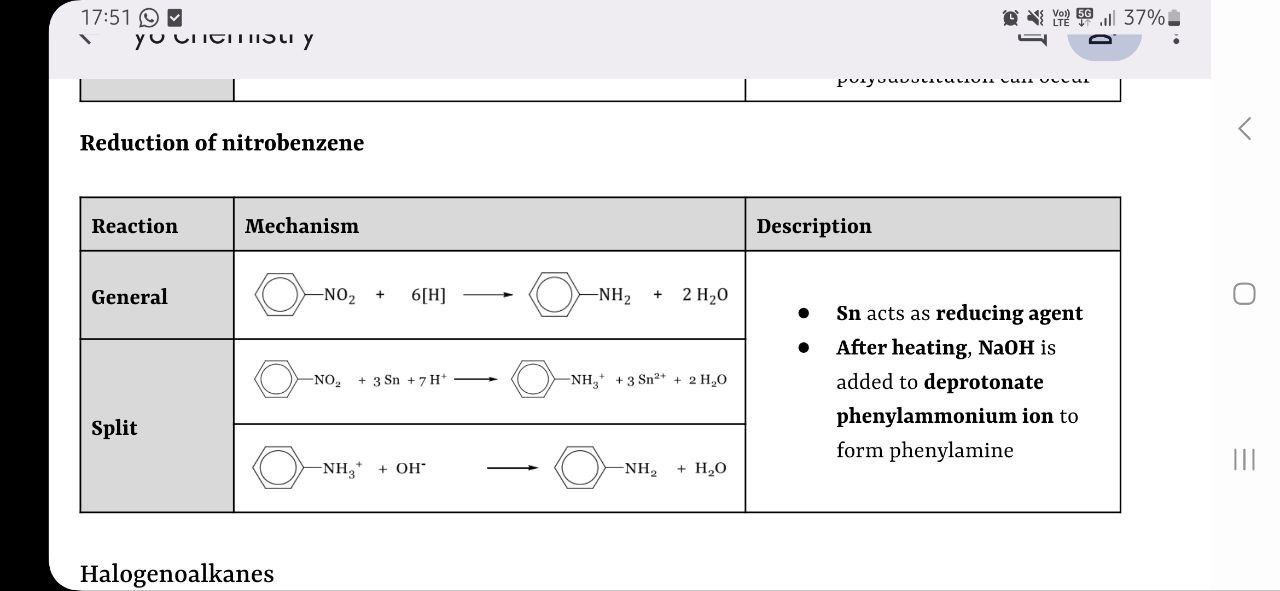
reduction of nitrobenzene
nitrobenzene → phenylamine
Sn (reducing agent) in conc HCl, heat under reflux followed by NaOH (aq) to neutralise (deprotonate the ion)
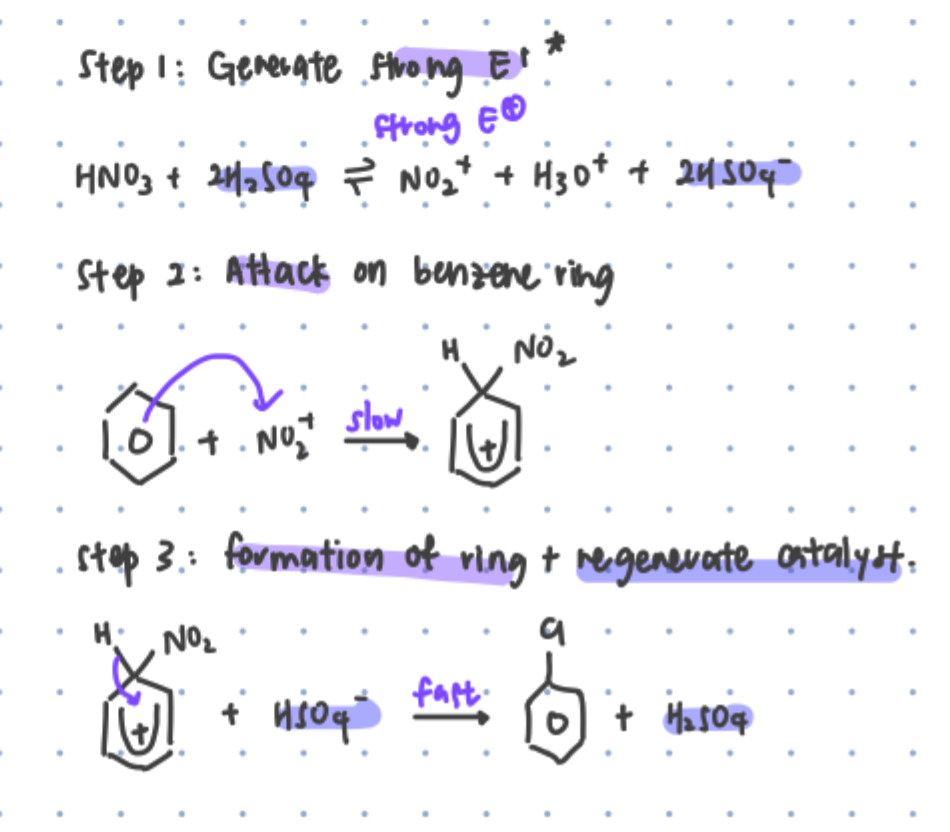
electrophilic substitution mechanism
generate positively-charged electrophile
H2SO4 catalyst protonates hydroxyl group in HNO3
N O bond breaks, forms H2O and NO2+
overall HNO3 + 2H2SO4 <=> NO2+ + H3O+ + 2HSO4-
benzene has high electron density, attacks electrophile NO2+ to form non-aromatic carbocation intermediate (SLOW STEP)
H-benzene bond attacks benzene carbocation to restore benzene ring’s delocalised pi electron ring system. lone pair from HSO4- attacks H, regenerate catalyst H2SO4
notes:
benzene resonance structure makes it stable → prefers substitution, doesn’t undergo addition
high activation energy since need to break resonance structure → needs lewis acid catalyst to generate strong electrophile
reactions for halogenoalkanes
nucleophilic substitution
with OH- → alcohol
with NH3 → amine
with CN- → nitrile
nucleophilic substitution reagents and conditions
formation of alcohol from halogenoalkane
halogenoalkane + OH- → alcohol + halogen anion
NaOH (aq), heat under reflux
MUST WRITE AQ
formation of amine from NH3
halogenoalkane + NH3 → amine + salt (eg NH4Cl)
ethanol, heat
formation of nitrile from CN-
halogenoalkane + KCN/NaCN → nitrile + salt (eg NaCl)
ethanol, heat under reflux
role of OH- ion in nucleophilic substitution
OH- is nucleophile, electron pair donor
attacks electron-deficient end of polar covalent bond (SN2) / attacks carbocation intermediate at positive carbon (SN1)
nucleophilic substitution SN1 mechanism
tertiary halogenoalkanes (tetrahedral)
polarised C-Br bond undergoes heterolytic fission → forms Br- and trigonal planar carbocation (slow step)
lone pair on nucleophile attacks carbocation, forms bond (fast step)
equal probability for nucleophile to attack from top/bottom of trigonal planar carbocation (to become tetrahedral again). therefore product is a racemic mixture of pair of enantiomers.
2-step reaction, rate = k[halogenoalkane], overall order of reaction 1
![<ul><li><p>tertiary halogenoalkanes (tetrahedral)</p><ol><li><p>polarised C-Br bond undergoes heterolytic fission → forms Br<sup>-</sup> and trigonal planar carbocation (slow step)</p></li><li><p>lone pair on nucleophile attacks carbocation, forms bond (fast step) </p></li></ol></li><li><p>equal probability for nucleophile to attack from top/bottom of trigonal planar carbocation (to become tetrahedral again). therefore product is a racemic mixture of pair of enantiomers.</p></li><li><p>2-step reaction, rate = k[halogenoalkane], overall order of reaction 1</p></li></ul><p></p>](https://knowt-user-attachments.s3.amazonaws.com/d309cddb-e29e-48f8-955e-6ebb7c3e20cd.png)
nucleophilic substitution SN2 mechanism
primary halogenoalkanes
nucleophile backside attack d+ C atom of C-Br bond
nucleophile attacks from back side because electron density mostly on halogen side
electrons from nucleophile repel d- halogen, lengthens C-Br bond
forms 5-membered trigonal bipyramidal transition state (negatively charged)
in transition state, both nucleophile and halogen are partially bonded to same C atom (represented by dotted lines)
note: transition state, not intermediate since only 1 step
concerted: nucleophile backside attack + halogen leaving group at same time, therefore 1 step reaction
1-step reaction, rate = k[halogenoalkane][nucleophile], overall order of reaction 2
DO NOT WRITE SLOW/FAST
![<ul><li><p>primary halogenoalkanes</p></li><li><p>nucleophile backside attack d+ C atom of C-Br bond</p><ul><li><p>nucleophile attacks from back side because electron density mostly on halogen side</p></li><li><p>electrons from nucleophile repel d- halogen, lengthens C-Br bond</p></li></ul></li><li><p>forms 5-membered trigonal bipyramidal transition state (negatively charged)</p><ul><li><p>in transition state, both nucleophile and halogen are partially bonded to same C atom (represented by dotted lines)</p></li><li><p>note: transition state, not intermediate since only 1 step</p></li></ul></li><li><p>concerted: nucleophile backside attack + halogen leaving group at same time, therefore 1 step reaction</p></li><li><p>1-step reaction, rate = k[halogenoalkane][nucleophile], overall order of reaction 2</p><ul><li><p>DO NOT WRITE SLOW/FAST</p></li></ul></li></ul><p></p>](https://knowt-user-attachments.s3.amazonaws.com/ef087cd0-c488-411c-b68d-0715daf36684.png)
why SN1 preferred?
in tertiary halogenoalkanes the three electron-donating R groups positive inductive effect will stabilise the C+ more
forms a more stable carbocation intermediate, therefore preferred mechanism SN1
steric hindrance
tertiary halogenoalkanes C+ surrounded by 3 bulky R groups
cannot proceed by SN2 mechanism because no room to accomodate 5 bulky groups in the transition state (note: transition state, not intermediate since only 1 step)
why SN2 preferred?
primary halogenoalkanes low steric hindrance: 2 groups are H atoms so there is room for other 3 groups
can have 5 groups surrounding central C in transition state, prefers SN2
factors affecting rate of nucleophilic substitution
class of halogenoalkane → rate eq of mechanism
type of halogen leaving group → reactivity of C-X bond → bond strength affected by resonance (unreactive) / bond enthalpy (lower, more reactive)
ONLY FOR SN2 type of nucleophile → availability of lone pair depends on charge (more negative, stronger nucleophile) + electronegativity (less negative, stronger nucleophile)
how does mechanism affect rate of nucleophilic substitution?
SN1 ‘transition state’ just lengthening bond vs SN2 transition state lengthen bond and increase crowdedness
SN1 ‘transition state’ thus has lower Ea
why don’t chlorobenzene/chloroalkenes undergo nucleophilic substitution?
p orbital of Cl overlaps with p orbitals of benzene ring / C=C
Cl part of resonance structure, C-Cl bond strong so breaks less easily
how does type of halide leaving group affect rate of nucleophilic substitution?
size of halogen increases, more diffused overlap of valence orbitals → bond length of C-halogen bond increases
bond strength decreases, bond enthalpy (energy needed to break bond) decreases, rate increases
rate: (fastest) iodoalkane > bromoalkane > chloroalkane (slowest)
how does reactivity of nucleophile affect rate of nucleophilic substitution?
anions > neutral
higher electron density, more reactive
within same charge, less electronegative > more electronegative
less electronegative, electrons held less strongly, donate electron pair more easily, more reactive
note: ONLY FOR SN2, rate determining step of SN1 doesn’t involve Nu-
preparation of alcohol
electrophilic addition of alkene
industrial: steam, heat with conc H3PO4 catalyst, 300C, 70atm
lab: conc H2SO4, heat with H2O
nucleophilic substitution of halogenoalkanes
NaOH/KOH (aq), heat under reflux
reduction of aldehydes, ketones, carboxylic acids (opposite of oxidation of alcohol)
aldehydes and carboxylic acids reduced to form primary alcohols, ketones reduced to form secondary alcohols
in order of reducing strength:
LiAlH4 in dry ether, followed by addition of dilute acid @ rtp
NaBH4 in dry ethanol followed by addition of dilute acid @ rtp (only for aldehydes and ketones)
heating with hydrogen gas and nickel catalyst (only for aldehydes and ketones)
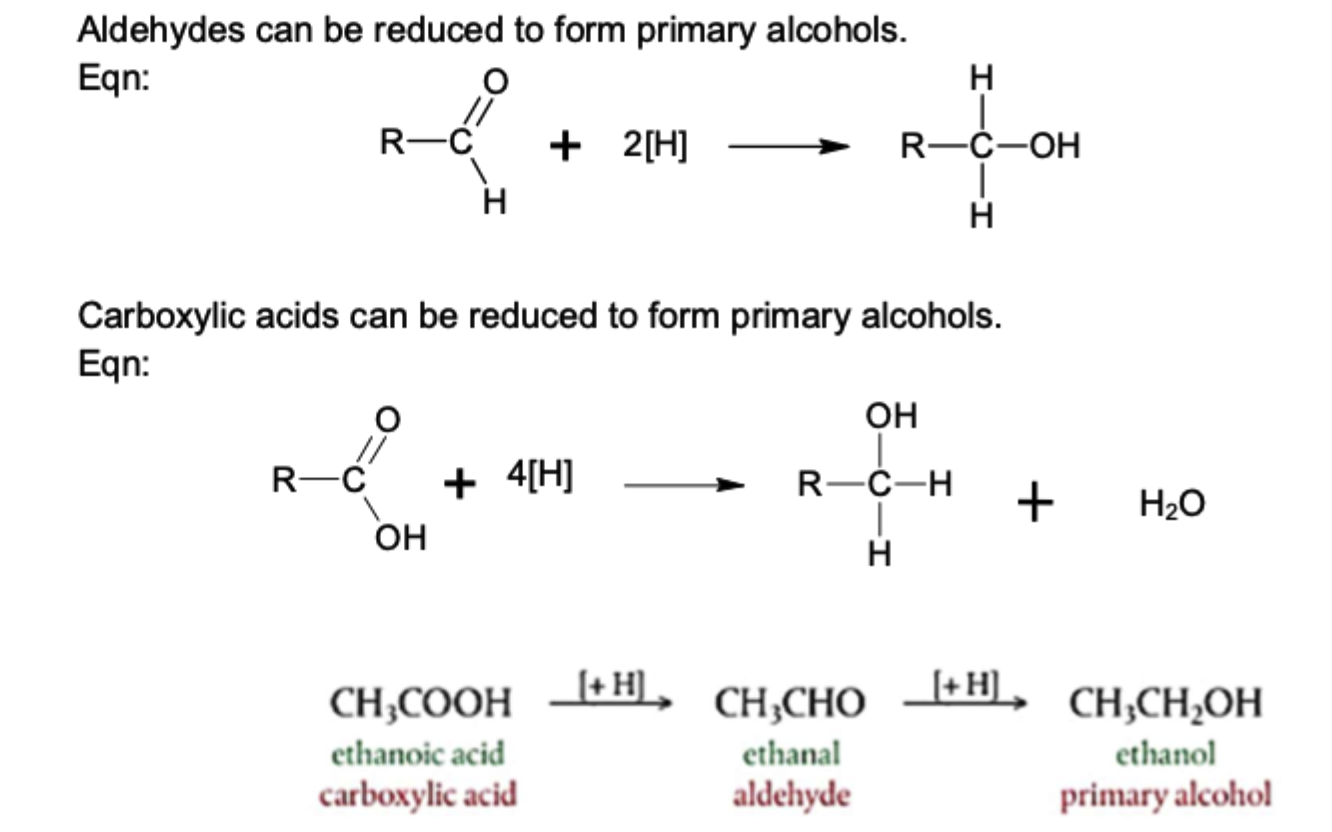
reduction of aldehydes
aldehyde + 2H → primary alcohol
C=O pi bond breaks, 2H added across
in order of reducing strength:
LiAH4 in dry ether, followed by addition of dilute acid
NaBH4 in dry ether followed by addition of dilute acid
heating with hydrogen gas and nickel catalyst
reduction of ketones
ketone + 2H → secondary alcohol
C=O pi bond breaks, 2H added across
in order of reducing strength:
LiAH4 in dry ether, followed by addition of dilute acid
NaBH4 in dry ether followed by addition of dilute acid
heating with hydrogen gas and nickel catalyst
reduction of carboxylic acids
carboxylic acid + H → aldehyde
aldehyde + H → primary alcohol
C-OH bond breaks, -OH lost as water by bonding with H, replaced by C-H bond (forms aldehyde)
C=O pi bond breaks, add 2H across (forms primary alcohol)
reagents and conditions
LiAH4 in dry ether, followed by addition of dilute acid
CANNOT use NaBH4 or hydrogen gas because resonance makes carboxylic acid stable
why NaBH4 cannot reduce carboxylic acid but LiAlH4 can?
bond length of Li-Al > Na-B
bond strength decrease, energy needed decrease, reactivity increase
carboxylic acid resonance makes it stable, need stronger reducing agent
therefore LiAlH4 stronger reducing agent than NaBH4, can reduce carboxylic acids, aldehydes and ketones
H2 vs NaBH4/LiAlH4
H2 is non-polar RA, so weaker. can reduce C=C and aldehyde/ketones
NaBH4/LiAlH4 are polar (hydride, H-) so stronger, BUT electron-rich C=C repels nucleophilic RA. aldehyde, ketone and carboxylic acid are polar, d+ C reacts with nucleophilic RA. (but acid only with LiAlH4)
reactions for alcohols
combustion
oxidation
condensation (esterification)
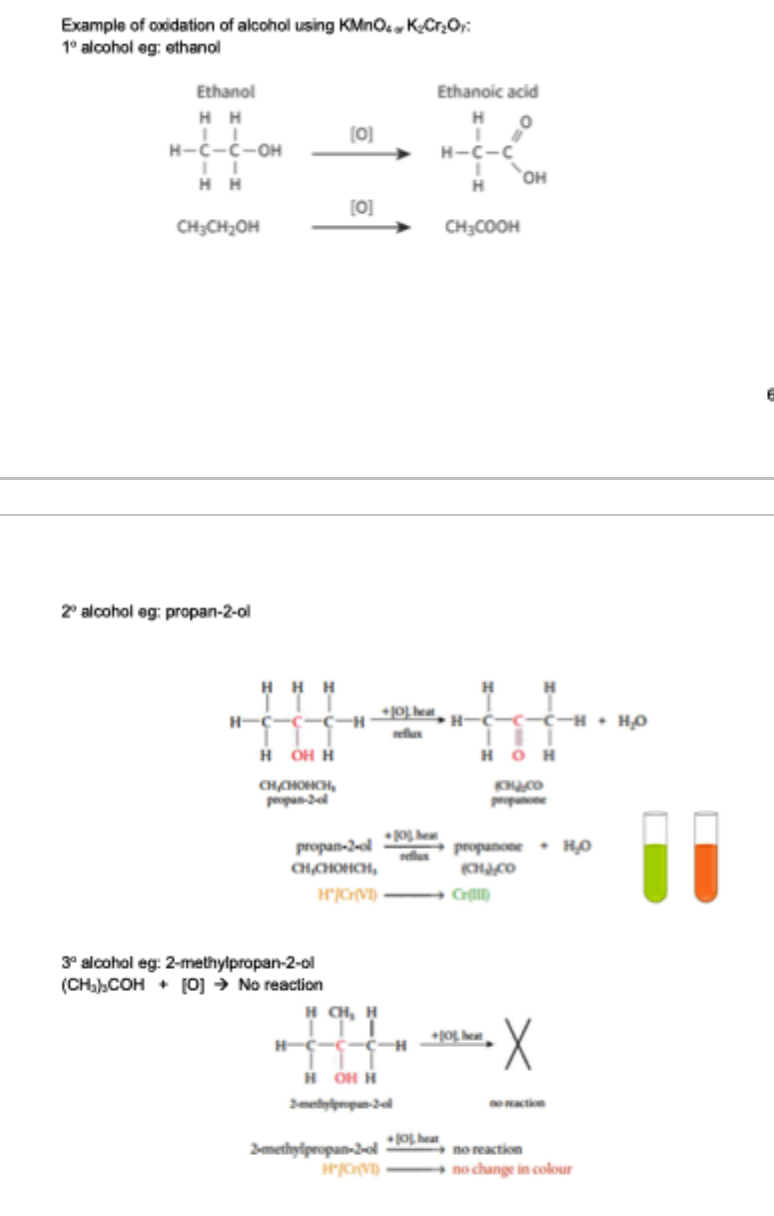
oxidation of alcohol
primary alcohols oxidised to aldehydes under controlled conditions, aldehydes further oxidised to carboxylic acids
has 2 alpha H → 2 step oxidation
secondary alcohols oxidised to ketones
has 1 alpha H → 1 step oxidation
tertiary alcohols do not undergo oxidation
no alpha H, cannot oxidise
formation of aldehyde
oxidation of primary alcohol
acidified (using H2SO4) weak OA: K2Cr2O7
heat with immediate distillation
so that doesn’t oxidise to become carboxylic acid
formation of carboxylic acid
2-step oxidation of primary alcohol
acidified (using H2SO4) excess strong OA: KMnO4 or K2Cr2O7
heat under reflux
prevent loss of volatile compound which would evaporate, by condensing it back into the mixture
alcohol oxidation mechanism
primary alcohols: 2 alpha hydrogen (2 H on the C attached to the -OH), 2 step oxidation
first alpha H and H from -OH are lost as water by bonding with O (from OA)
C forms pi bond with O (C-O → C=O)
second alpha C-H bond is oxidised by adding O (from OA)
secondary alcohols: 1 alpha hydrogen, 1 step oxidation
alpha H and H from -OH are lost as water by bonding with O (C-O → C=O)
tertiary alcohols: no alpha hydrogen, no oxidation
condensation of alcohol
alcohol + carboxylic acid <=> ester + water
conditions
conc H2SO4 → catalyst + dehydrating agent to remove water and favour forward reaction
heat under reflux
H from alcohol and OH from ethanol lost to form H2O, ester bond formed between RO- and ROC-
hydrolysis of ester
reverse of condensation of alcohols
ester + water <=> alcohol + carboxylic acid
conditions
H2SO4 (aq) → aqueous to provide H2O
heat under reflux → prevent loss of volatile compounds
H2O breaks ester bond, H attaches to RO- to form alcohol, OH attaches to ROC- to form RCOOR ester
transesterification
replace alcohol (-OR) of ester (RCOOR) with a different alcohol
RCOOR + R’OH <=> RCOOR’ + ROH
reversible reaction, excess R’OH alcohol used to shift POE towards products
conditions: strong acid/base catalyst (H2SO4, NaOH etc)
application: forming biodesel from plant oils
vegetable oils cannot be used directly in engine because too viscous + solidify at low temperature
vegetable oils: formed from 3 fatty acids (separate carboxylic acids) reacting with glycerol (triol) → triglycerides
transesterification: each fatty acid molecule bonds with methanol
methanol react with NaOH to form CH3ONa which is then reacted with the vegetable oils
since reversible reaction, excess alcohol used
biodiesel must be separated from glycerol (denser and sinks) and purified before use
addition polymerisation
alkene monomers, pi bond breaks to form repeating unit
no by-product, 100% atom economy
ethene and propene obtained from cracking
poly__ (monomer name)
polyethene
polychloroethene / polyvinylchloride (PVC) → ethene with 1 H replaced by Cl
polyvinylalcohol (PVA) → ethene with H replaced by OH
polytetrafluoroethene (PTFE) → ethene with 4 F instead of H
polysytrene (PS) → ethene with 1 H replaced by benzene ring
polypropene (PP) → propene
condensation polymerisation for polyesters
two monomers react to form polyesters and water/HCl
R’OH + RCOOH <=> RCOOR’ + H2O (note formation of ester bond)
conditions: conc H2SO4 catalyst (and dehydrating agent), heat under reflux
two cases
2 monomers: dicarboxylic acid + diol
1 monomer with 2 diff functional groups on each side
note: if n is a even number, both cases would look the same, cannot tell what the monomers are (puzzle analogy)
condensation polymerisation for polyamides
two monomers react to form polyesters and water/HCl
two cases
2 monomers: diamine + dicarboxylic acid → polyamide + (2n-1)H2O
n is no. of repeating units
nylon 6,6 → made up of 2 6-C carbon chains
note: if n is a even number, both cases would look the same, cannot tell what the monomers are (puzzle analogy)
1 monomer with 2 diff functional groups on each side
conditions: high temperature, high pressure, presence of catalyst
intermolecular interaction between polymer chains: H atom on N-H forms hydrogen bond with C=O on other chain
nitrobenzene → phenylamine 2-stage reaction
nitrobenzene reduced with Sn in acidic environment to form intermediate ion and Sn2+ ions
intermediate ion converted to phenylamine in the presence of OH-
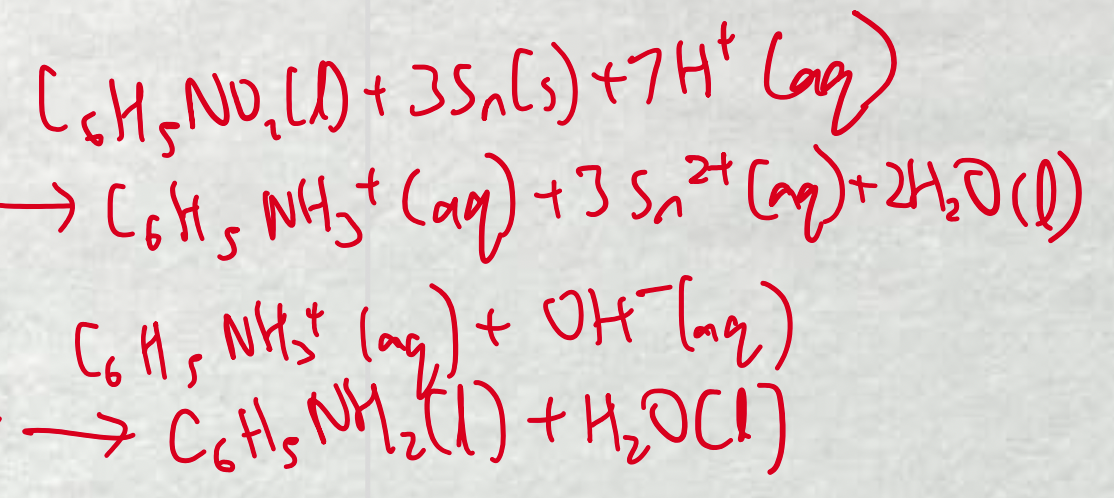
why for nucleophilic substitution polar solvent more suitable?
polar solvent will stabilise the carbocation → lower Ea, faster rate