Inorganic materials - structural properties
1/36
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
37 Terms
Outline the structure of 1. Wurtzite (ZnS) and CdI2
Wurzite: Zn2+ occupies half of the Td holes in a HCP structure of S2-. 4:4 coordination.
Cadmium Iodide: Cd2+ occupies alternate layers of octahedral holes in a HCP structure of I-. 6: 3 coordination of Cd:I.
Outline the structure of NaCl and Fluorite
NaCl: Na in all Oh holes of a CCP of Cl. 6:6 coordination in the unit cell.
CaF2: F in all tetrahedral layers of Ca CCP structure. 8:4 coordination of F:Cu.
Outline the structure of perovskite
ABO3 structure. B sits Oh holes of oxygens, with A/O close packed layers.
Outline the structure of spinel
E.g. MgAl2O4 CCP oxides ions, with Mg in 1/8th Td holes, and Al in ½ Oh holes.
What are the 3 different point defects?
vacancy
interstitial defect (atoms sitting in a site it would not normally occupy e.g. a hole).
Substitutional (where a foreign isovalent or aliovalent atom adopts a site in the structure of a pure element).
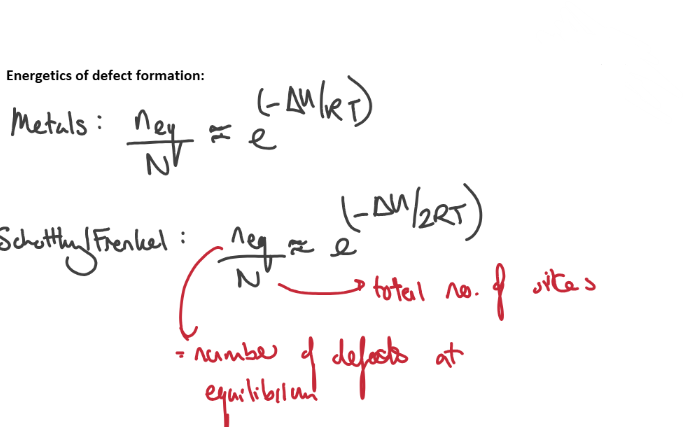
Explain what the driving force is behind vacancy formations - why will the number of vacancy defects stop increasing eventually?
The main driving force behind vacancy formation is the increase in configurational entropy.
There is also a smaller vibrational contribution to the entropy upon vacancy formation.
The favourable entropic increase is counteracted by positive enthalpy of vacancy formation (bond breaking > bond forming at the surface site)
As the number of defect sites keeps increasing, the entropy eventually stops increasing and begins to decrease, at which point defect formation becomes unfavoured.
Give the equation for determining the number of defects at equilibrium.
What is the difference between intrinsic and extrinsic defects?
Intrinsic: defects that occur in pure materials.
Extrinsic: defects that involve new chemical species.
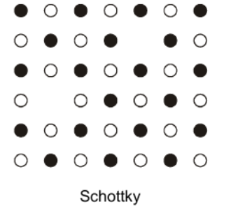
Outline Schottky defects
A cation vacancy, compensated by an appropriate number of anion vacancies. Negative and positive charges within a local vacancy attract one another → vacancy clustering. E.g. in NaCl
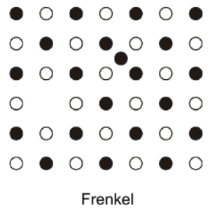
Outline Frenkel defects
A vacancy and a corresponding interstitial ion of the same charge to maintain charge balance. E.g. in AgI, CaF2.
Outline F-centres.
Electron sits in a normal anion site e.g. in calcite.
Compare isovalent and aliovalent substitution as extrinsic defects.
Isovalent: Substitutional defect where a cation or anion is replaced by another cation or anion of the same oxidation state.
Aliovalent: An atom is replaced by one with a different oxidation state. E.g. the introduction of a lower-valent cation means that the substituted site has a formal negative charge that must be compensated e.g. by introducing anionic vacancies.
How is conductivity related to the activation energy/energy barrier for vacancy/interstitial diffusion of an ion for EXTRINSIC defects?
conductivity is proportional to e-Em/RT, where Em = activation energy.
For pure intrinsic conductivity, how does the activation energy change?
Two activation energies are involved:
migration activation energy Em
Thermal energy required to form defects, Ed
Draw the full graph for ln(conductivity x T) vs 1/T
For intrinsic, slope = -(0.5Ed+Em/R), for extrinsic slope = -Em/R. Extrinsic dominates at low temperatures since not enough energy to generate the intrinsic defects.
What makes AgI a good cation superionic conductor? outline its structure at room temperature and at 420K.
Room temp: beta-AgI: Ag+ occupies half Td holes of hcp of I-.
420K: alpha-Agi, iodide anions adopt a bcc packing, conductivity increases. → lots of sites for Ag+ ions to occupy. Large number of mobiles ion and sites for Ag+ to hop through.
Low charge on the ion (+1) allowing for higher mobility.
Very low associated Em (3kJ mol-1)
I- is large and polarisable ('floppy'): can deform their electron clouds to allow movements of Ag+ easily.
Explain how the sodium beta alumina structure allows it to act as a good cation conductor.
Spinel structure with some oxygen ions missing every 5th layer. The Na+ ions locate in these anion deficient layers.
Na+ locate at 9 coordinate sites but can migrate through 8 and 5 coordinate sites, providing a lower energy site network for Na+ movement.
Give an example of where Sodium beta-alumina may be used
Use in development of high power, sulfur rechargebale battery. 2Na + 5S → Na2S5. The Na+ formed at anode migrates through the beta-alumina electrolyte to the cathode where they combine with liquid S and electrons from external circuit.
Explain these differences
PbF2 shows a gradual increase in conductivity in solid states and almost no change in its liquid state. Increasing number of Frenkel defects with temperature.
NaCl shows a sharp increase upon melting. Arising from Schottky defects.
AgI shows a sharp change going from the beta to alpha structure.
All converge at melting points/molten state
How/why can CSZ be used in fuel cells
Cubic fluorite structure of ZrO2 at high temperatures can be doped with Ca2+ → oxide vacancy, stabilising the structure at room temperature.
CSZ used to allow oxygen diffusion through, conducting oxygen ions to the anode, combining with hydrogen at the anode. Electrons move via an external circuit. H, requires high temperatures. Also used in oxygen sensors.
Why is graphite good at intercalation chemistry as a host?
Uptakes guest species between graphite layers that move apart to accommodate.
Graphite will interact with both electron donors (e.g. K) and electron acceptors (e.g. Br2). Lies in the fact that graphite is a semi-metal (see band structure)
K is ionise to K+ and electrons are donated to the empty conduction band.
Br2: Electrons from the valence band partially reduce the guest, leaving holes at the top of the valence band.
Why do we not use Li metal as the anode in batteries?
due to safety -> Li+ ions do not redeposit at the same location, leading to dendritic growth of Li metal at the anode surface, which can short circuit the cell and lead to ignition.
Why do we use Co4+/Co3+ in the cathode of modern Li batteries?
Graphite, at the anode instead of Li metal. This stabilises Li, and eliminates dendrimers from forming. However, leads to a loss of Ecell.
Hence to increase Ecell, we need to lower the chemical potential of the cathode. Requirs low energy sites to adopt Li+ ions. We can achieve this using the Co4+/Co3+ couple in an oxide e.g. Li1-xCoO2. -> Greatest negative ΔG by lowering the energy of the cathode.
Explain the structure of LixCoO2
Ordered rock salt type structure
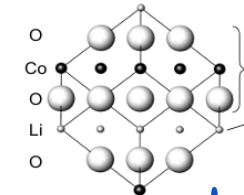
Why are only ½ the Li ions used in Li batteries?
Due to stability: when you remove Li, Co must oxidise from Co3+ to Co4+ → over oxidation can lead to instability and safety concerns.
Outline the structure of zeolites
Corner-sharing network of SiO4 and AlO4 tetrahedra, containing charge balancing cations, anion and/or other guest molecules in the framework pores.
What are zeotypes?
Materials with similar structures but containing other elements on the tetrahedral sites.
How can zeolites be synthesised?
a silica source and an alumina source are combined with water, template molecules and guest cations (metal) to produce a gel, which is heated to its crystalline phase with removal of water.

What is the structure of a sodalite? How can this be used to produce different zeolite structures.
sodalite cage = 24 tetrahedra.
Hexagonal faces and square faces (6T-rings and 4T-rings)
Sodalites can be linked by a T-O-T bridge between 4T-rings -> Zeolite A or linked by 6T-rings -> Zeolite X or Y.
Zeolite X/Y has larger cavities than Zeolite A.
Directly sharing 4T-rings leads to a primitive cubic structure, where the cavity itself is a sodalite unit.
How can templates control the Si:Al in zeolites?
Each cage owns 6 T (Al or Si) atoms. Amount of counterion that can fit in the cage determines the amount of Al required to charge compensate.
Na+ is small -> can fit 3 per cage -> Al3Si3 therefore 1:1 ratio of Al:Si.
NMe4+ is larger -> can only fit 1 per cage -> AlSi5 -> 1:5 ratio of Al:Si.
How can templates control the size of the cavity?
Templates can act a structure-directing agents that increase the likelihood of a certain framework topology. The use of a larger template results in a larger cavity forming, which forms around the template.
Outline how zeolites can act as molecular sieves
Different zeolites have different number of T atoms in rings and hence different size of guests can diffuse, allowing for some molecules to absorb and some to not/more slowly. E.g straight chain alkanes v branched chain alkane
Outline how zeolites can act as catalysts
cations in zeolites can be exchanged for protons by heating ammonium-exchanged form of the zeolite or by calcination of organic template containing zeolites. These protons can act as Bronsted acid sites for acid catalysis e.g. in isomerisation reactions of alkenes. Can give shape-selectivity.
Give the 3 types of shape selective catalyst reactions
Reactant-size selectivity: access to the catalytically active site of the zeolite is controlled by the size of the framework pores. Can lead to only selective cracking of straight chain alkanes as it diffuses more readily, leading to higher rates of cracking compared to the branched-isomer.
Product-size selectivity: relies on the fact that different products have vastly different diffusion rates out of the zeolite.
TS-selectivity: where the TS of the undesired product is sterically disfavoured in the pores, leading to preferential production of the desired isomer.
Outline how zeolites can help with ion exchange
E.g. for zeolite A, Ca2+ is preferred over Na+ and Ca2+ will undergo full ion exchange with Na+.
Used in laundry detergents to remove Ca2+ to soften water.
Outline how zeolites can act as drying agents