Lysosomal Storage Disorders - Biochem Genetics
1/40
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
41 Terms
Lysosomes
Simple lipid membranes surrounding degradative enzymes
Found in all mammalian cells except RBCs
MACROPHAGES IN PARTICULAR have high number of lysosomes
Clinical features of lysomal dieases depdends on
Distribution/expression of he deficient lysosomal Enzyme
Distribution of the substrate expression
Effects of substrate accumulation/End-product defeicieny
Monocyte AMcrophages
when macrophages in the body are deficient in lysosomes, the substrates they scavenged CANNOT BE degraded and accumulate:
Bone Marrow --
Liver
Brain: Neurodegeneration
Spleen: Splenomegaly
Lung
Lysosomal Storage Disorder Categoreis
Sphingolipidoeses: white matter neorudegrantion and organ dysfucntion
Mucopolysaccharidoses: numverous body tissues + skeletal dysostosis
Oligosaccharidoses: involving numerous tissues with skeltal dystosis
Glycogenoses: Progressive glycogen accumualtion in the muscle leades to myopathy and weakness
Lysomomal Storage Disorders: Typically, Progressive Disease
most affected individuals appear normal at birth
clinical signs/symptoms develop in weeks/months
organomegaly and organ dysfunction
loss of motor and cognitive skills
cherry red macule
Often present with lethal/infantile onset but milder/later onset (attenuated) variants exist
Lysosomal Storage Disorders: Phenotypic Variability
All LSDs, regardless of Age of Onset, are Progressive Disorders: MOST are asymptomatic at birth
There is a progressive ACUMULATION OFSUBSTRATE over the lifetime of the affected patient
Many LSDs have subtypes:
Earlier Onset, Severe Phenotype
Later Onset, Mild/Attenuated Phenotype
Many have CNS degradation
Fabry Disease: Metabolic Pathway
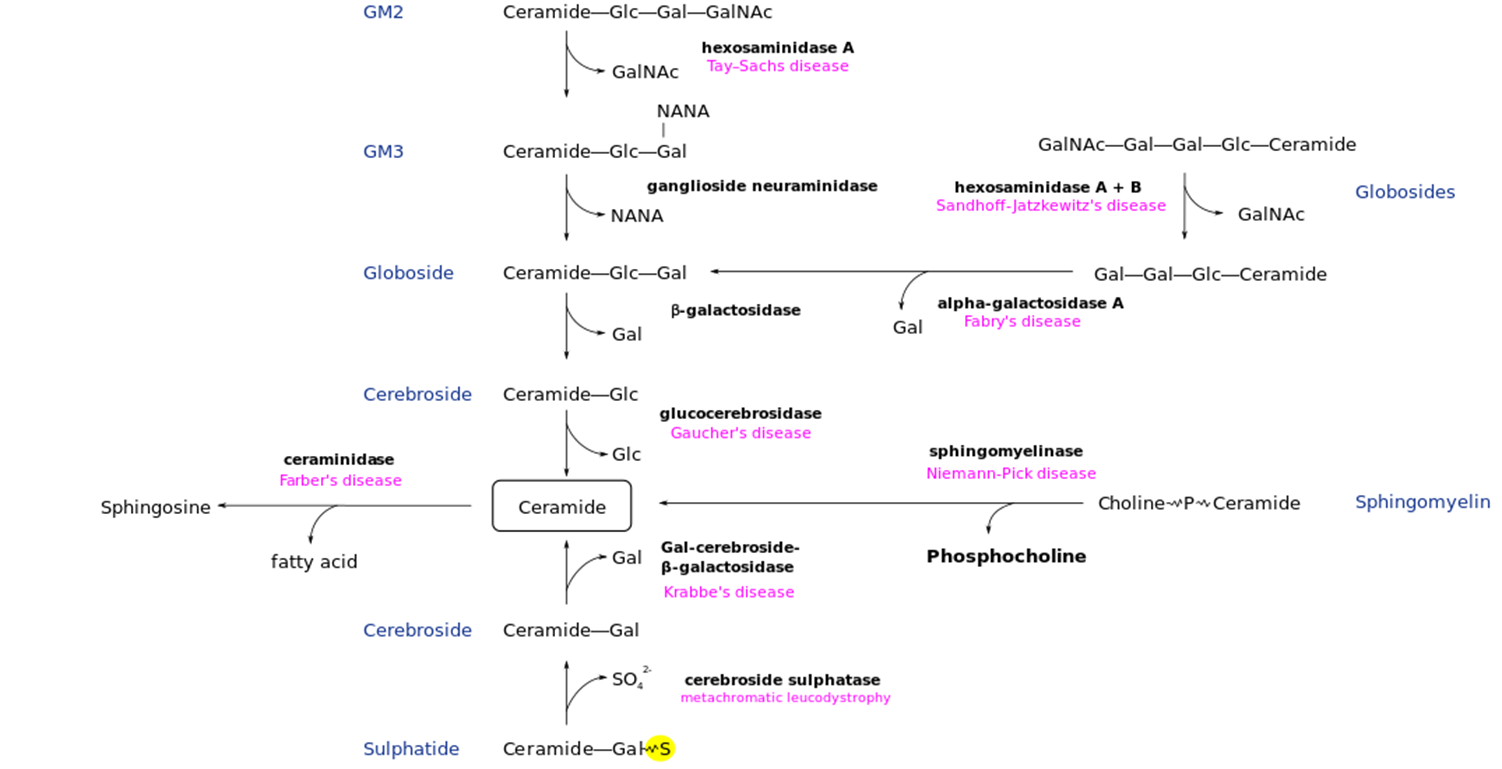
One of the few non-neuronopathic LDS
Deficiency of Alpha-Galactosidase → Accumulation of Globotriaosylceramide (GL-3)
Fabry Disease: General
Cardiac, Renal and Cutaneous affect
Inheritance: X-Linked Recessive
Genetic Defect: Alpha-Galactosidase (GLA) Gene Deficiency
GB3 accumulates in vascular endothelial cells: Kidney, Heart, Skin Nerves/Brain (but no damage)
Fabry Disease: Untreated Features
Classic: Typically, Males with <1% Enzyme Activity —> Onset 4-8yo, but diagnosed much later in life
Burning Painful episodes (Acropathies): in extremities, stress triggered
Angiokeratomas (small red/purple skin spots): bellybutton to knees, more w/ age
Abnormally increased or decreased sweating
Ocular Opacities: corneal haze/opacity, cataracts (even in carrier females)
***Renal Failure: progressive, end-stage renal diease 30-50s yo
***Cardiovascular: HTN, Hypertrophic Cardiomyopathy, CAD)
***Cerebrovascular Disease: Strokes
Fabry Disease: Variants
Cardiac Variant (Males >1-20% enzyme activity)
Renal Variant: presents with relatively isolated renal disease
Heterozygous Females: varies from asymptomatic to classic
Typically milder with later age of onset
Rarely progresses to End Stage Renal Diase
Fabry Diease: Diagnosis
NBS: not done in every state
Diagnostic test:
measure of A-Galactosidase Enzyme activity (Plasma. WBC, Fibroblast)
GB3 and Lyso-GB3 levels
GLA genotyping: del/dup 100% in positives
Prenatal Tests
Molecular testing
GLA enzyme activity on Amnio/CVS
Other Routine Tests
Renal function: BUN/Creatine, urinalysis
Cardiac Function: Echocardiogram, EKG, stress test
Vision Hearing: eye and ear testing
Cerebrovascular function: Brain MRI/MRA
Fabry Disease: Treatment
Preventative
Recombinant enzyme Replacement Therapy (ERT) → shows increased GL03 clearance and decreased symptoms
Symptomatic
pain meds
Ace inhibitors
Dialysis/Renal transplantation
Routine care if cardiac/cerebrovascular complication
Gaucher Disease: Metabolic Pathway
One of the most Common
Deficney of gluccocerebrosidase → Build-up of Glucocerebroside in tissue
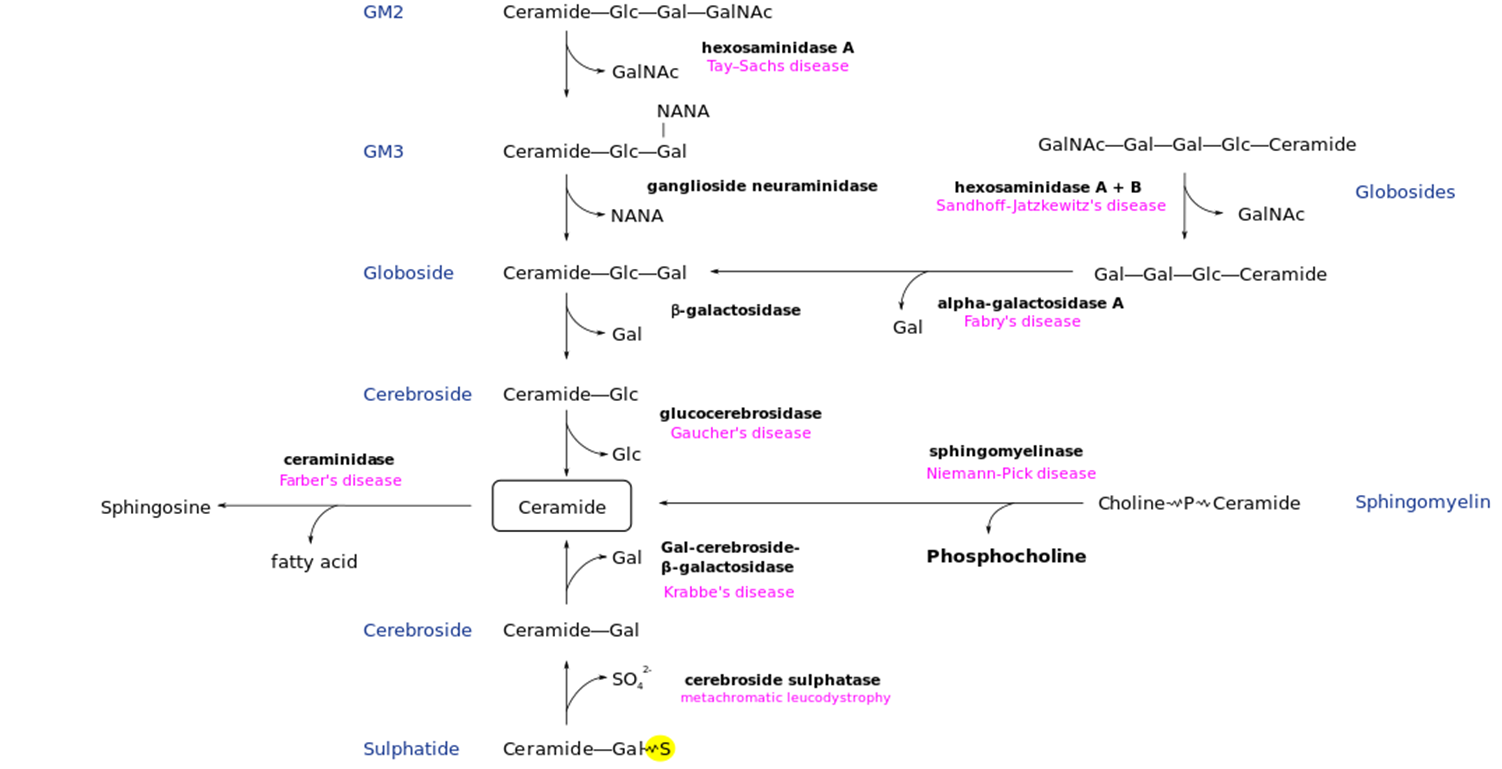
Gaucher Disease: Overview
Glucosylceramides Deficiency
Inheritance: Autosomal Recessive
Incidence: seen highly in Ashkenazi Ancestry (Type I)
Genetic Defect: GBA gene deficiency
Build-up of Glucocerebroside in monocyte macrophages: Liver, Spleen, Bone Marrow, Lungs Brain
Gaucher Disease: Untreated
Type 1 (Non-Neuronopathic) most common in Ashkenazi
Onset from Childhood to adult (some remain asymptomatic)
Bone Disease: bone pain
Hepatosplenomegaly: swelling of spleen and liver
Cytopenia decreased RBC/WBC
Pulmonary involvement: plumonary hypertension
Parkinsons DieaseL 20-30x increased risk
Miscellaneous
depression, gallstone, possibly myeloma/lymphoma/liver carcinoma
No primary Early Neurodegeneration
Gaucher Disease: non-type 1
Type II/III (Neuronopathic):
Type II: onset before 2yo, progressive, lethal neurodegeneration
Type III: Children at any age, more slowly progressive, death by 20-30yo
Perinatal Letha Variant (RARE)
Hydrops Fetalis, Ichthyotic Skin changes
Cardiovascular Variant (Atypical)
Mild Symptoms but Prominet Valvular heart Disease
Gaucher Disease: Diagnosis
NBS: not in all states
Diagnostic Tests
Glucocerbrosidae Enzyme Activity (WBC, Fibroblast)
GBA Genotyping
4 mutations cover 90% of alleles in Ashkenazi (N370S, L444P, 84GG, IVS2+1)
N370S allele limits disease to Type I Non-Neuronopathic Type
Prenatal Tests: Molecular, GBA Enzyme Activity on Amnio/CVS
Gaucher Disease: Treatment
Preventative (Not in type II)
Recombinant Enzyme Replacement Therapy (ERT): prevents type 1 complications, no effect in Type II, mixed benefit for Type III
Oral Substrate Reduction Therapy (SRT) → inhibits glucocerbroside formation
Bone marrow transplantation
Symptomatic
Splenectomy
Tranplation
Pain meds/joint replacment for bone complications
Niemann-Pick Disease: Metabolic Pathway
Has Neuropathic and Non-neuropathic forms
Deficiency of slingomaylonase enzyme → buildup of Phosphocholine
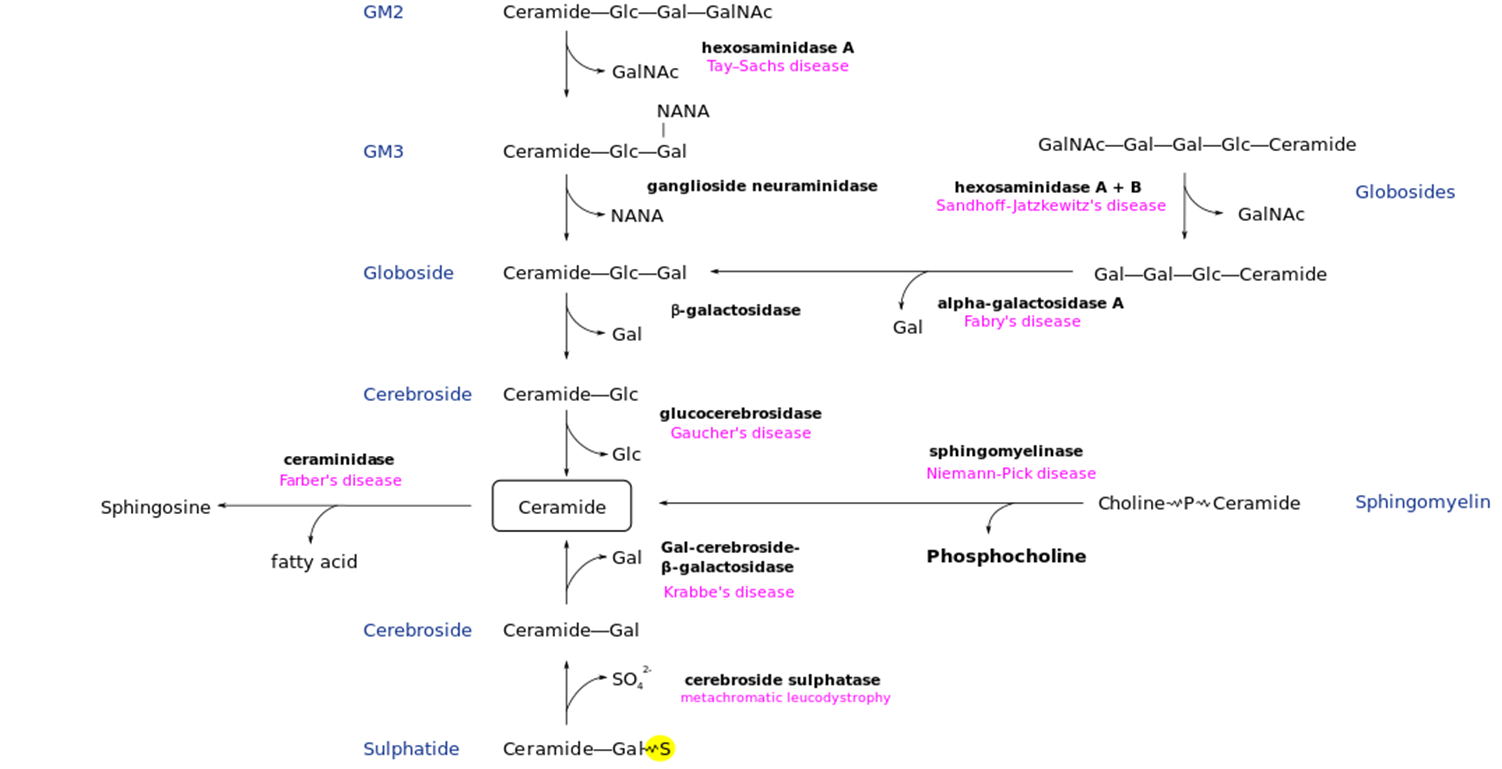
Neimann-Pick Disease: Overview
Two Types:
Type A (Neuronopathic)
Type B( Non-Neuronopathic)
Type C does exist, but non LSD
Inheritance: Autosomal Recessive
Incidence: Higher prevalence (Type A) in Ashkenazi
Genetic Defect
Sphingomyelin Phosphodiesterase 1 (SMPD1) Gene
Sphingomyelin accumulation on monocyte macrophage cells in Brain, Liver, Spleen, Lung
Neiman Pick Disease: Untreated, Type A
Neuronopathic form of Neiman Pick: similar to the Neuronopathic Form of Gaucher Diease
Onset within 1st year, death usually before 3-4 years of age
Neurodegeneration: typical onset between 6-12 months of age
Hepatosplenomegaly (HSM): present by 3 months of age
Pulmonary Involvement
Miscellaneous
Cherry red spot on Macual of Eye ****UNIQUE TO NB but NOT GAUCHER****
Neiman Pick Disease: Untreated, Type B
Non-Neuronopathic form of Neiman Pick, less common
Later onset and milder variable symptoms, survival to adulthood
More organ swelling: massive hepatosplenomegaly
Deterioration of pulmonary function
Miscellaneous: Abnormal Lipids (low HDLc/high LDL)
Neiman Pick Disease: Diagnosis
NBS not done for all states
Diagnostic Tests
Acid Sphingomyelinase Enzyme Activity (WBC, Fibroblast)
SMPD1 Genotyping
3 mutations (L302P, R496L, fsP330) in 90% of alleles in Ashkenazim Type A
R610del very common Type B allele
Prenatal Tests
Molecular, SMPD1 enzyme activity on Amnio/CVS
Other Routine Tests
Pulmonary Assessment, Lipid/Cholesterol levels, blood counts
Liver function tests, Bone Density, Opthalmogic Evalutation
Neimann Pick Disease: Treatment
Preventative (Not Type A)
Bone marrow transplant:
Recombinant Enzyme Replacement therapy (ERT)
Symptomatic
Platelet transfusion
Avoid splenectomy
Cholesterol lowering meds
Supplemental oxygen and respiratory support
Tay-Sachs Disease: Metabolic Pathway
Deficiency of Hexosaminidase A → Accumulation of GalNAc
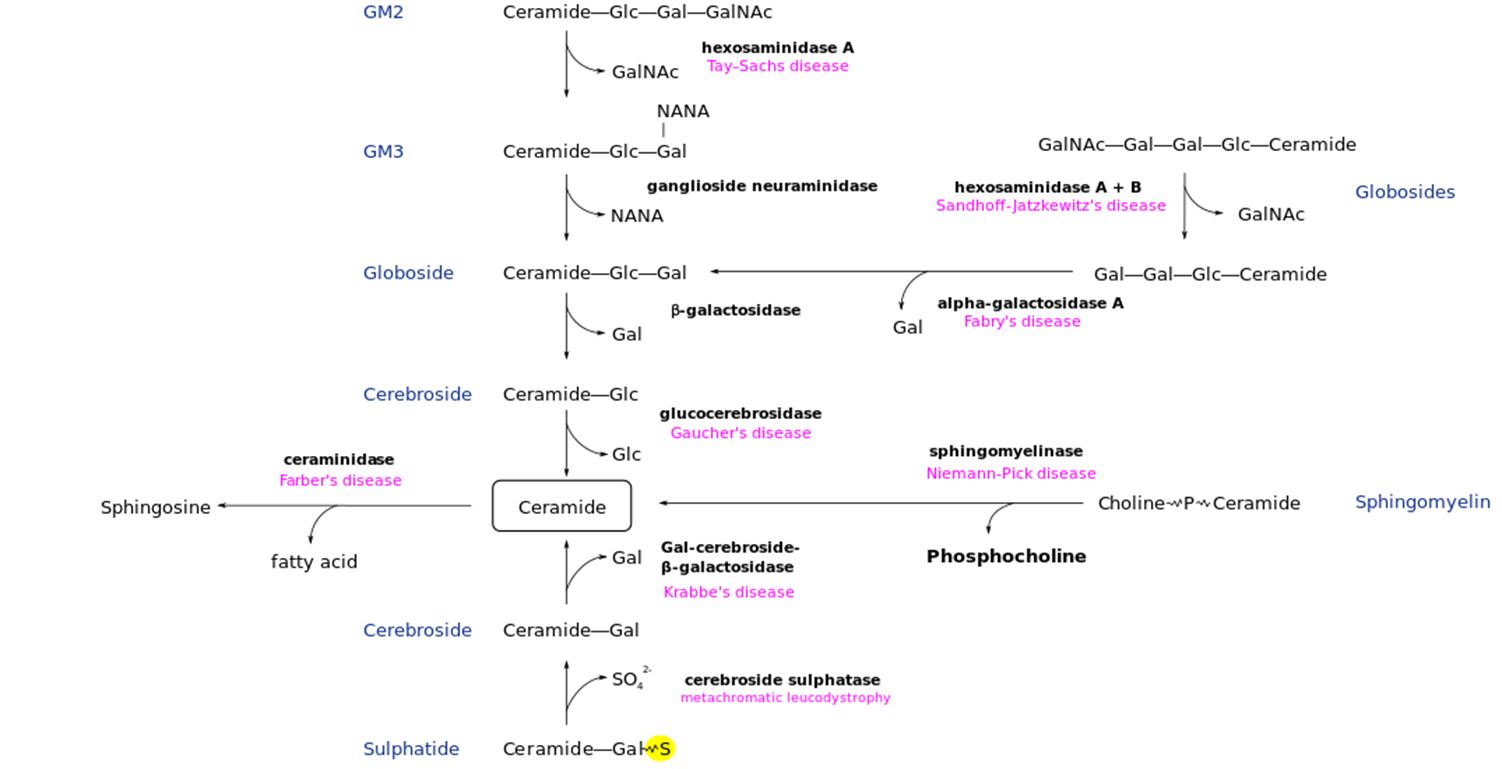
Tay-Sachs Disease: Overview
Most common: early onset, neuropathic form
Inheritance: Autosomal Recessive
Incidence: High in Ashkenazi and French Candain
Genetic Defect
Hexosaminidase A (HEXA) Gene → GM2 ganglioside accumulation primarily in CNS (neurodegeneration)
HEX A: 1a and 1b subunit, HEXB contain 2 beta subunits
Tay-Sachs: Infantile Form, Untreated
0-5% HEX A activity
Onset by 6 months, Death usally by 2-4 years of Age
Neurodegeneration - Starting 3-6 months of age
Decreased attentiveness with increased startle repsonse
Tay-Sachs: Juvenile+Adult Form, Untreated
5-15% HEX A activity
Later onset and slower progression
Progressive dystonia,
Speech and cognitive decline
Death
Juvenile: 10-15 years old
Adult: variable
Tay-Sachs: Diagnosis
NBS: Not done
Diagnostic Testing
Hexosaminidase A Enzyme Activity (serum, WBC, Fibroblast)
Reduced HEX A, but elevated HEX B activity
HEX A Genotyping
Null genes
Pseudodeficieny mutations: there is decreased enzyme activity in the lab but not IN VIVIO (in the actual body) : can cause positive carrier screening result → appear to be disease carrier but actually just a pseduo-deficiency carrier which does not cause the disease
Prenatal: molecular, HEX A Enzyme activity on Amnio/CVS
Brain MRI shows atrophy and white matter demyelination
Tay-Sachs: Treatment
NO PREVENATIVE OR CURATIVE CARE
Supportive care only: little effect on mortality
Antiepileptics, Respiratory support, Supplemental nutrition
Krabbe Disease: Metabolic Pathway
A common leukodystrophy;
Deficiency of Gal-cerebroside-B-galactosidase → Galactosylceramide accumulation
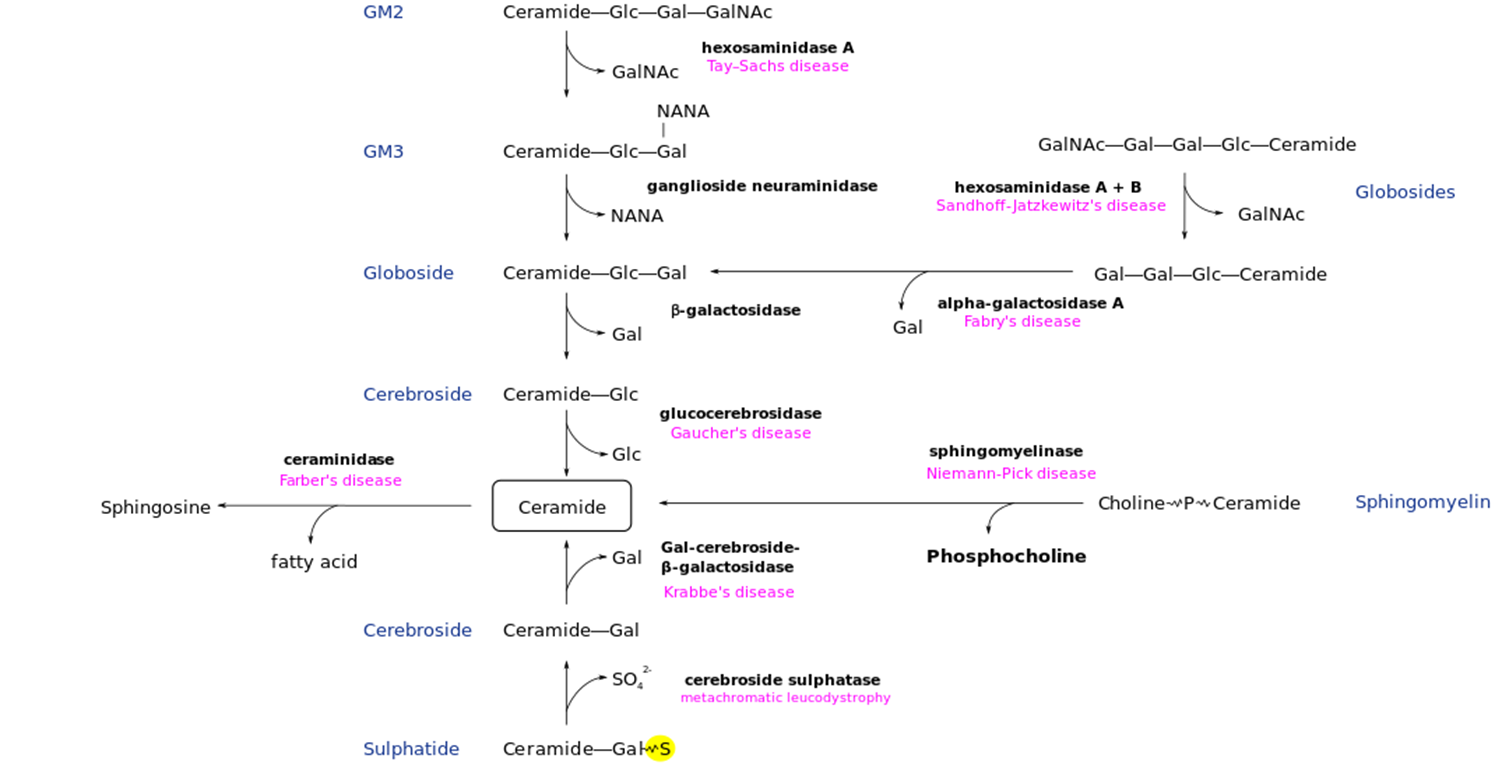
Krabbe Disease: Overview
Inheritance: Autosomal Recessive
two forms
90% Infantile Leukodystrophy
10 Late Onset Leukodystrophy
Genetic Defect
Beta-Galactosidase (GALC) Gene → Accumulation and deposition of galactosylceramide and pyschosine primarily in brain leading to oligodendoglial destruction and white matter demyelination (leukodystrophy)
Krabbe: Infantile, untreated
Onset by 6 months, death by 2yo
Unlike neuronopathic Neiman Pick type A, or Tay Sach Disease → No Cherry Red Spot
More similar to Type II Gauches: BUT no organ enlargement
Initial (Stage I):
Initial (Stage II):
Intial (Stage III):
Krabbe: Later onset, untreated
Variable onset, severity and progression of:
Weakness, ataxia, neuropathy
Visual deterioration
Psychomotor regression ± death when earlier onset
Krabbe: Diagnosis
NBS: tested in many state -→ Low GALC Activity leads to targeted DNA analysis ± reflex to sequencing
Diagnostic Tests
Beta-Galactosidase Enzyme Activity (WBC, Fibroblast)
GALC Genotyping
specific deletion = infantile Krabbe (45%)
857G>A results in late onset Krabbe (50%)
Prenatal Tests
Molecular, GALC enzyme activity on Amnio/CVS
Other Routine Tests
Brain neuroimaging (atrophy and white matter changes)
Elevated CSF Protein, abnormal NCV/EMG
Krabbe Disease: Treatment
Preventative:
Hematopoietic Stem Cell Transplantation:
only benefits presymptomatic infants and mildly symptomatic late-onset cases
Can preserve function and slow down progression
Hromeon Replacement therapy not shown to be affective in human
Symptomatic
directed at maximizing/Prolonging Function and Minimizing Discomfort/Suffering
Metachromatic Leukodystrophy: Metabolic Pathway
Cerebroside Sulphatase deficiency → Sulphatide accumulation
Metachromatic Leukodystrophy: Overview
3 sub-type: Late-Infantile, Juvenile and Adult
Inheritance: Autosomal Recessive
Genetic Defect
Arylsulfatase A (ARSA) Gene
Accumulation of sulfogalactosylcermide and other sulfatides in brain and Kidney → White Matter Demyelination (Leukodystrophy)
Untreated Metachromatic Leukodystrophy: Late Infantile
Onset between 1-2 yo, death in 3-10 years
Initial: 6-12 month normal development
Followed by regression, neurodegeneration, neuropathy,
Leading to prolonged: blindness,, unaware, decerabte posturing, Death
No organomegaly, cherry-red eye spot
Untreated Metachromatic Leukodystrophy: Juvenile + Adult
Juvenlie: Onset 4-14yo
Adult: Onset after 14yo
Initial: De;cine in school or job prefmeona, behcviour/perosnality changes
Followed by: deterioting gait, speech, behaviour, neuropathy, coginton : with or without death
Diagnosing Metachromatic Leukodystrophy
No NBS
Diagnostic
Arylsufacte A Enzyme activity (WBC, Fibroblast)
ARSE Genotyping
Null/nonsense genes
Psuedodefiencney alleles → low ARSA enzyme activity (5-20%) but no disease
Prenatal test: molecular + enzyme activity (if no pseudodeficiney)
Other routine testing
Brain neuroimaging (atrophy and white matter change)
Elevated CSF protein
Treating Metachromatic Leukodystrophy
Preventive
Hematopoietic stem cell transplantation
Best outcomes when presymptomatic → no universally affective
Progression after a time in some patient
Symptomatic
Treatment directed at maximizing/prolonging function and minimizing discomfort/suffering