Med Chem II Final Exam - Li
1/127
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
128 Terms
drugs used for mild-to-moderate pain (step 1)
- nonopioids such as acetaminophen or NSAIDs
drugs used for mild-to-moderate pain uncontrolled after step 1 (step 2)
- short-acting opioid as required +/- nonopioid
- morphine, oxycodone, or hydromorphone should be added to acetaminophen or NSAID
moderate-to-severe pain or pain uncontrolled after step 2 (step 3)
- sustained-release/long-acting opioid or continuous infusion short-acting opioid as required +/- nonopioid
- sustained-release morphine, oxycodone, or hydromorphone or transdermal fentanyl as indicated
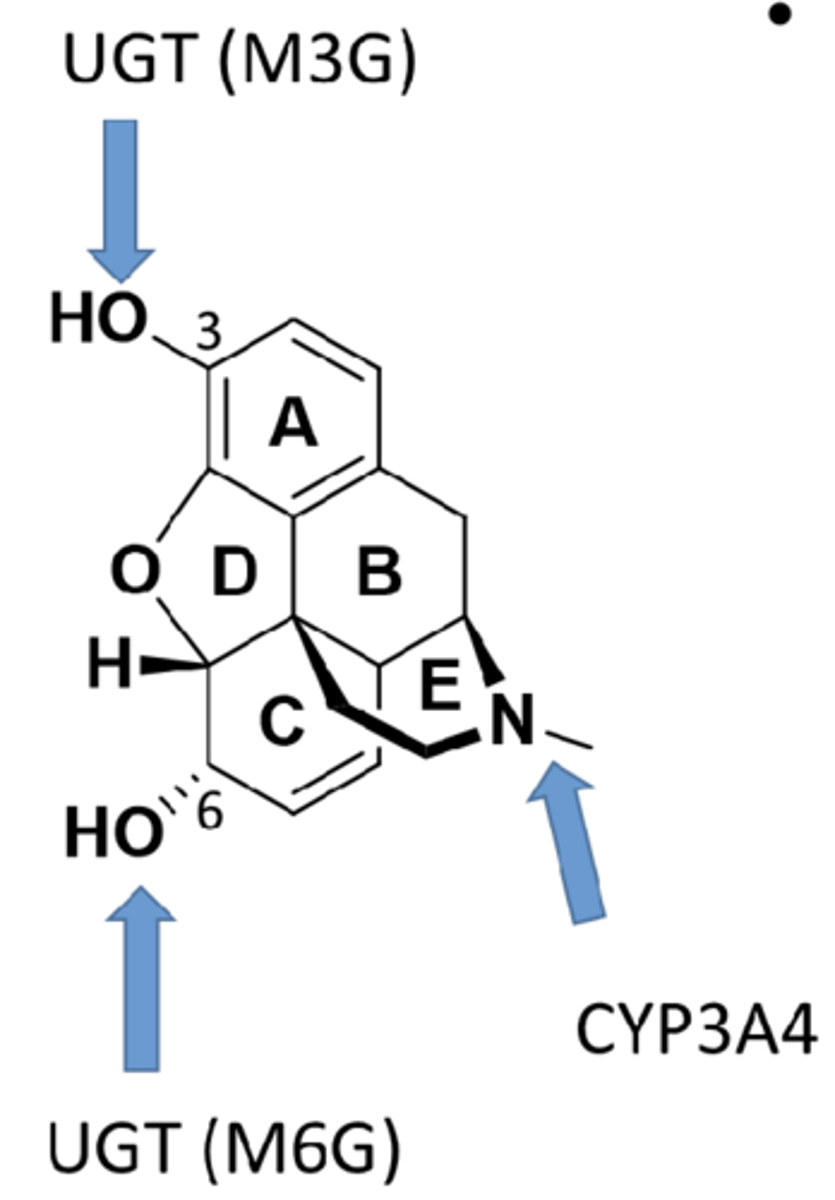
metabolism of opioids
- first pass metabolism in liver
- major metabolic reactions include phase II metabolism and glucuronidation (M6G active and M3G inactive metabolites)
- phase I reaction includes demethylation (non-morphine, less-active metabolites)
adverse effects of opioids
- tolerance and dependence
- abuse and addiction
- respiratory depression
- constipation
respiratory depression with opioids
- MOR expressed on respiratory neurons in brainstem regulates breathing
- activating MOR depresses ventilation
- life threatening
constipation with opioids
- MOR in GI tract regulates GI motility
- activating MOR in GI delays transit and stimulates non-propulsive motility, segmentation, and tone
SAR of 3-phenolic OH in opioids
- phenolic OH is crucial for activity
- masking the phenolic OH loses binding affinity and dramatically decreases analgesic activity
- as affinity to MOR decreases, analgesic activity decreases
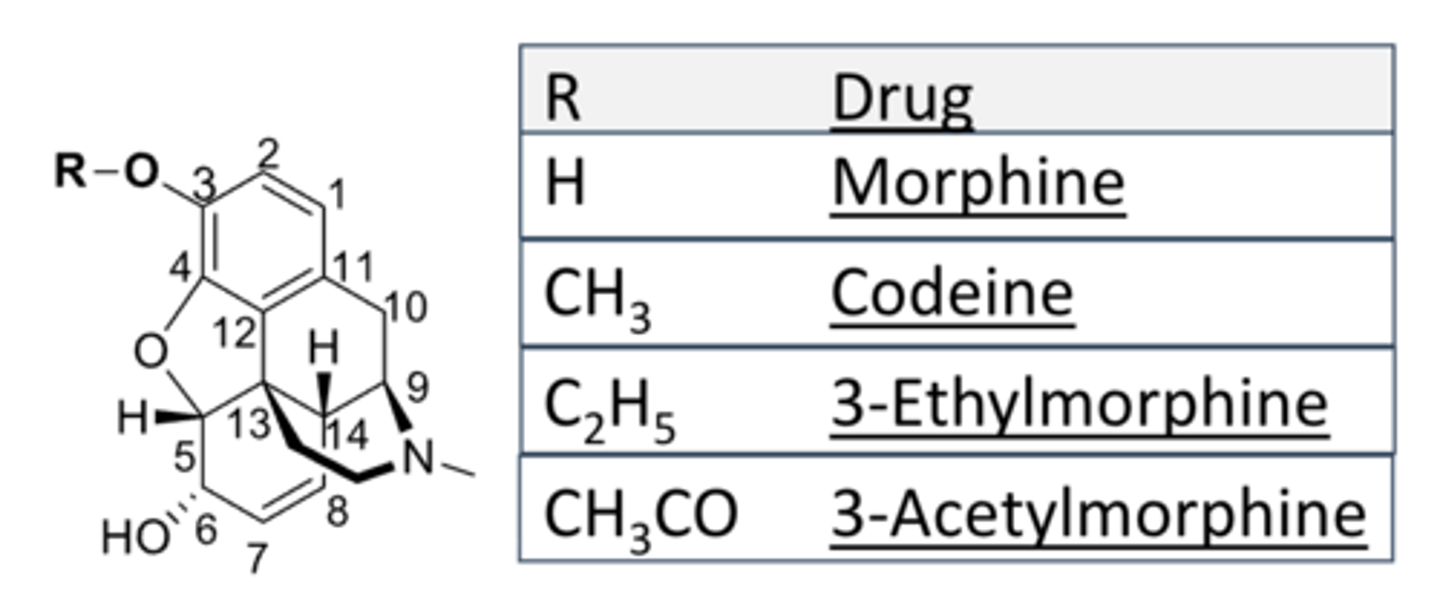
SAR of 6-alcoholic OH in opioids
- alcoholic OH is not important for activity
- masking 6-OH does not decrease analgesic activity, it often increases activity due to increased hydrophobicity
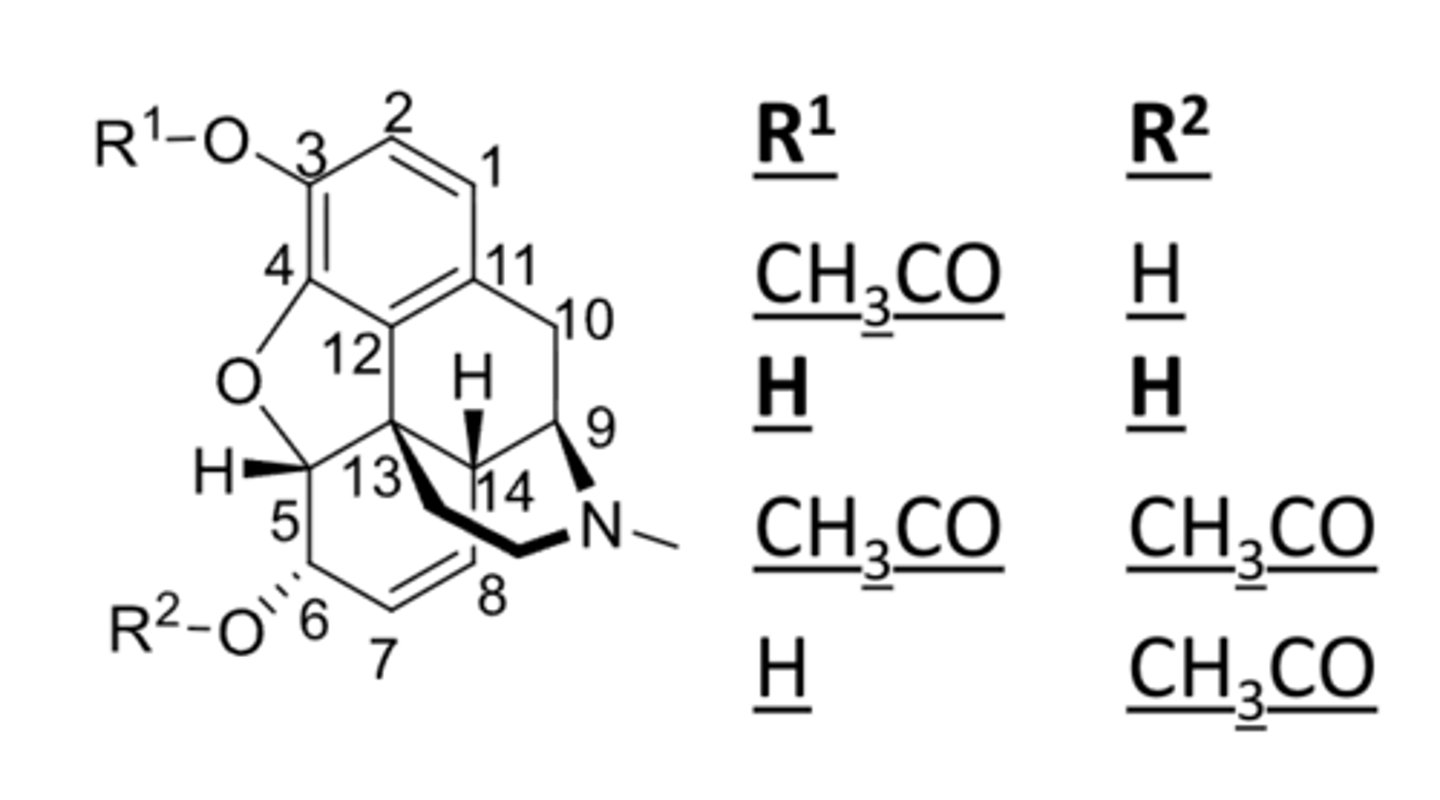
SAR of ring E in opioids
- ring E, particularly the basic N, is crucial for activity
- removing E results in complete loss of activity
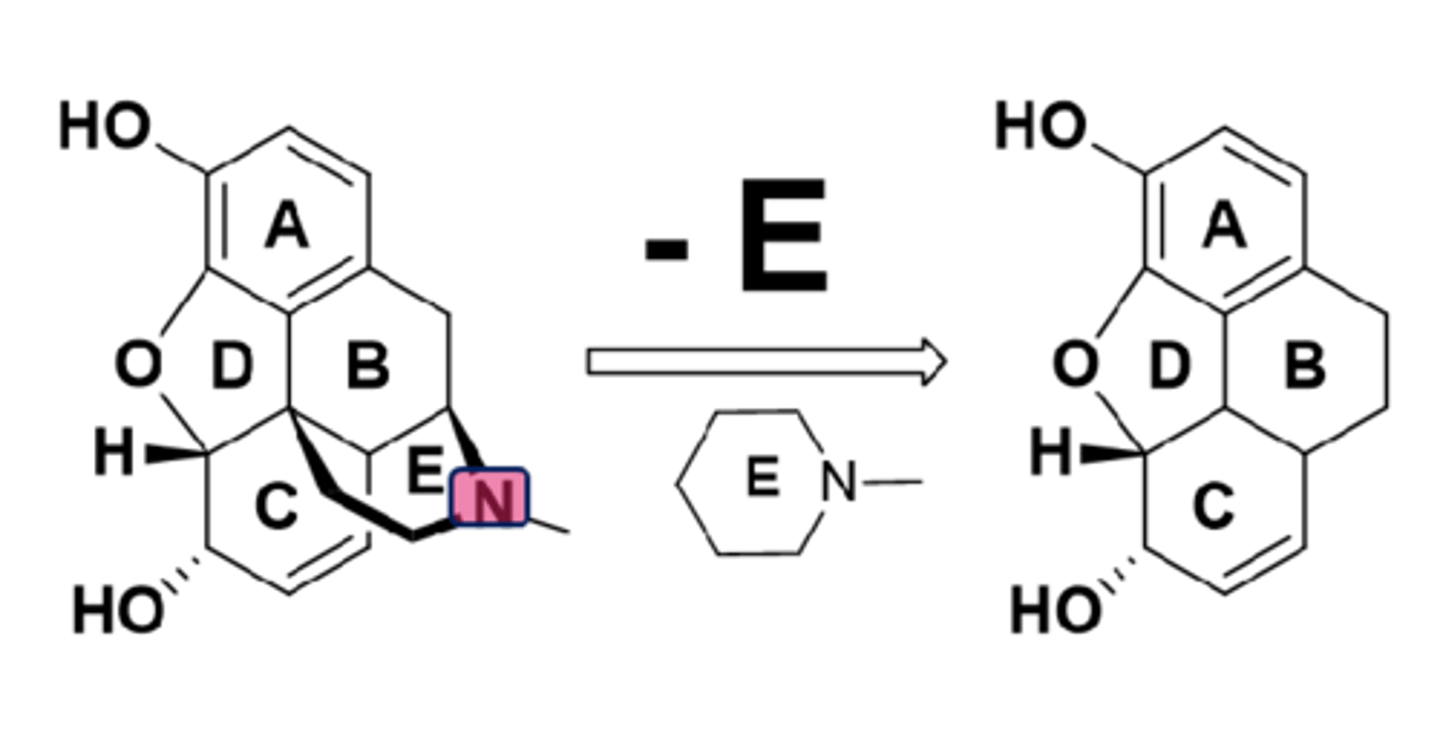
SAR of ring D in opioids
- ring D is not essential for activity
- activity retains
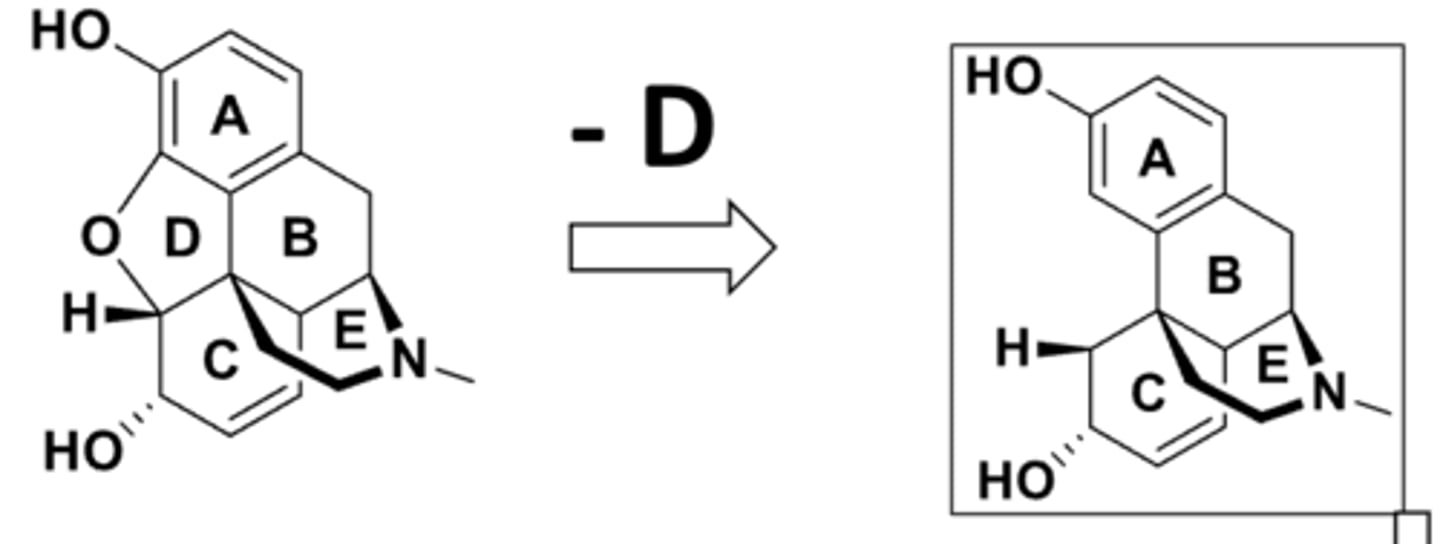
SAR of ring C in opioids
- ring C is not essential for activity
- metazocine is as potent as morphine
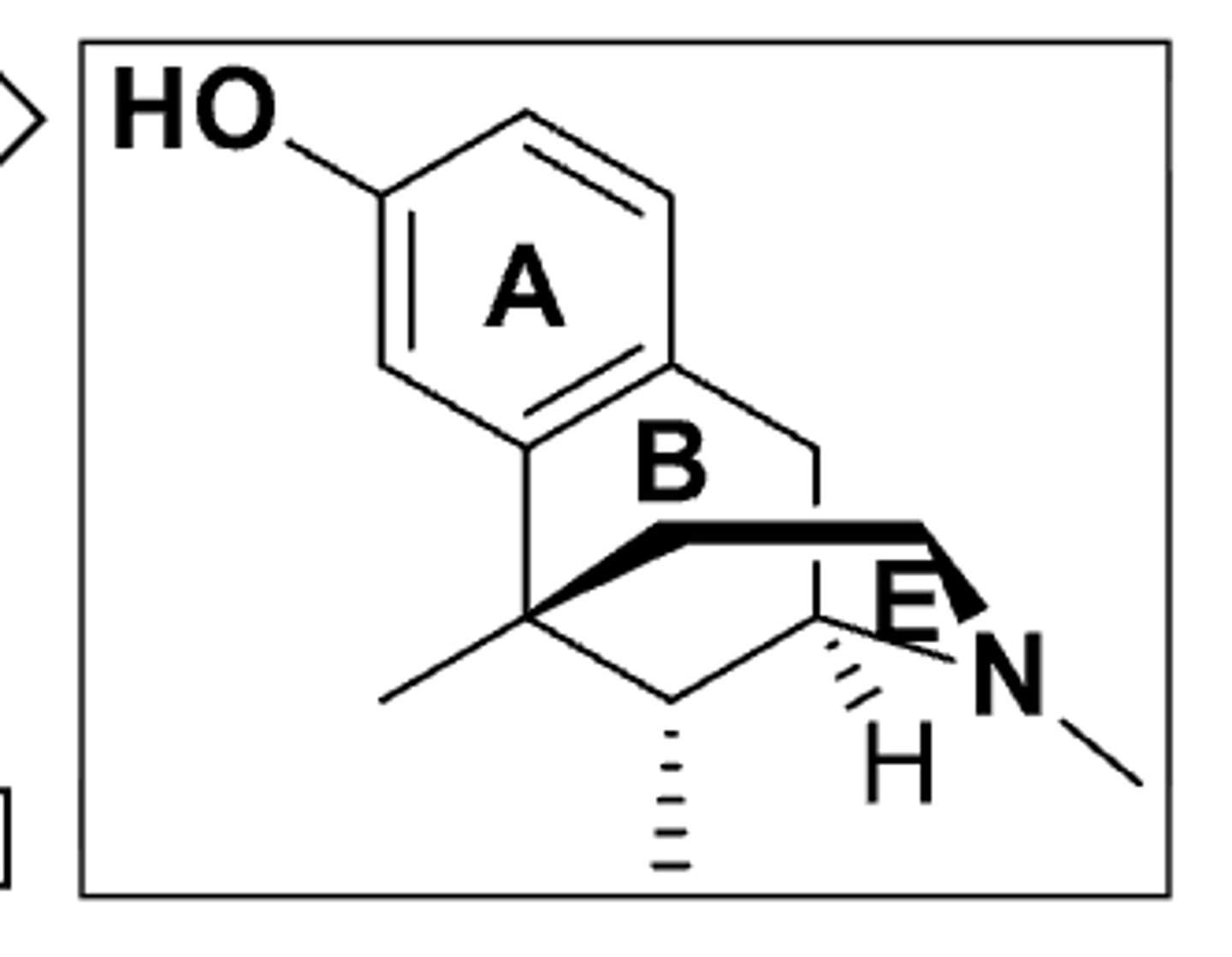
SAR of ring B in opioids
- ring B is not essential for activity
- pethidine has 20% of the activity of morphine
morphine structure and activity properties
- 5 fused ring system (increases selectivity and affinity to receptor)
- rigid T shape (increases brain penetration)
- 2 hydroxyl groups (phenolic OH is crucial for activity)
- 5 chiral centers (crucial for activity but only (-) is active)
- a basic tertiary amine (basic N (alkaloid) is crucial for activity)
codeine
- morphine opioid analgesic that is less potent than morphine and is used in pain and cough
- prodrug of morphine
- binding affinity is only 0.1% that of morphine and resulting analgesic effect is 20% that of morphine
- metabolized by CYP2D6 in the liver into morphine to produce analgesia (O-demethylation and UGTs)
- CYP2D6 is highly polymorphic and considerable variation exists in the efficiency and amount of CYP2D6 enzyme produced between individuals
- individual patient's response to codeine varies from no effect to high sensitivity
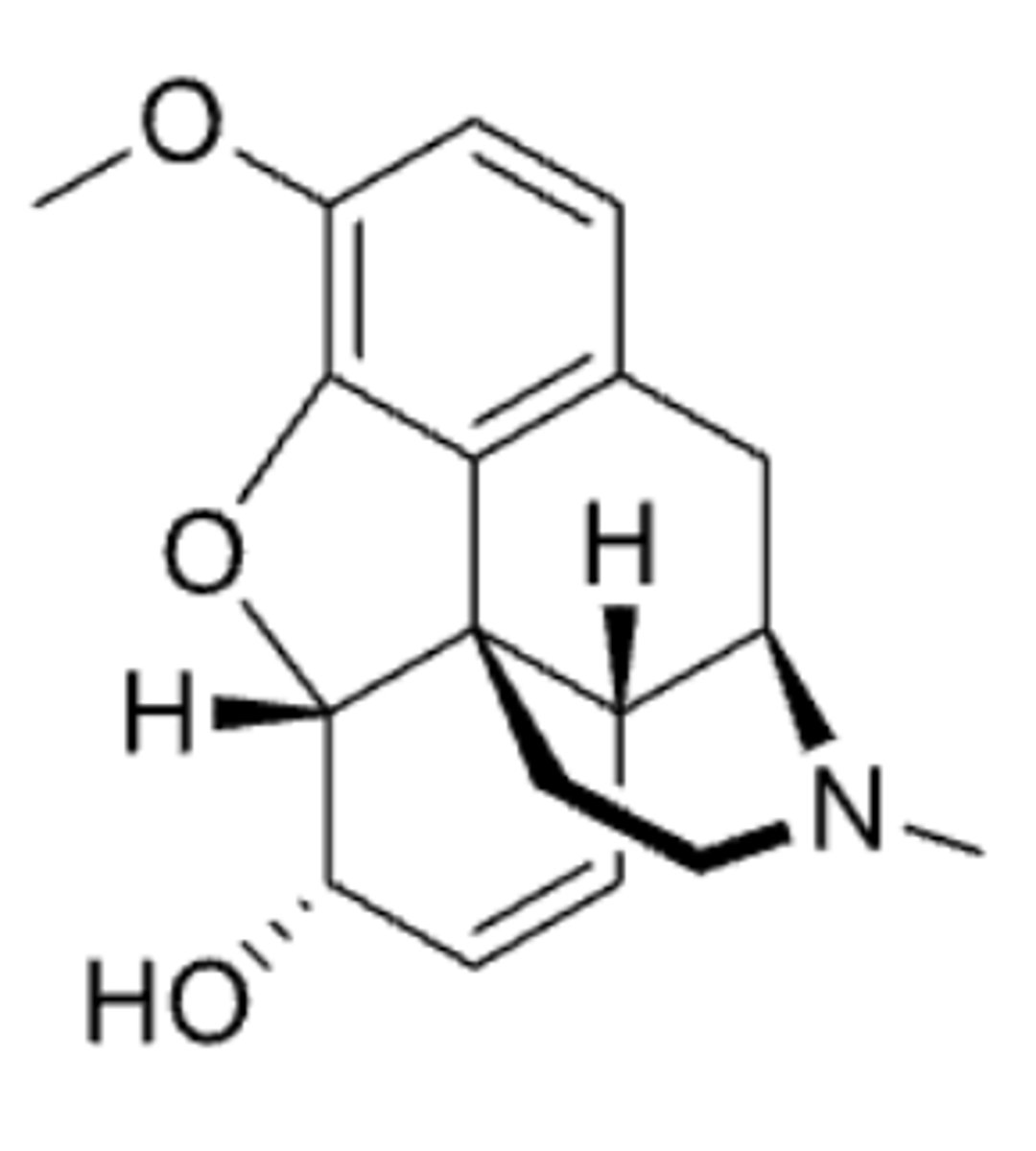
hydromorphone
- morphine opioid analgesic that is more potent than morphine and is used in pain
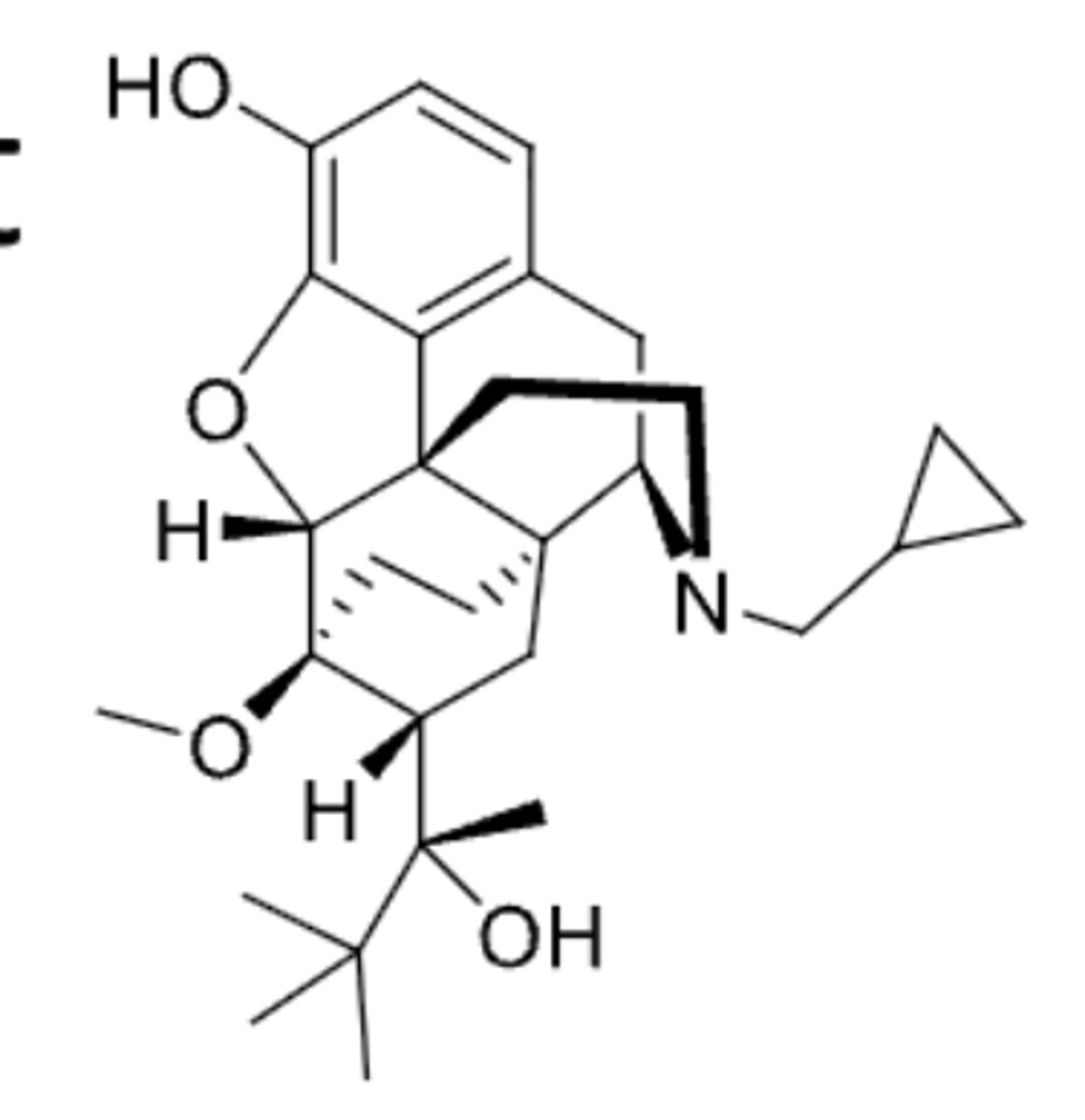
buprenorphine
- morphine opioid analgesic that is a partial agonist and is used in pain and withdrawal symptoms
- occupies MOR but activates MOR at a lower efficacy than morphine and has a "ceiling" on analgesia and side effects
- lower risk of respiratory depression than a full agonist morphine
- tolerance development with chronic use
- low-dose injection and transdermal patches for moderate-severe chronic pain
- high-dose SL tablets also containing naloxone are used for the treatment of opioid addictions
- medication for opioid use disorder
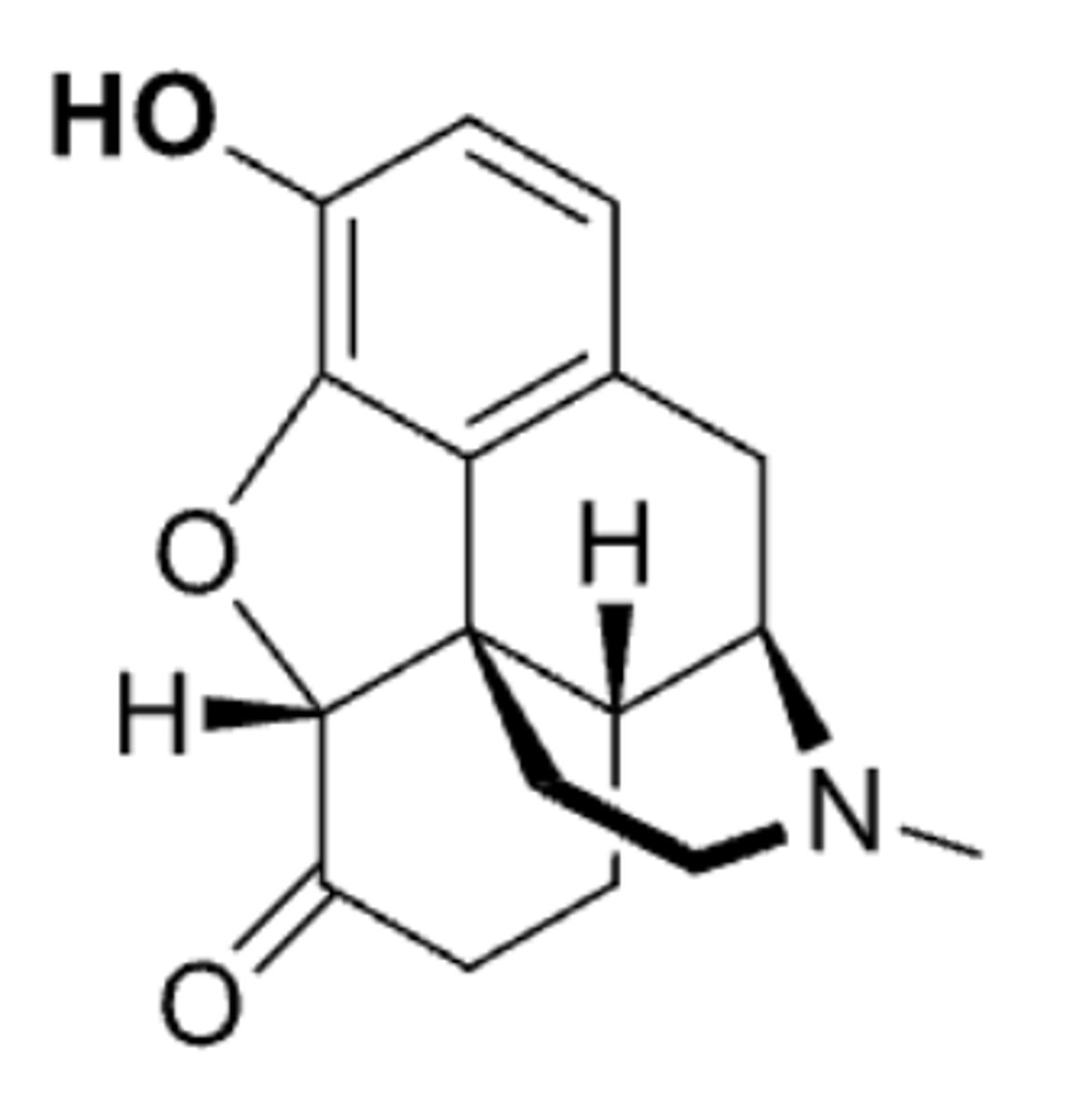
oxycodone
- morphine opioid analgesic that is more potent than codeine and is used in pain
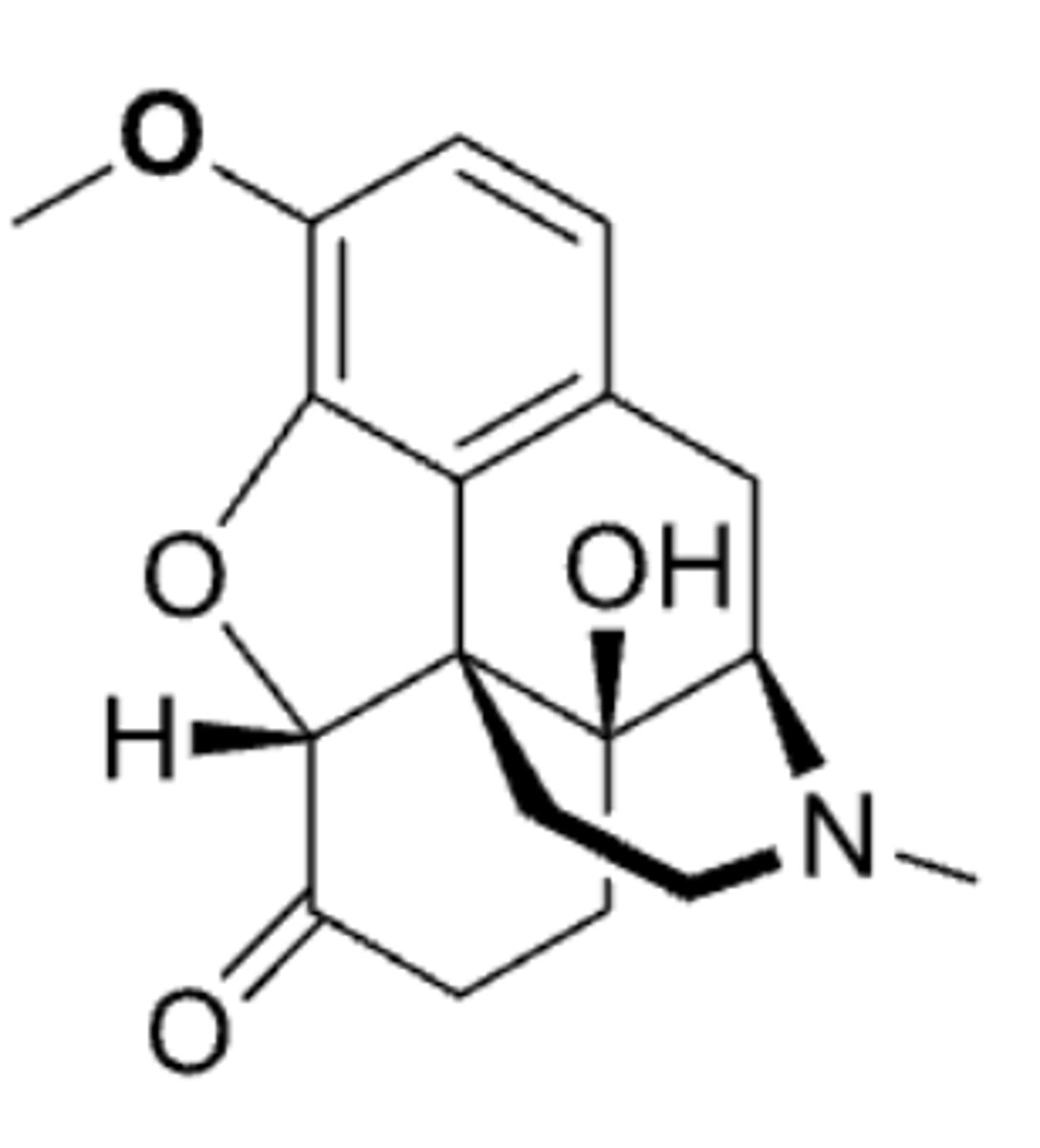
hydrocodone
- morphine opioid analgesic that is used in pain and cough
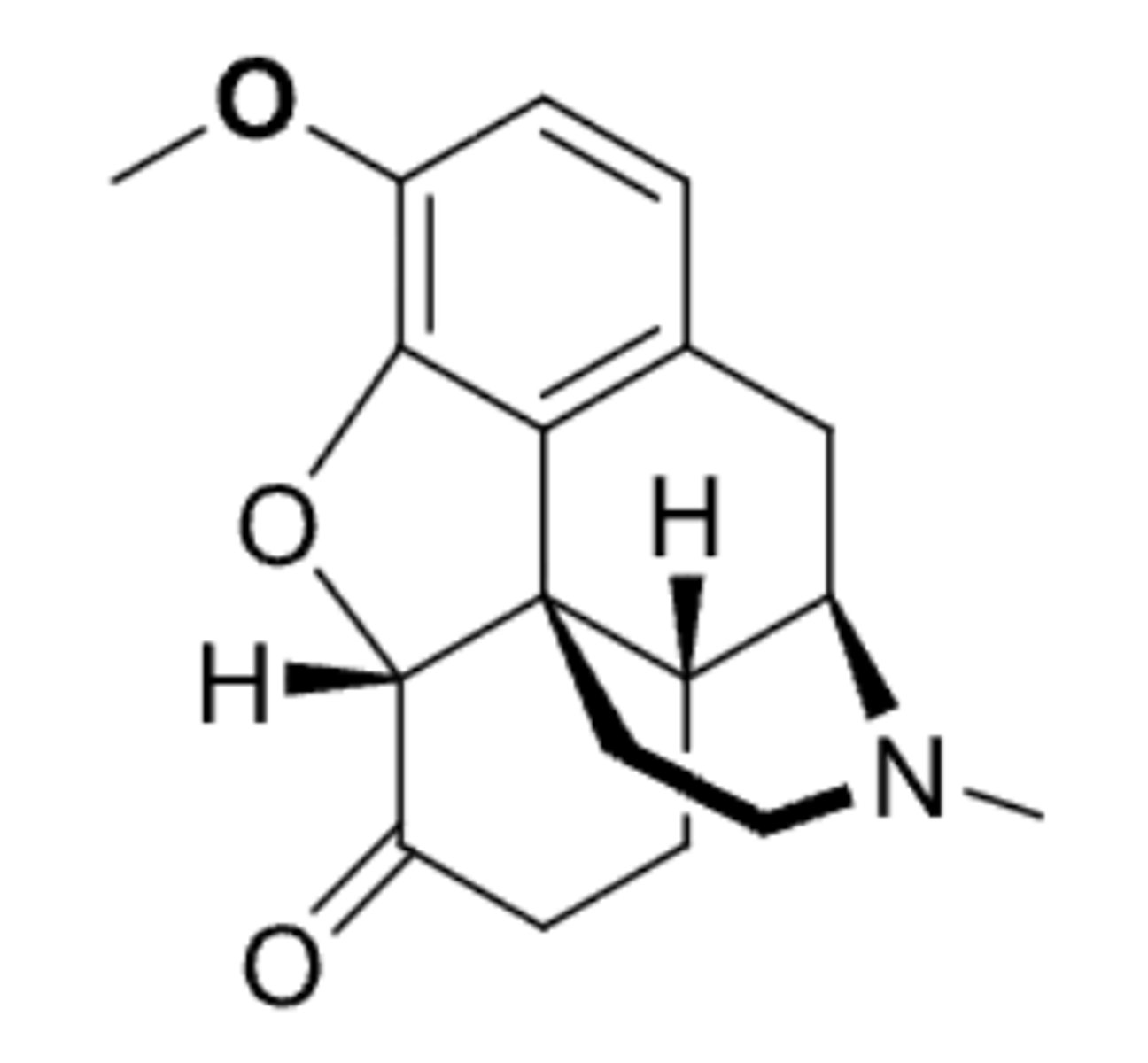
tramadol
- synthetic opioid analgesic
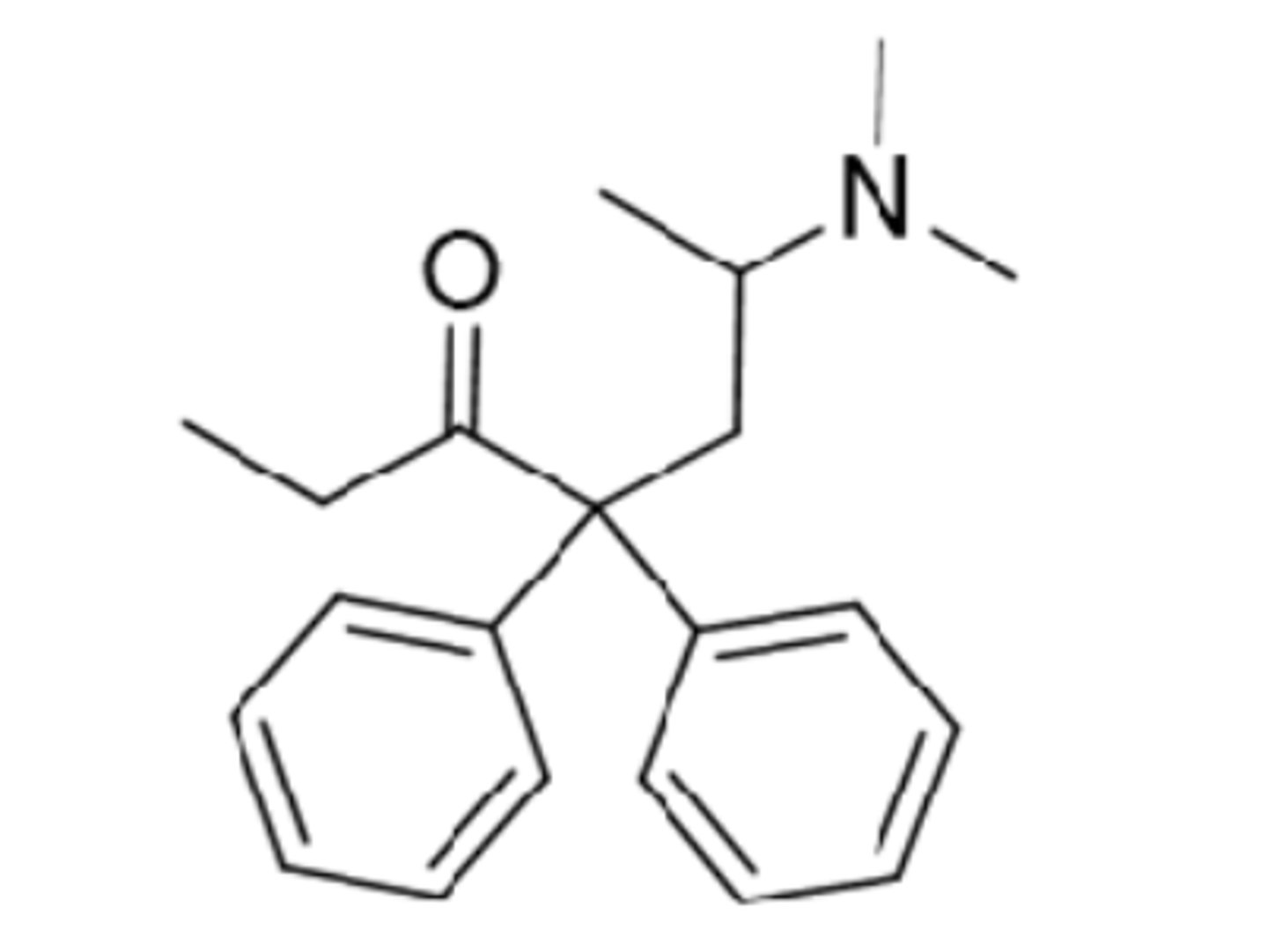
fentanyl
- synthetic opioid analgesic that is more potent than morphine and is used in severe pain and sedation
- full agonist at MOR and ~50-100 times more potent than morphine after systemic delivery (extremely potent analgesic)
- accounts for the rapid increasing rates of overdose death
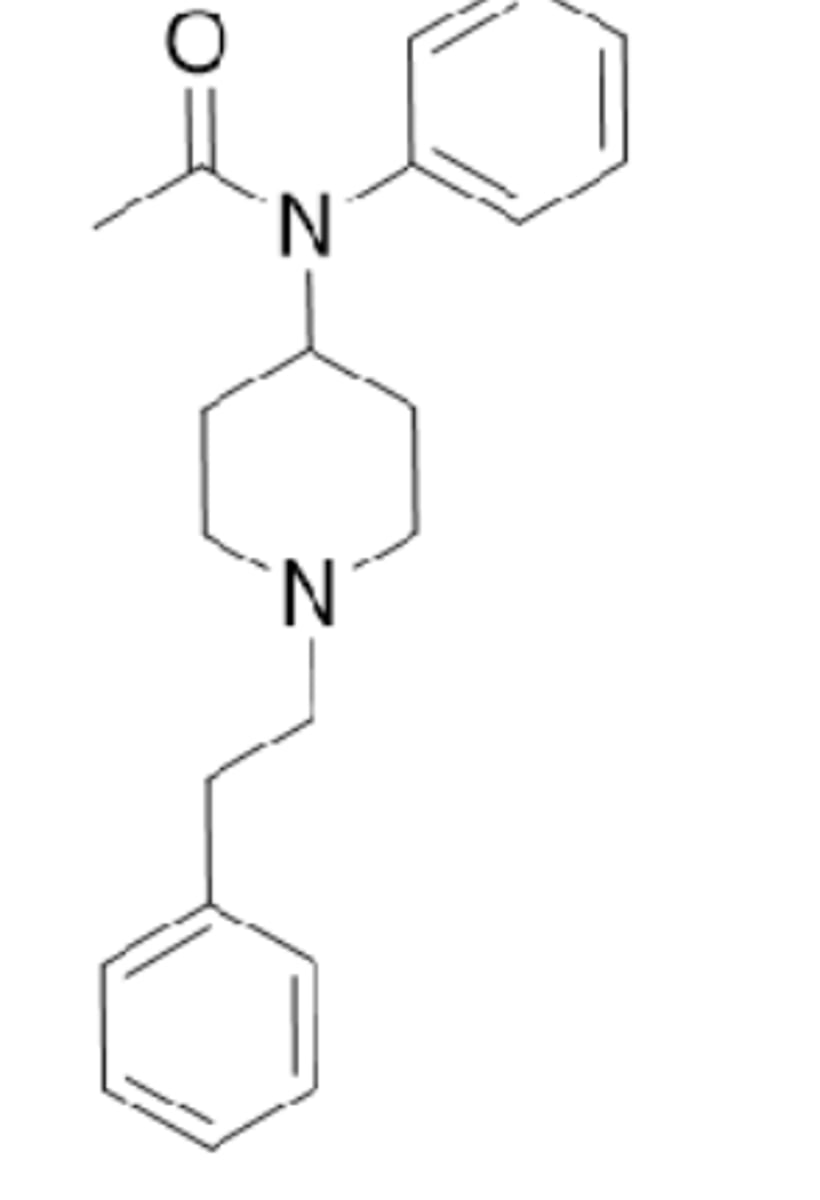
methadone
- synthetic opioid analgesic that is used in opioid dependence (less craving) and pain
- mu agonist that is more selective to mu than morphine
- can reduce symptoms of withdrawal and reduce craving
- one of the most common medicines used to treat opioid dependence
current strategies for safer opioids
- G protein-based and B-arrestin-biased agonists that allow for receptor internalization
- biased agonism produces more precise therapeutic effects
- identification of biased ligands at GPCRs became a standard approach in GPCRs drug discovery
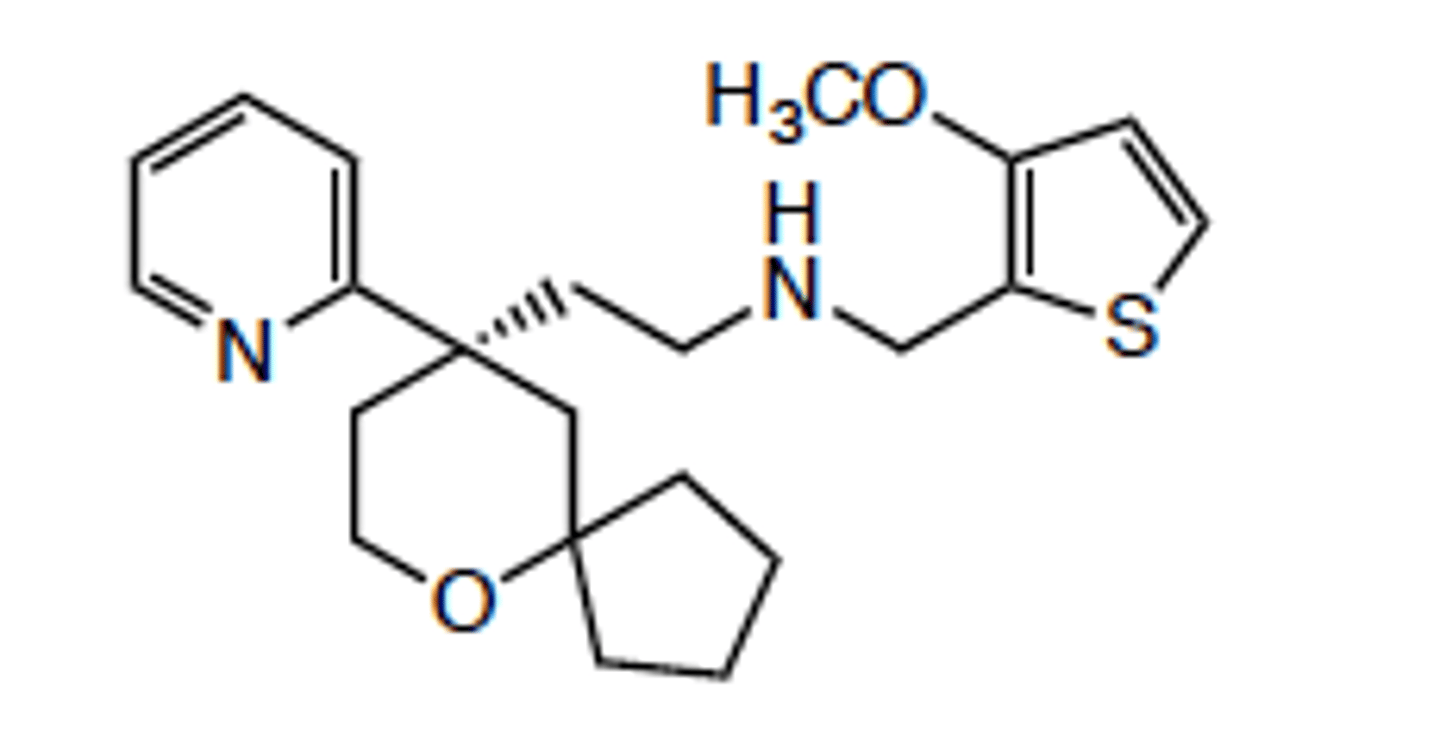
Olinvyk (oliceridine)
- a G protein based mu-opioid agonist
- approved for acute pain management in certain adults for short-term IV use in hospitals and other controlled clinical settings
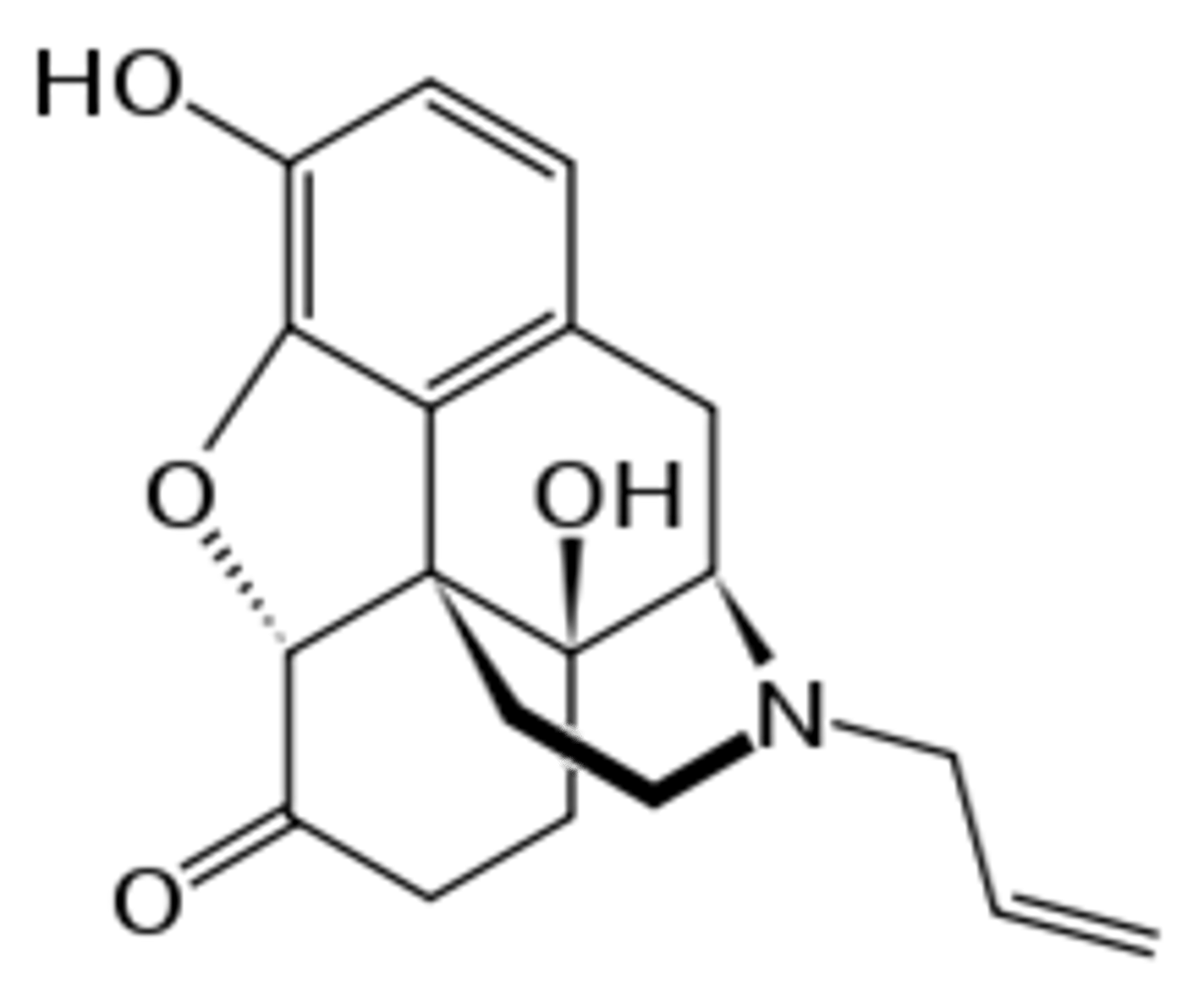
naloxone
- mu-opioid receptor antagonist/competitive MOR antagonist
- CNS acting
- rapid metabolism
- acts within minutes and lasts ~1 hour
- used for treating acute opioid overdose and alcoholism opioid abuse
- can NOT reverse fentanyl overdose - need repeated doses
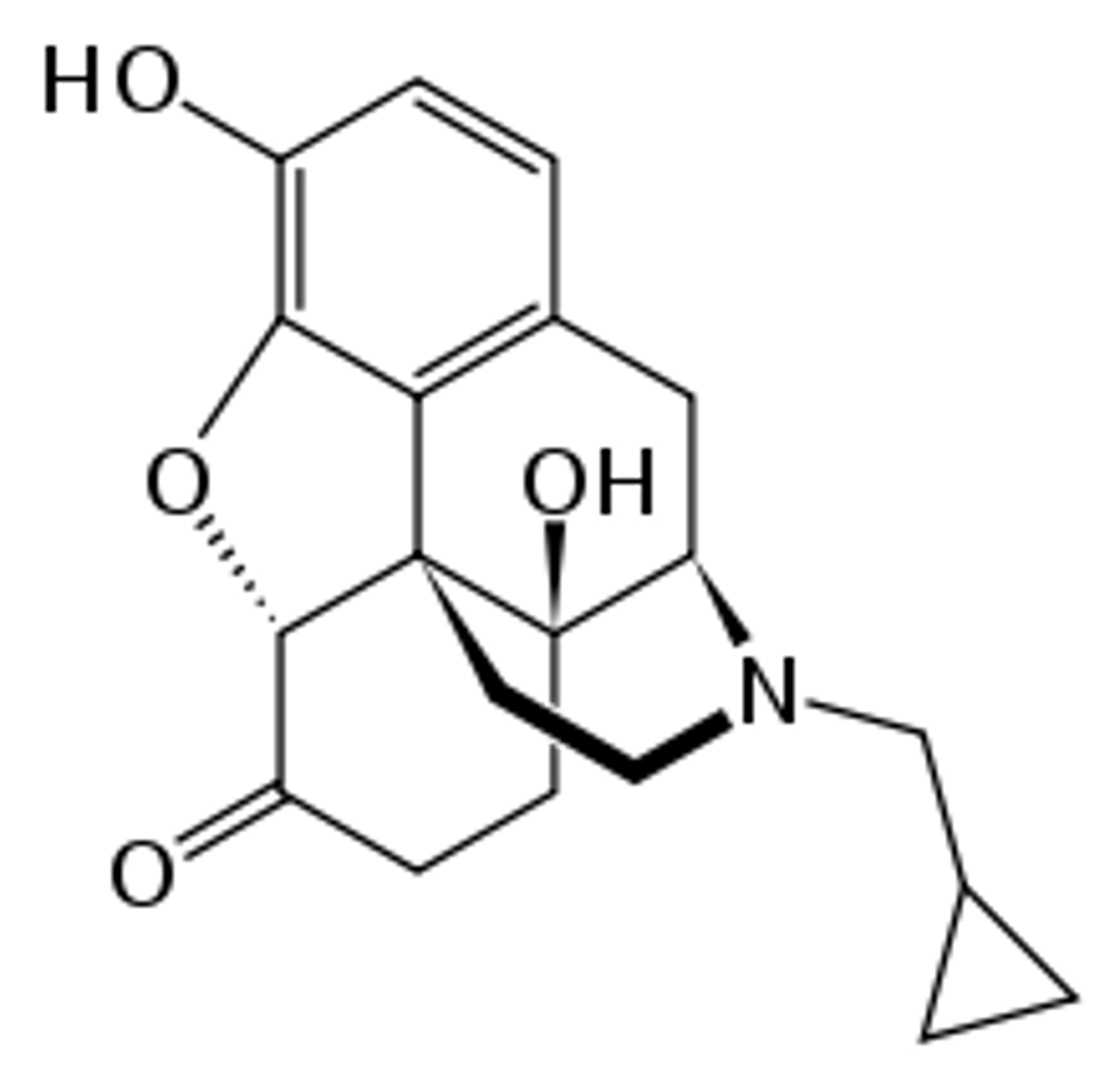
naltrexone
- mu-opioid receptor antagonist/competitive MOR antagonist
- CNS acting
- first-pass liver metabolite
- acts in ~30 minutes and lasts a day
- can NOT treat acute opioid overdose
- used for treating alcoholism and drug craving
MOR ligand agonist
- morphine
- used for analgesia
- high risk of adverse effects including respiratory depression, constipation, and addiction
MOR ligand partial agonist
- buprenorphine
- used for analgesia and the treatment of addiction
- lower risk of adverse effects
MOR ligands antagonist
- naloxone
- used for counteracting drug overdose
- no opioid analgesic effect or adverse effects
neurons (nerve cells)
- electrically excitable cells that express a variety of ion channels and ion transporters that allow them to conduct impulses or action potentials that ultimately trigger release of neurotransmitters during chemical neurotransmission
- consist of 3 parts = dendrite, cell body (soma), axon
dendrites
- part of neuron that recieves input from other cells
- recieves electrical stimulation
soma
- cell body of neuron
- contains nucleus
axon
- part of neuron through which action potential is generated and travels the length of to cause the release of neurotransmitters
- responsible for transmitting electrical impulses away from nerve cell body
axon terminal
- part of neuron which contains vesicles that store neurotransmitters for release at the synapse
communication between neurons
- an electrical signal travels down the axon, chemical neurotransmitter molecules are released, neurotransmitter molecules bind to receptor sites
- signal is picked up by the 2nd neuron and is either passed along or halted
- signal is also picked up by the 1st neuron causing reuptake of some of the released neurotransmitter
criteria of NTs
- chemical must be synthesized in the neuron
- when neuron is active the chemical must be released and produce a response in some target
- same response must be obtained when the chemical is experimentally placed on the target
- a mechanism must exist for removing the chemical from its site of activation after its work is done
types of NTs
- classified by the nature of material = amino acids, peptides, monoamines
- classified by function/effect = excitatory (glutamate), inhibitory (GABA), other psychological function (dopamine, serotonin, norepinephrine)
excitatory NTs
- glutamate (>90% of the synapses)
inhibitory NTs
- GABA (>90% of the synapses that don't use glutamate)
serotonin
- NT found in the enteric nervous system (ENS) and central nervous system (CNS)
- biosynthesis from tryptophan through a 2-step pathway
- 90% is synthesized and stored in GI tract and blood platelets
- also synthesized in brain where it performs its primary functions
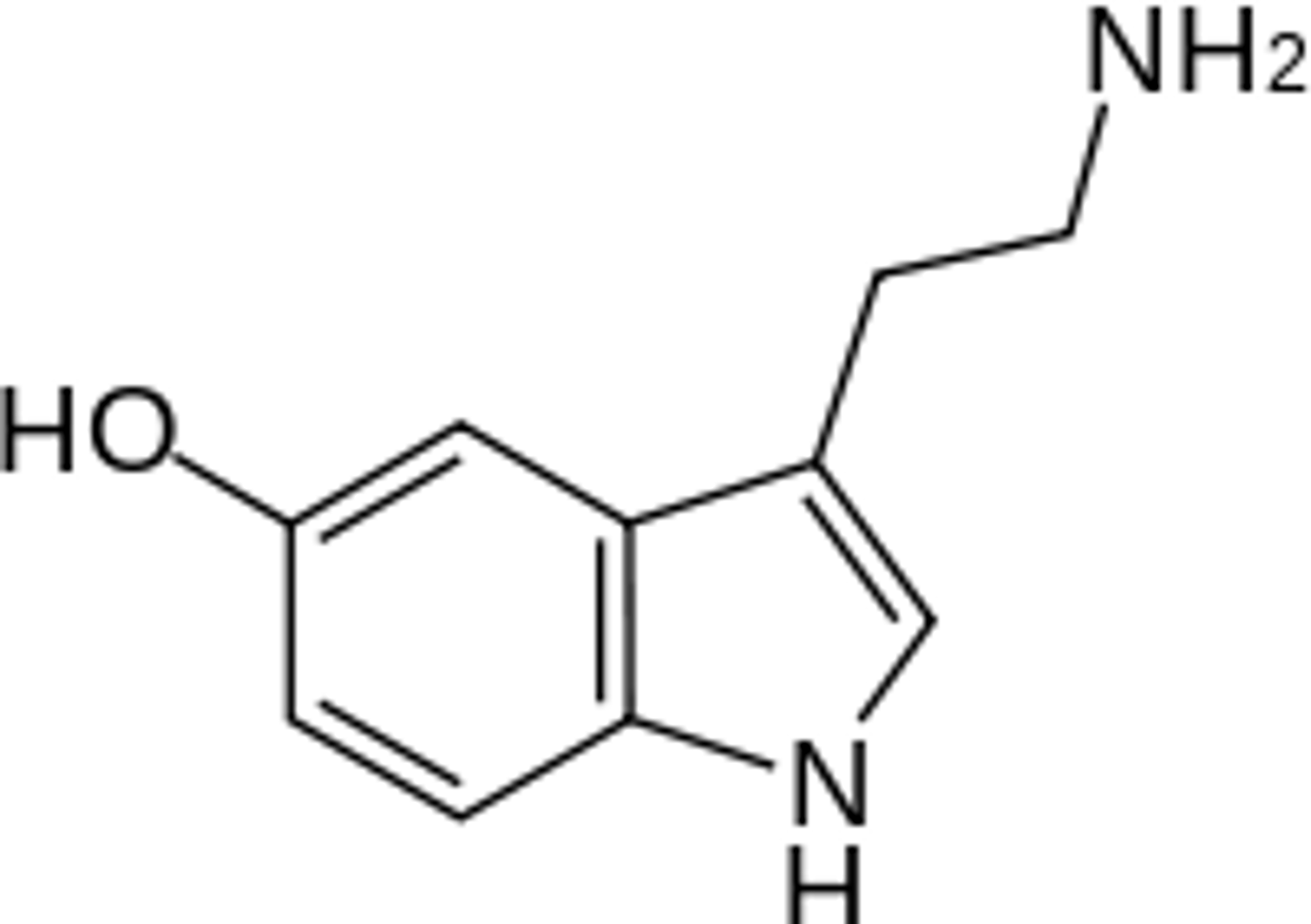
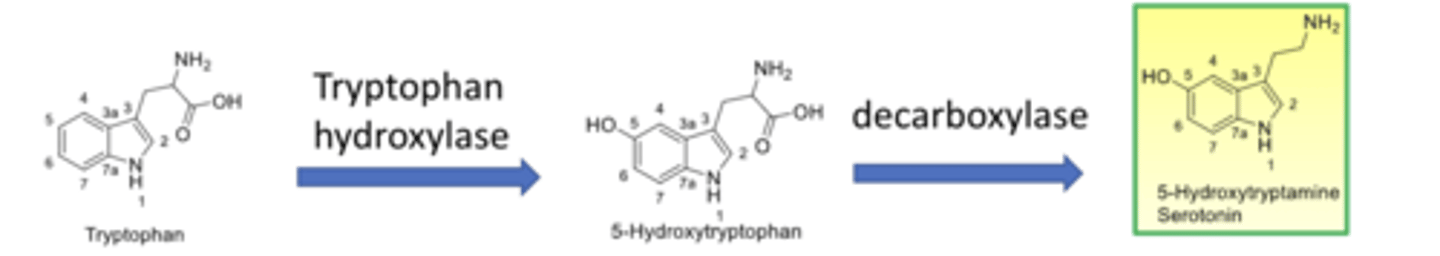
serotonin biosynthesis
- tryptophan converted to 5-hydroxytryptophan by tryptophan hydroxylase
- 5-hydroxytryptophan converted to 5-HT/serotonin by decarboxylase
serotonin metabolism
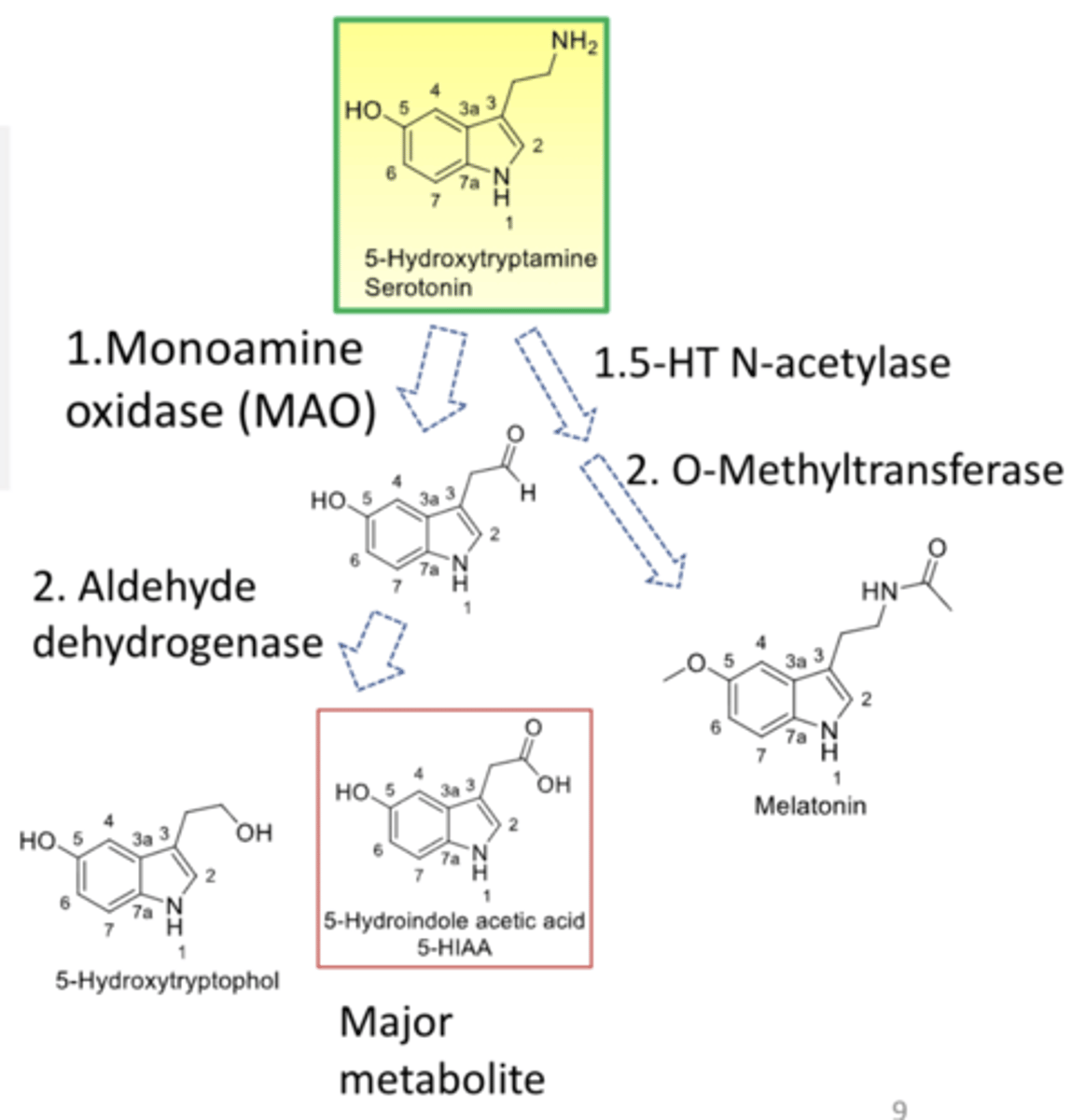
- mainly is metabolized in the liver to 5-HIAA by oxidative deamination by MAO and the aldehyde is then converted to 5-HIAA by aldehyde dehydrogenase or reduced to an alcohol
- melatonin is a metabolite by N-acetylation and O-methylation that serves a role in regulating circadian rhythms and shows promise in the treatment of sleep disturbances
- metabolites are excreted from urine
serotonin primary functions performed in brain
- influences a multitude of functions including sleep, cognition, sensory perception, motor activity, temperature regulation, nociception, mood, appetite, sexual behavior, and hormone secretion
- 5-HT neurons are principally in raphe nuclei of the brainstem and project throughout the brain and spinal cord
- control sleep-wake cycle, aggression and impulsivity, appetite and obesity
5-HT and 5-HT receptors in brain
- biosynthesized from tryptophan and packed in vesicle in presynaptic neuron
- released into synapse and activates cell-specific postsynaptic receptors leading to signal transduction
- taken up by 5-HT transporter (SERT) to terminate 5-HT action
- reuptake of 5-HT allows it to be either repacked in vesicle or metabolized by monoamine oxidase
5-HT receptors and functions
- 7 general 5-HT receptor families including 14 serotonin receptors
- all serotonin receptors are GPCRs except 5-HT3 which is a ligand-gated Na+ and K+ ion channel
serotonin signaling and depression and anxiety
- in general an imbalance/deficiency of the monoamine NTs (serotonin, norepinephrine, dopamine), particularly serotonin, may lead to depression
clinical significance of affecting 5-HT signaling
- act at monoamine oxidase (in presynaptic)
- act at 5-HT transporters (on presynaptic)
- act at 5-HT receptors (on postsynaptic)
pharmacotherapy for depression
- selective serotonin reuptake inhibitors (SSRIs)
- serotonin-norepinephrine reuptake inhibitors (SNRIs)
- tricyclic antidepressants (TCAs)
- serotonin antagonists and reuptake inhibitors (SARIs)
- serotonin modulators and stimulators (SMSs)
- norepinephrine reuptake inhibtors (NRIs)
- norepinephrine-dopamine reuptake inhibitors (NDRIs)
- monoamine oxidase inhibitors (MAOIs)
SSRIs
- reuptake of 5-HT into presynaptic terminals is mediated by SERT
- neuronal uptake is the primary process by which neurotransmission via 5-HT is terminated
- block reuptake of 5-HT to enhance and prolong serotonergic neurotransmission
- effective in treating major depressive disorder (MDD) and also used as anxiolytics in the treatment of generalized anxiety, panic, social anxiety, and OCD
- first-line drug for depression due to effectiveness and a lower risk of side effects compared to older antidepressants
- FDA black box warning = antidepressants may increase the risk of suicide in patients under 25
citalopram
- SSRI
- has a chiral center, 2 isoforms, sold as racemic mixture
- S-citalopram (escitalopram) less selective for SERT than citalopram
- used in treating major depressive disorder (MDD)
- common side effects = sexual dysfunction and abnormal heart rhythm
- serious side effect = increased risk of suicide in patients under 25
- discontinuation symptoms = electric shock sensations (brain shivers), dizziness, acute depression
- overdose may result in sedation, dizziness, tremors; deaths have occurred
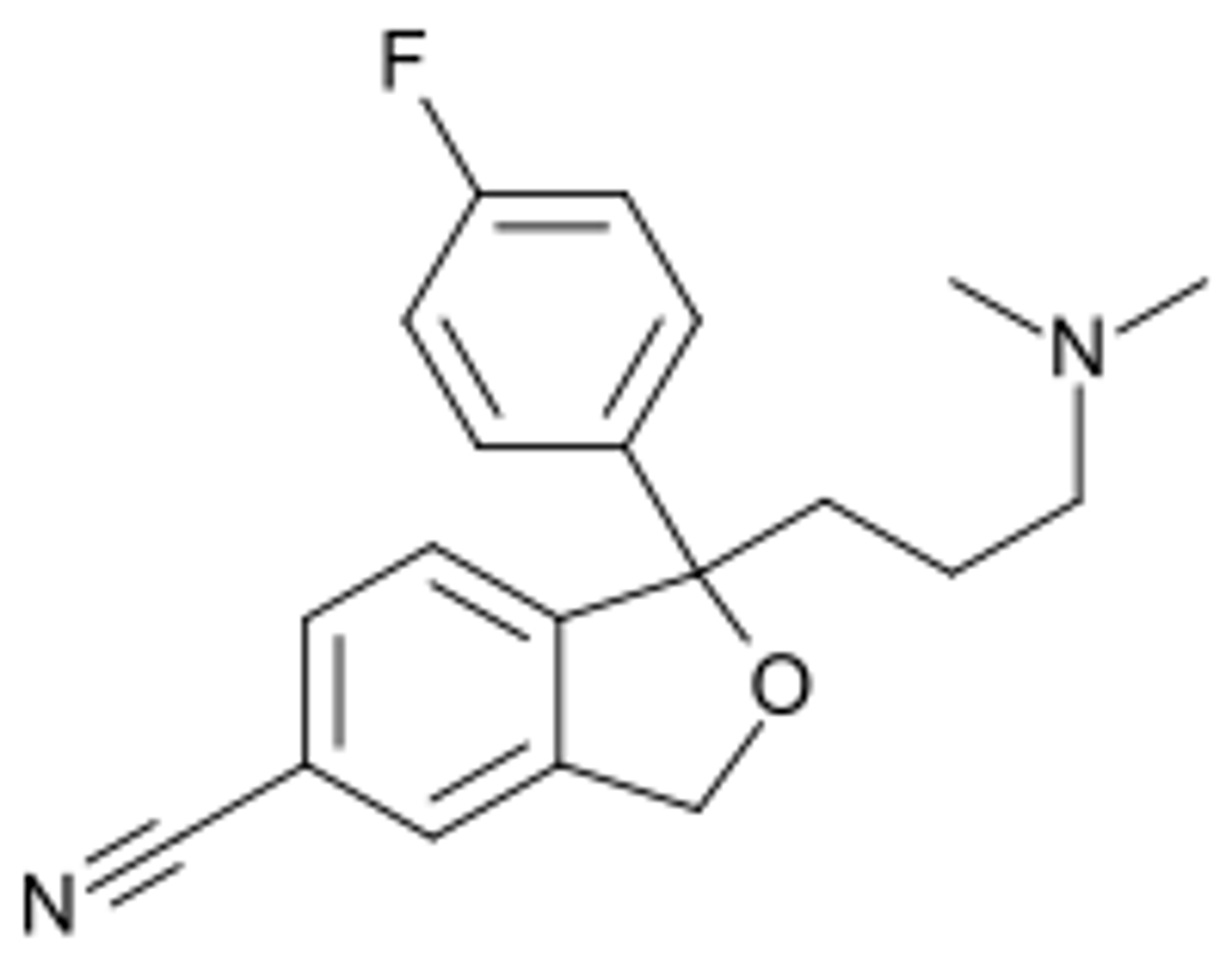
citalopram metabolism
- metabolized in the liver mainly by CYP2C19 and 3A4/2D6
- 80% cleared by the liver and 20% cleared by the kidney
citalopram drug interactions of
- may increase risk of bleeding if using together with NSAIDs, warfarin, and other blood thinners
- lead to serotonin syndrome (high body temp, agitation, etc.) if taken with St. John's wort (inhibits 5-HT reuptake) or other SSRIs
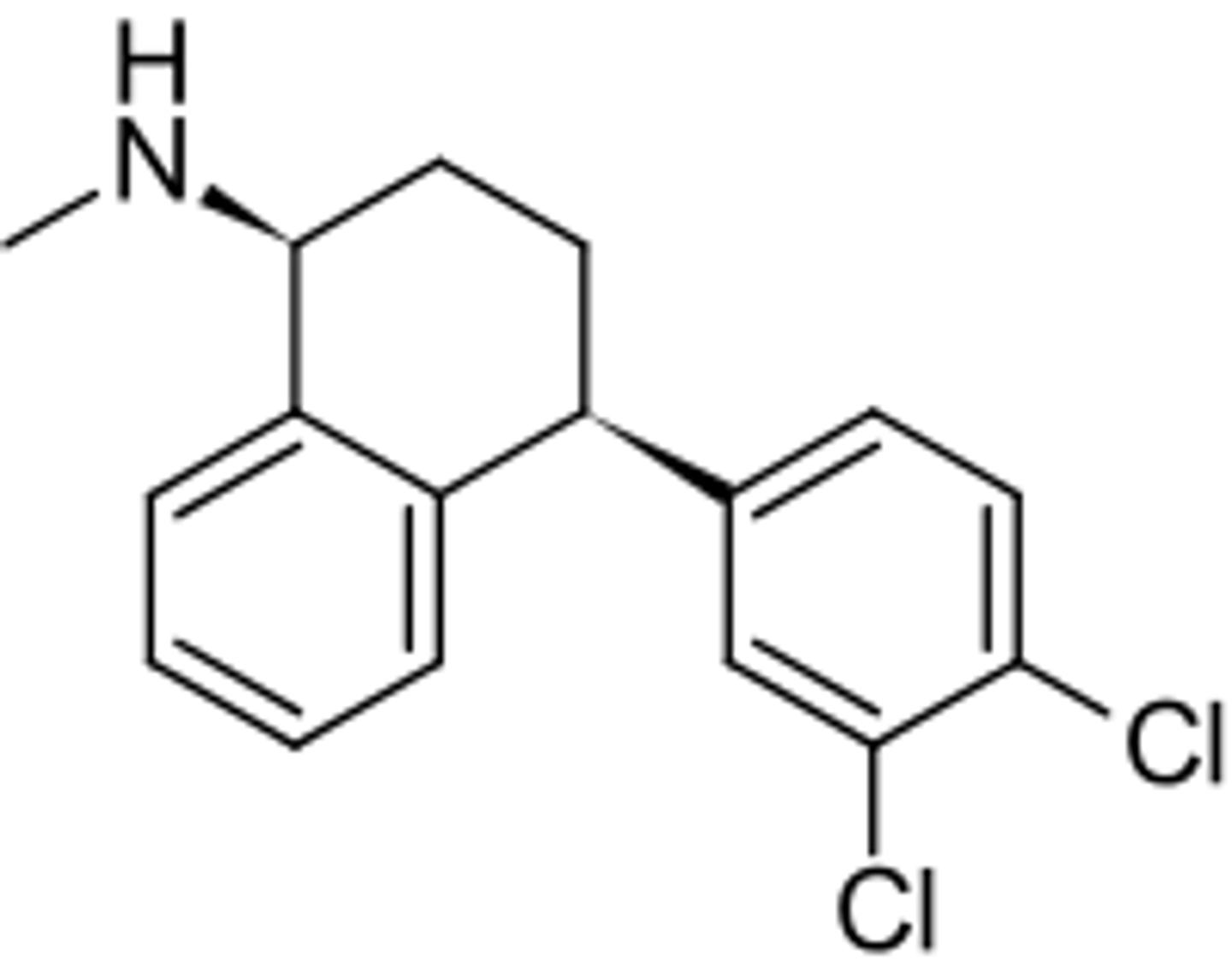
sertraline
- SSRI
- most commonly prescribed psychiatric medicine in the US
- treatment of MDD, OCD, PTSD, panic disorder, social anxiety disorder, etc.
- common side effects = sexual dysfunction and abnormal heart rhythm
- serious side effect = increased risk of suicide in patients under 25
- discontinuation symptoms
- its toxicity in overdose is considered relatively low as with most other SSRIs
- major metabolite norsertraline is less active than sertraline
paroxetine
- SSRI
- metabolized in liver by CYP2D6, 3A4, and 1A2 via demethylation and dealkylation
- same common side effects as other SSRIs
- increased risk of suicide in patients under 25
- discontinuation symptoms
- overdose may result in nausea, vomiting, sedation, dizziness, sweating, seizures
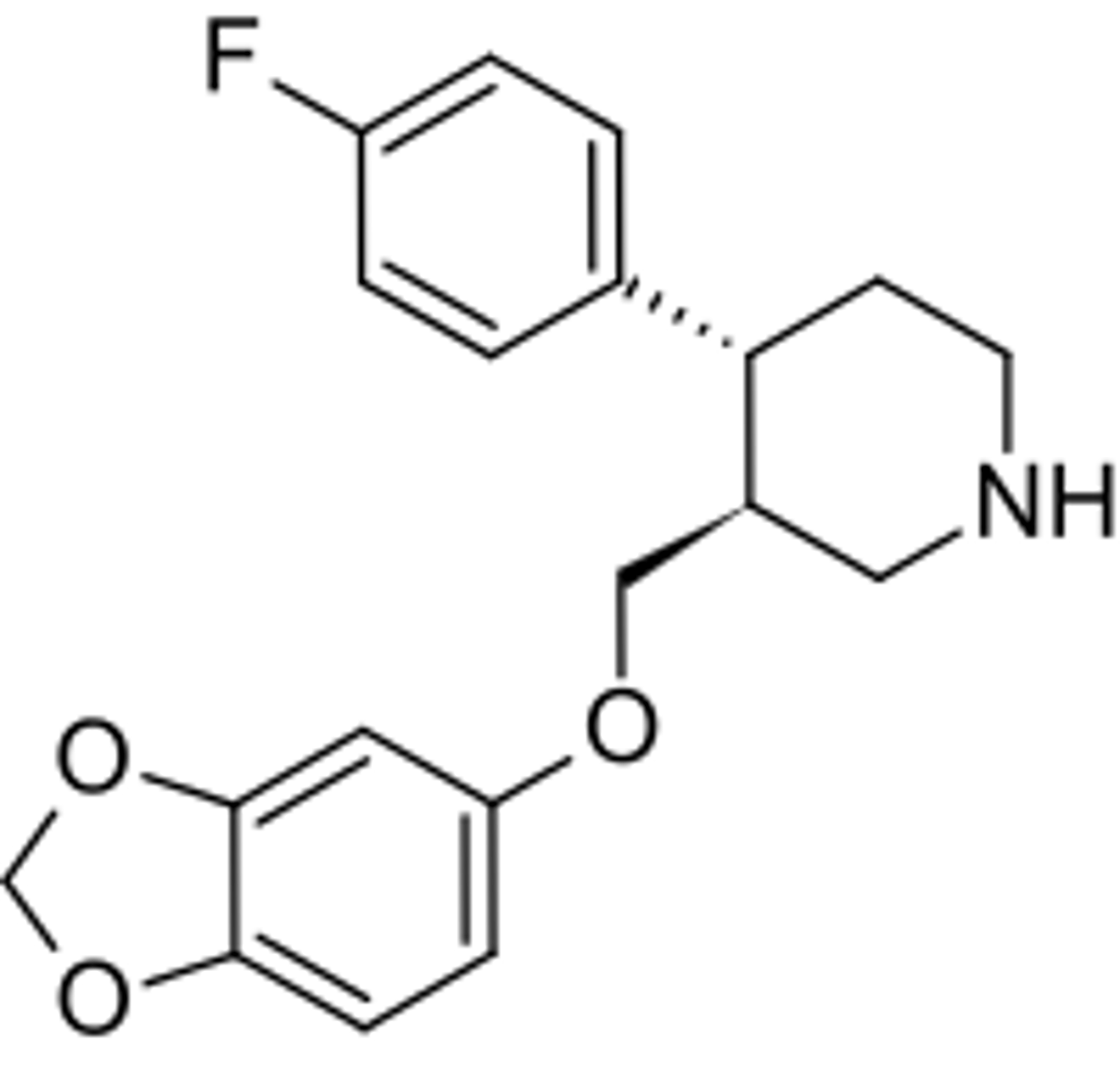
fluvoxamine
- SSRI
- first SSRI approved for the treatment of OCD; also for the treatment of depression and anxiety disorders
- has E configuration
- same common side effects as other SSRIs
- has more GI side effects than other SSRIs
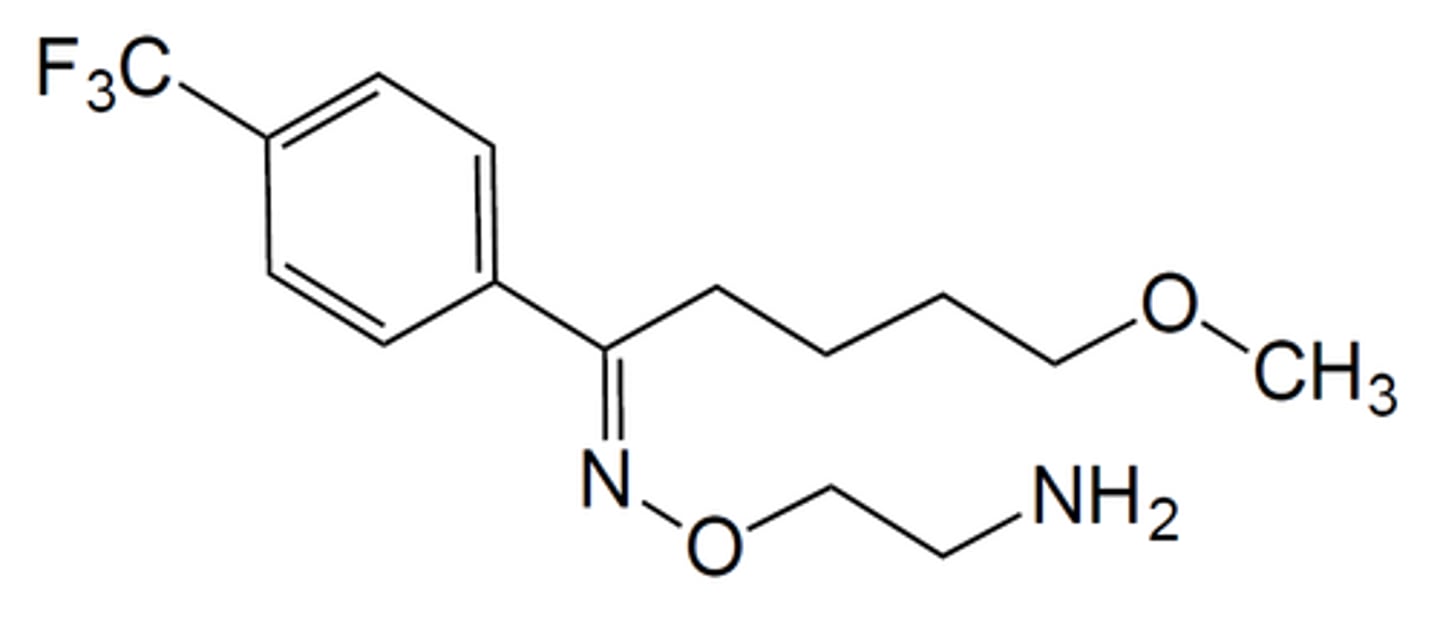
fluoxetine
- SSRI
- sold as racemic mixture
- metabolized in the liver by CYP2D6
- major metabolite is demethylated product norfluoxetine
- both fluoxetine and norfluoxetine inhibit CYP2D6
- has extremely long half-life which makes it less common to develop discontinuation syndrome following cessation of therapy compared to other SSRIs (elimination in 1-3 days)
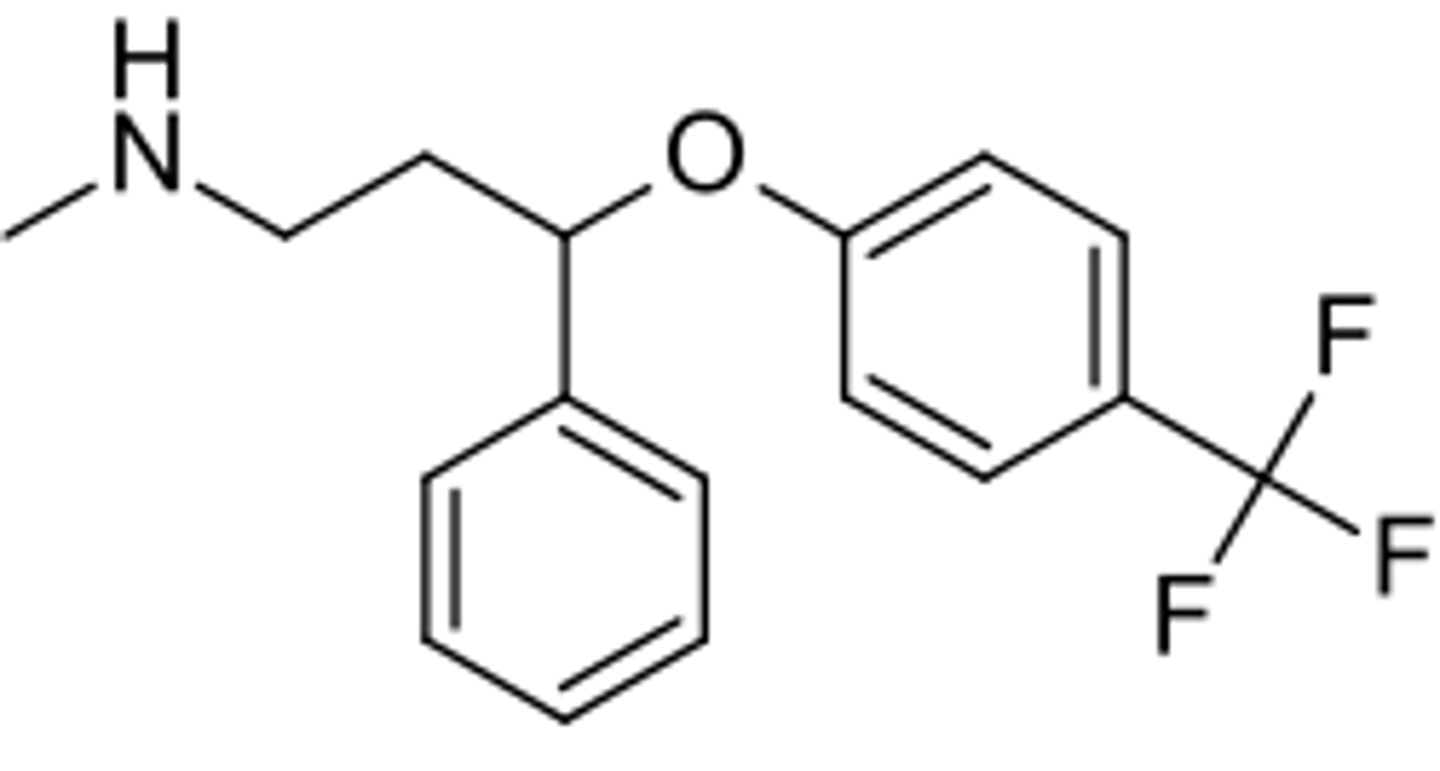
SNRIs
- medications with a non-tricyclic structure that inhibit the reuptake of both 5-HT and NE approved for treatment of depression, anxiety disorders, pain, etc.
- FDA black box warning = antidepressants may increase the risk of suicide in patients under 25
- inhibit both SERT and NET and cause serotonergic or noradrenergic neurotransmission
- may improve overall treatment response compared to SSRIs
- repeated treatment reduces the expression of SERT or NET resulting in reduced neurotransmitter clearance and increased serotonergic or noradrenergic neurotransmission
venlafaxine
- SNRI
- sold as racemic mixture
- reserved as second-line treatment for depression due to a combination of its superior efficacy to the first-line SSRI treatments and greater frequency of side effects
- patients taking venlafaxine had significantly higher risk of completed suicide than the ones taking SSRIs
- metabolized in the liver by CYP2D6
- major metabolite is desvenlafaxine which is as potent as venlafaxine
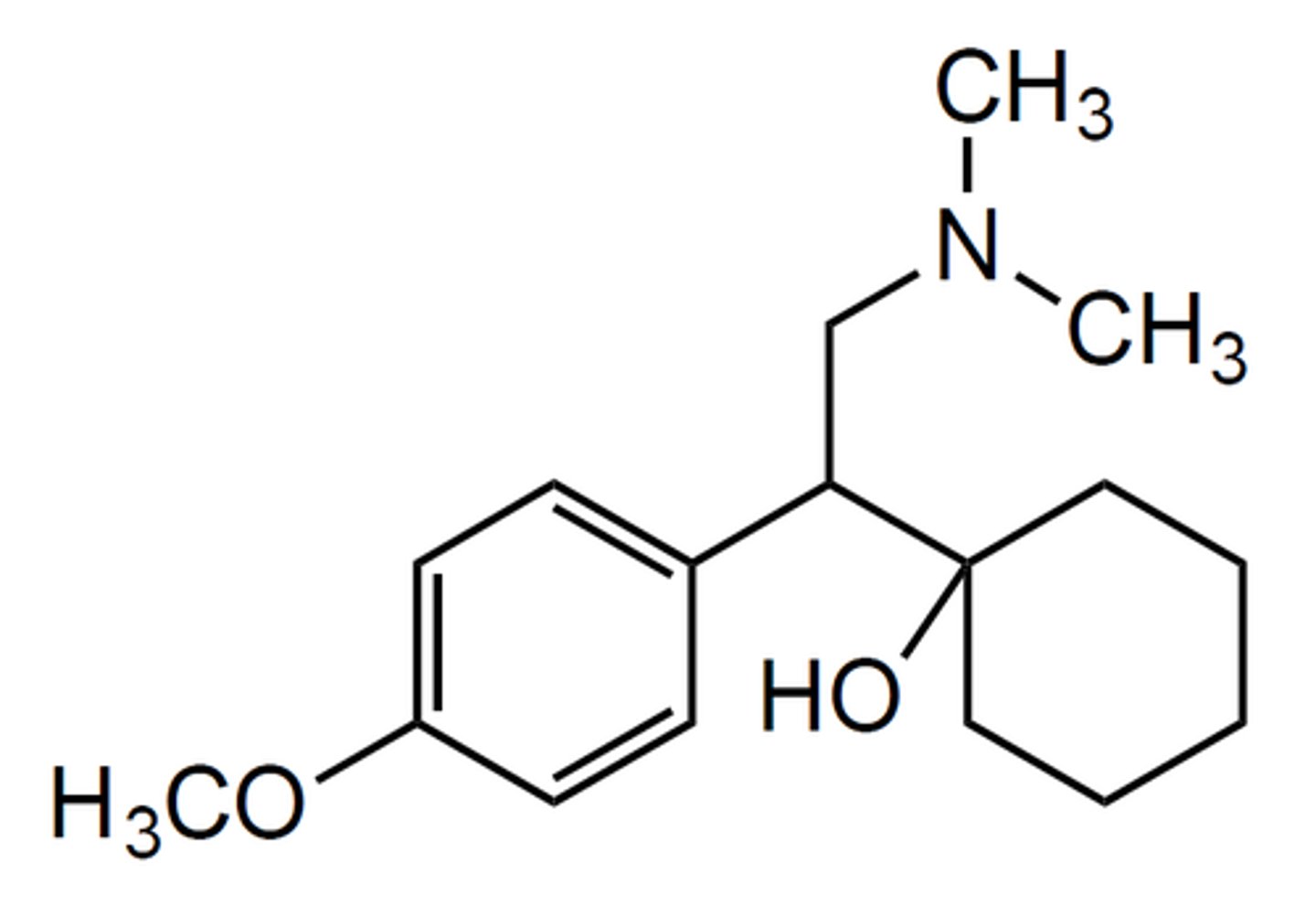
desvenlafaxine
- SNRI
- major metabolite of venlafaxine that is more potent (higher binding affinity to both SERT and NET) and is able to be continually metabolized
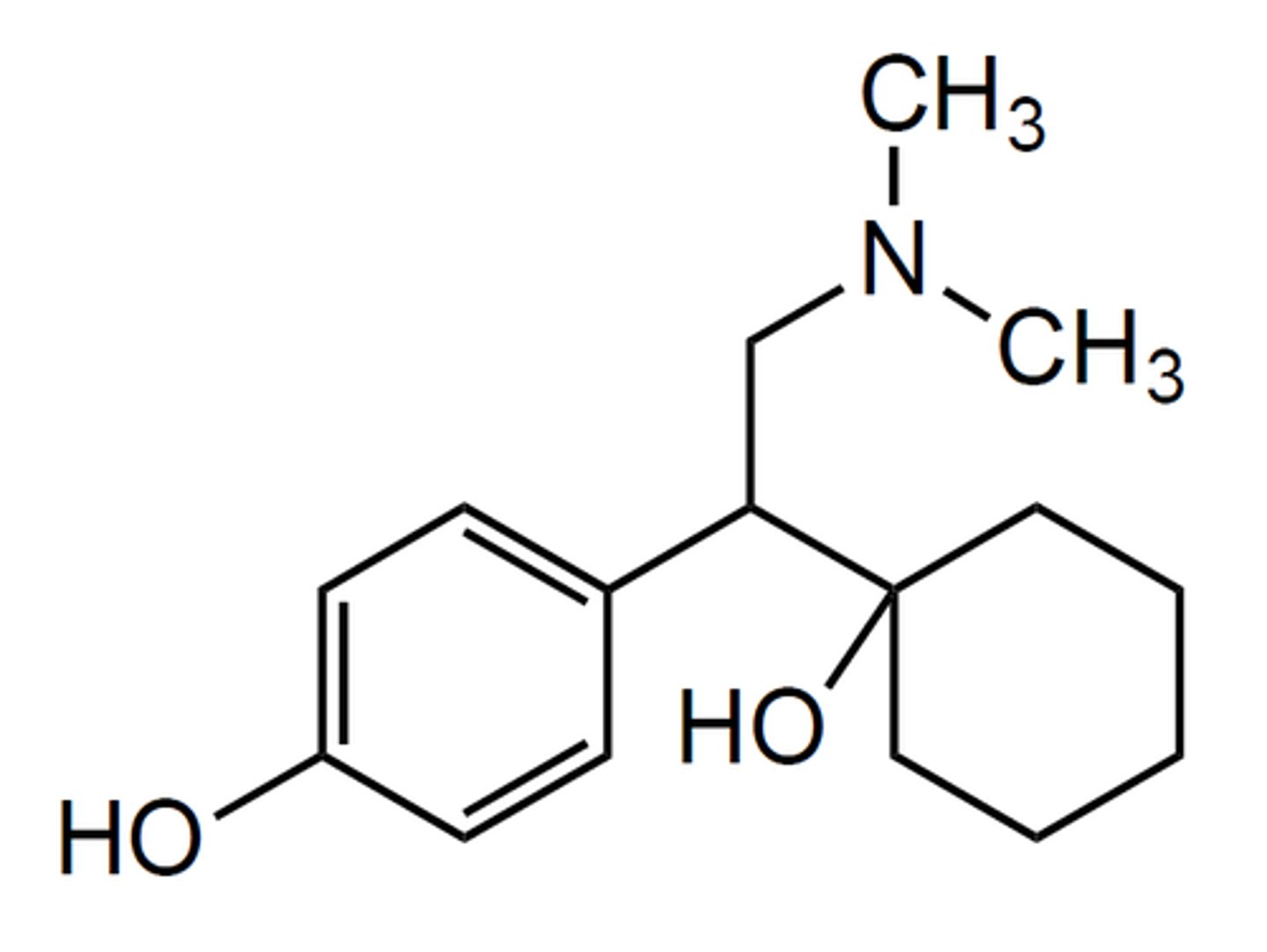
duloxetine
- SNRI
- S-enantiomer inhibits serotonin reuptake to twice the degree of R-enantiomer
- metabolized in liver by CYP2D6 and 1A2
- major metabolites are inactive
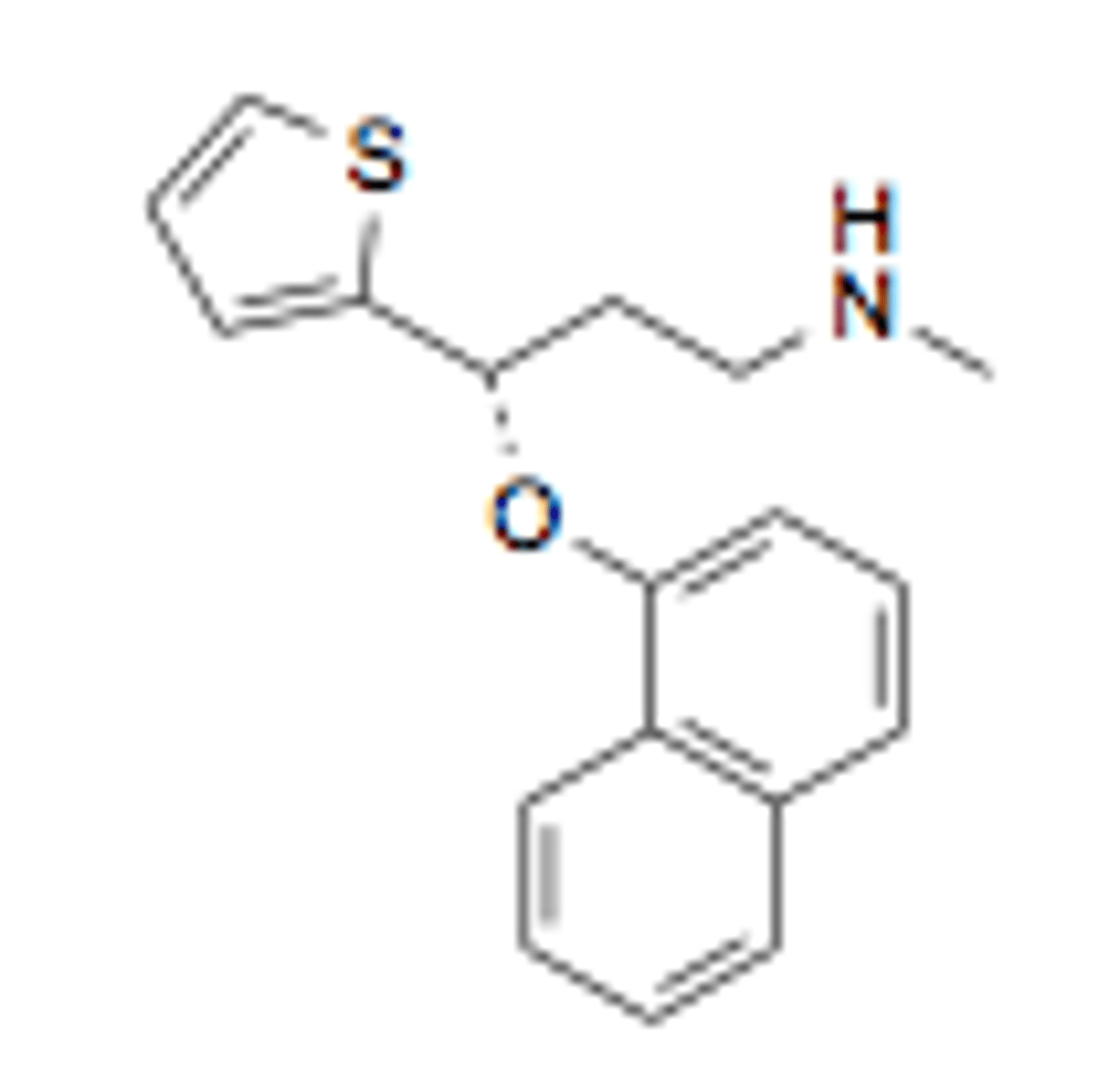
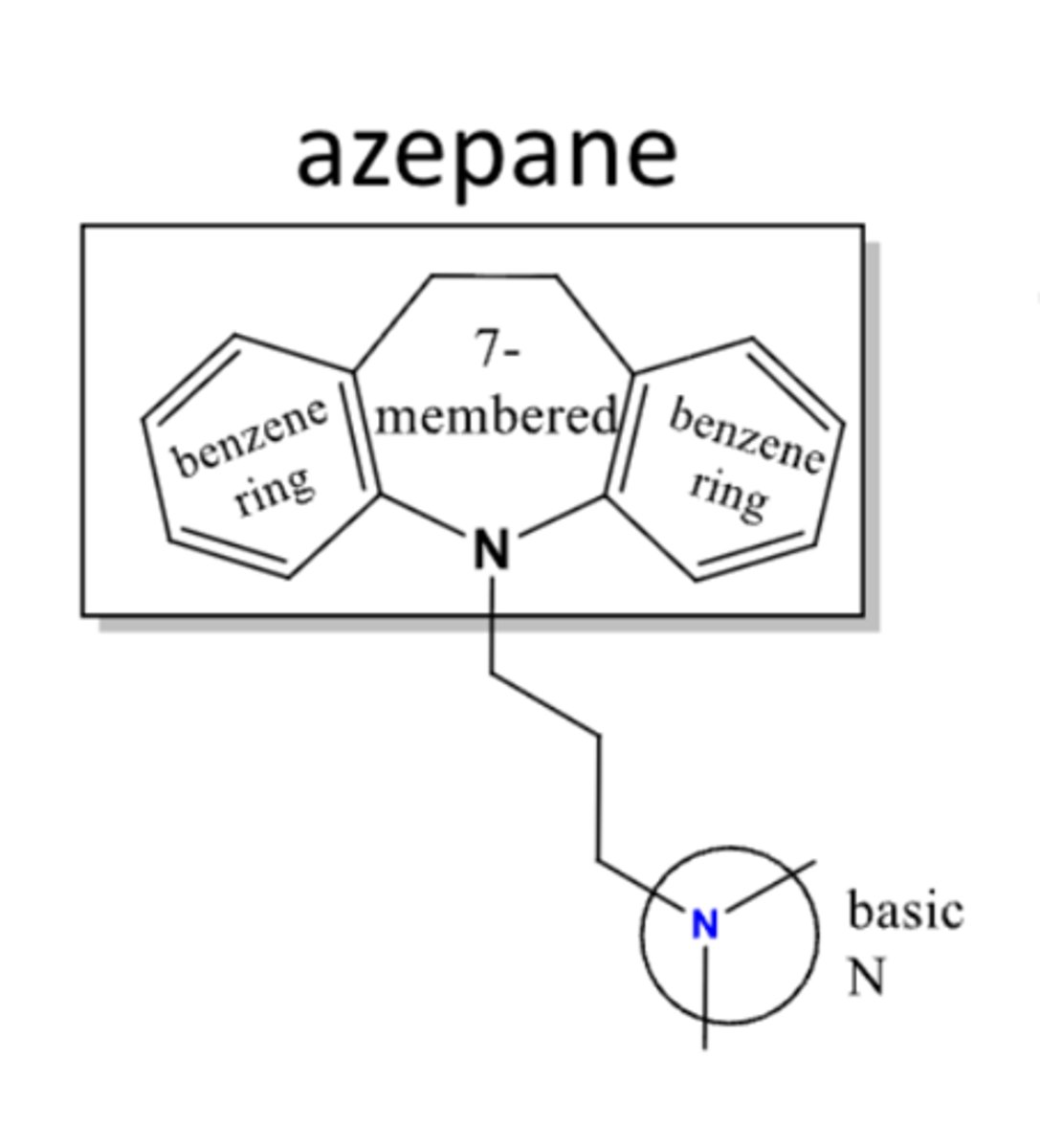
tricyclic antidepressants (TCAs)
- act primarily as SNRIs by blocking SERT and NET but lack selectivity to at SERT and NET
- many also have high affinity as antagonists at 5-HT2, 5-HT6, 5-HT7, a-adrenergic, and NMDA receptors, and as agonists at the o receptors
- replaced by newer antidepressants such as SSRIs, SNRIs, and NRIs
- primarily used for the treatment of mood disorder, anxiety, panic disorder, PTSD, ADHD, Parkinson's, chronic pain, etc.
- structure composed of 7-membered ring with 2 benzene rings and a 3-C linker that leads to a basic amine attached
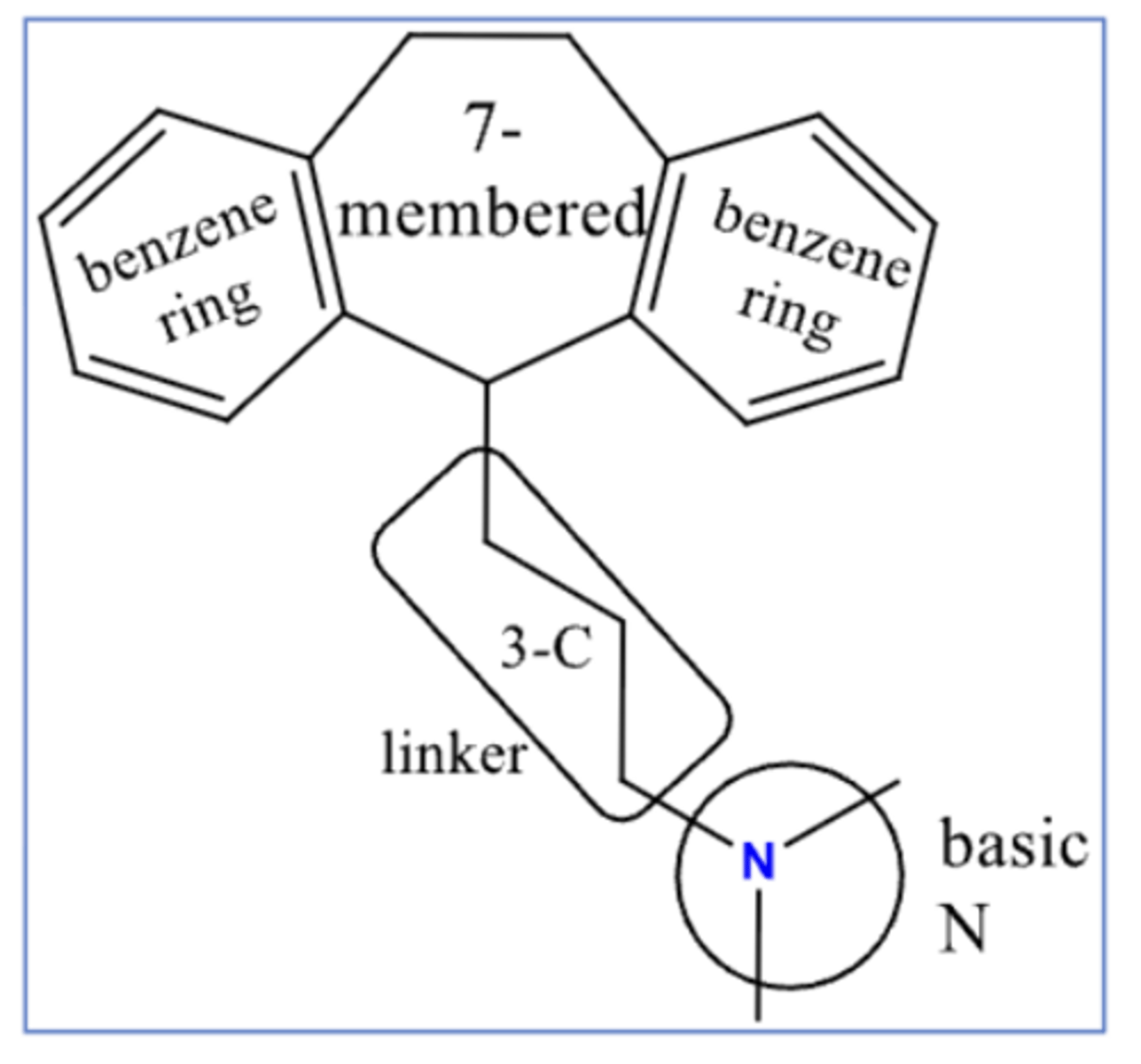
dibenzazepines
- TCAs that end in -ipramine
- structure contains "N" on 7-membered ring that attaches to 3-C linker
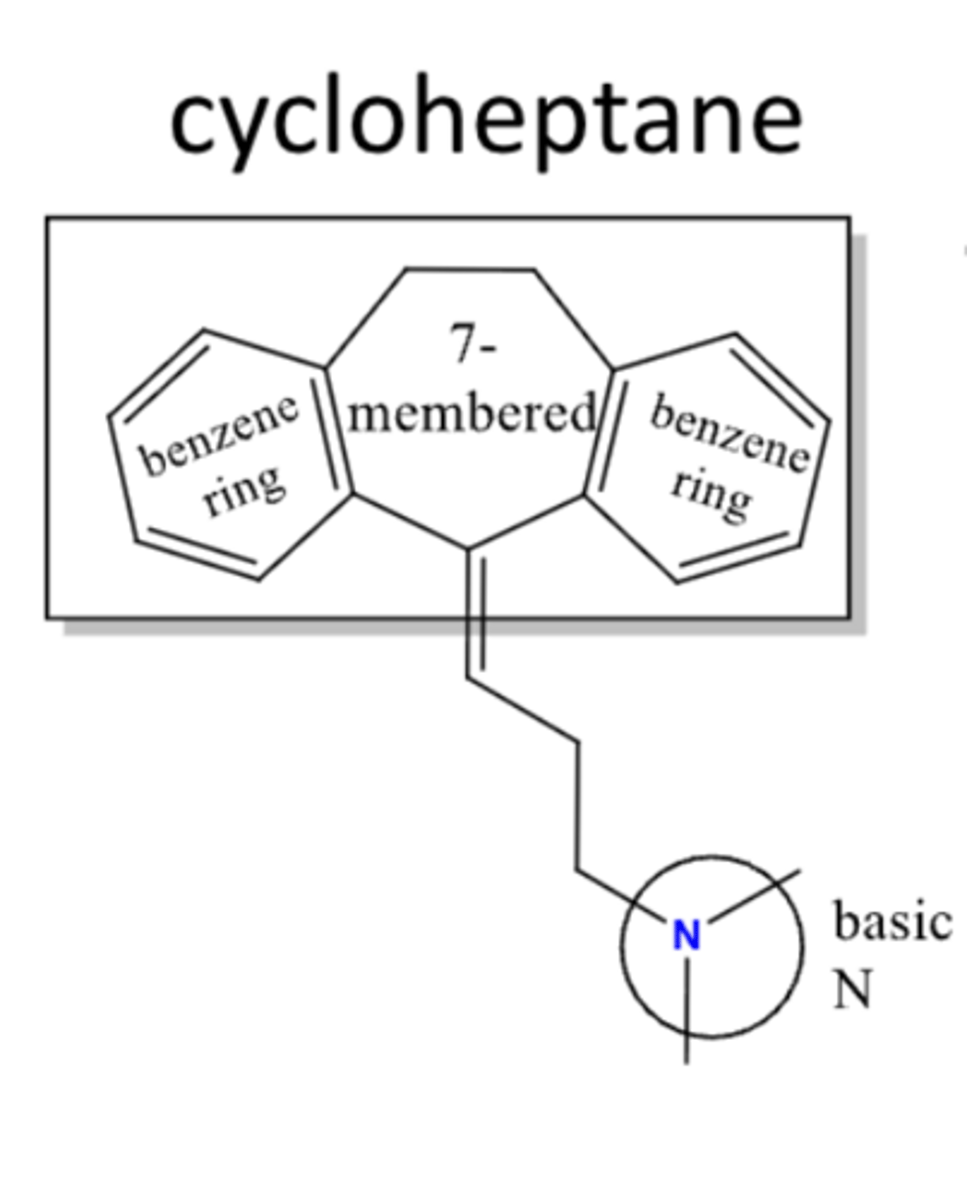
dibenzocycloheptadines
- TCAs that end in -triptyline
- structure contains double bond on 3-C linker attached to 7-membered ring
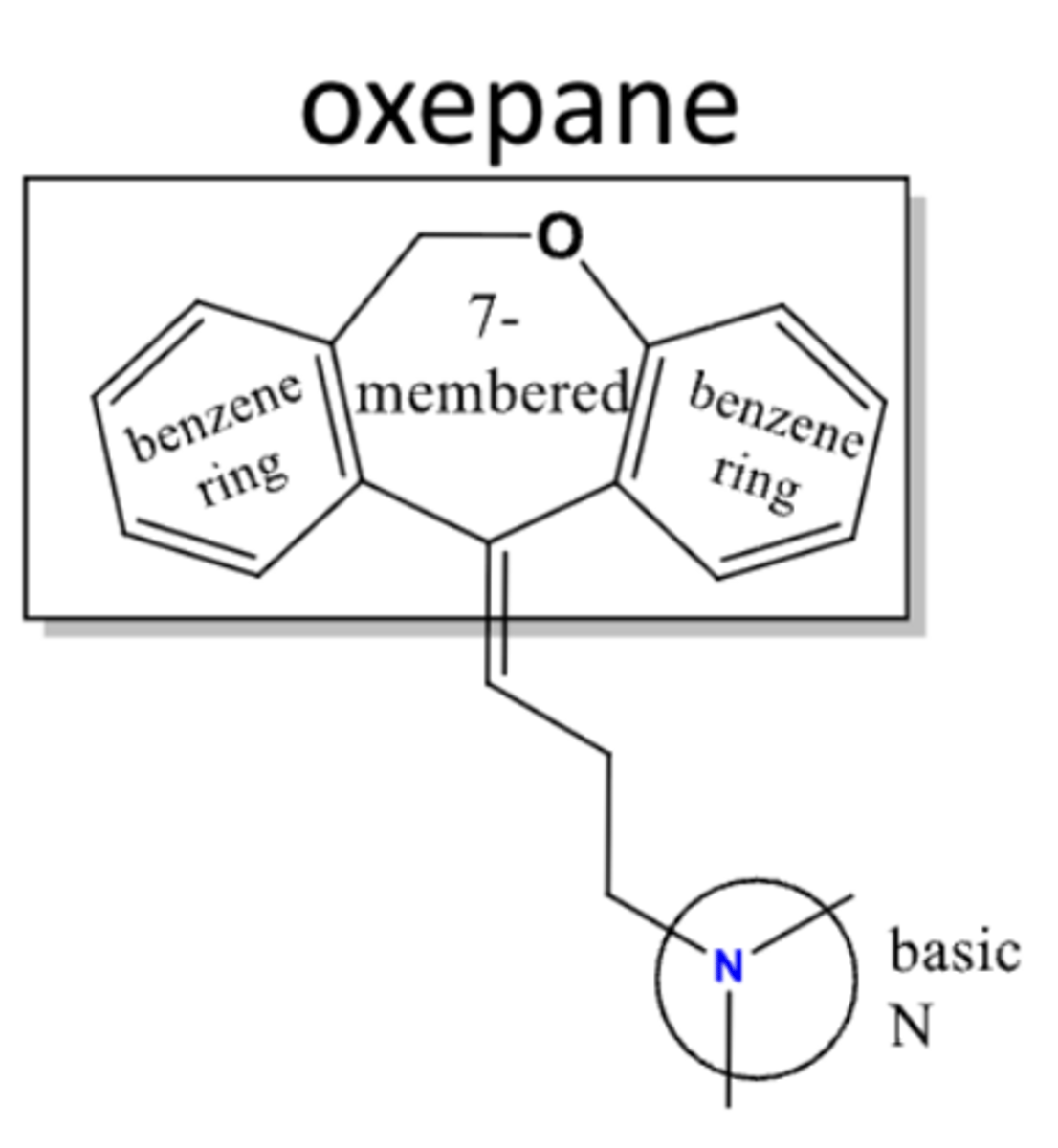
dibenzoxepines
- TCA = doxepin
- structure contains "O" on 7-membered ring adjacent to a benzene ring (not attached to 3-C linker)
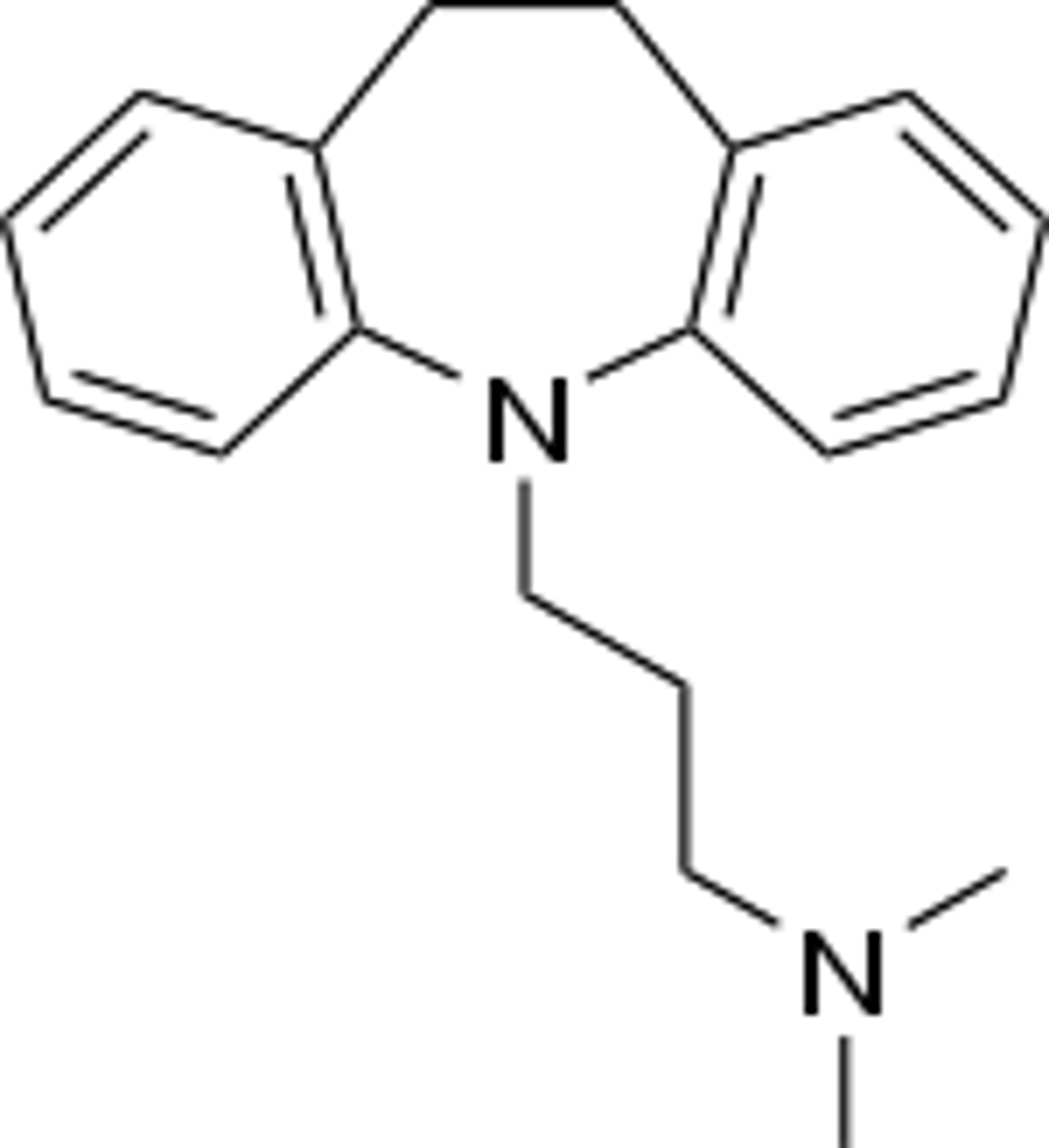
imipramine
- TCA
- inhibits SERT and NET
- antagonize a1-adrenergic receptors
- primarily used to treat major depressive disorder (2nd line antidepressant)
- metabolized to desipramine, a 5-HT and NE reuptake inhibitor
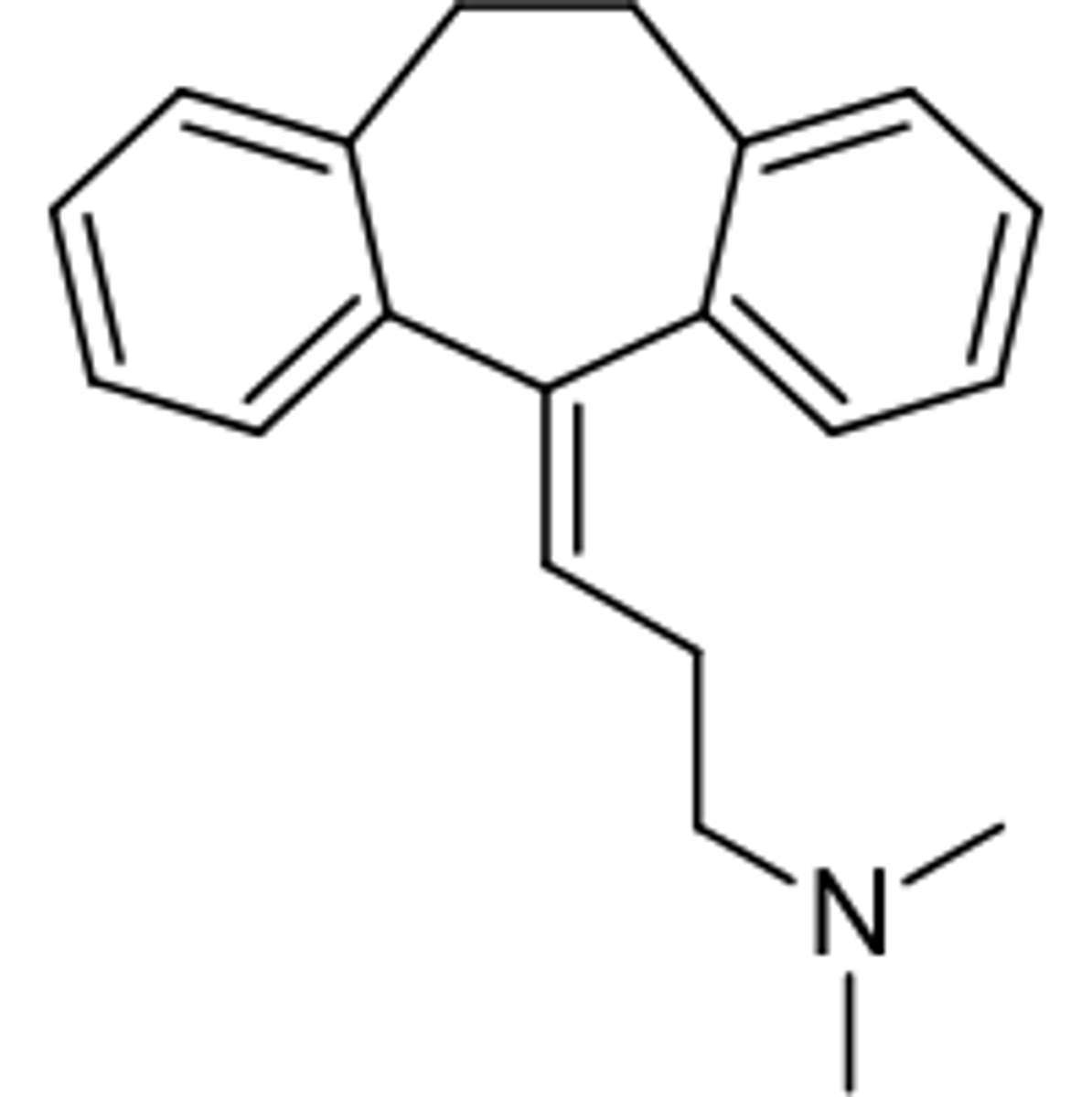
amitriptyline
- TCA
- inhibits SERT and NET
- potent non-selectively block multiple ion channels
- primarily used to treat major depressive disorder (2nd line antidepressant) and used to treat neuropathic pain
- metabolized to nortriptyline, a stronger NE reuptake inhibitor
tetracyclic antidepressants
- atypical, 4-ringed antidepressants
- inhibits the central presynaptic a-2-adrenergic receptors to cause an increased release of 5-HT and NE
- mirtazapine
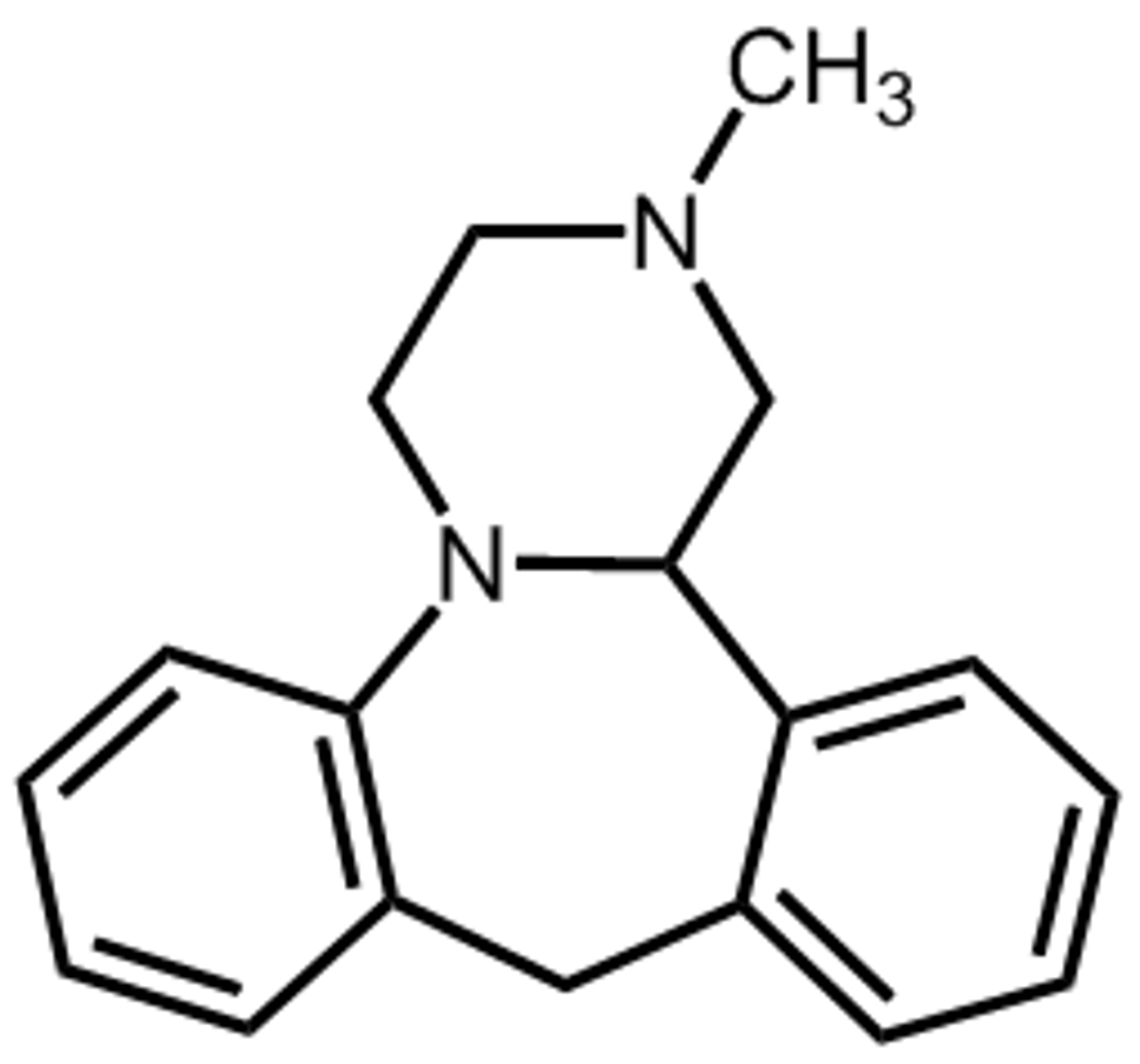
serotonin antagonists and reuptake inhibitors (SARIs)
- antagonize 5-HT2 (primarily 5-HT2A) to mediate its therapeutic benefits against anxiety and depression
- also weakly inhibit the reuptake of 5-HT
- effective drugs for depression and anxiety disorder and alcohol dependence
- efficacy may be more limited than that of SSRIs
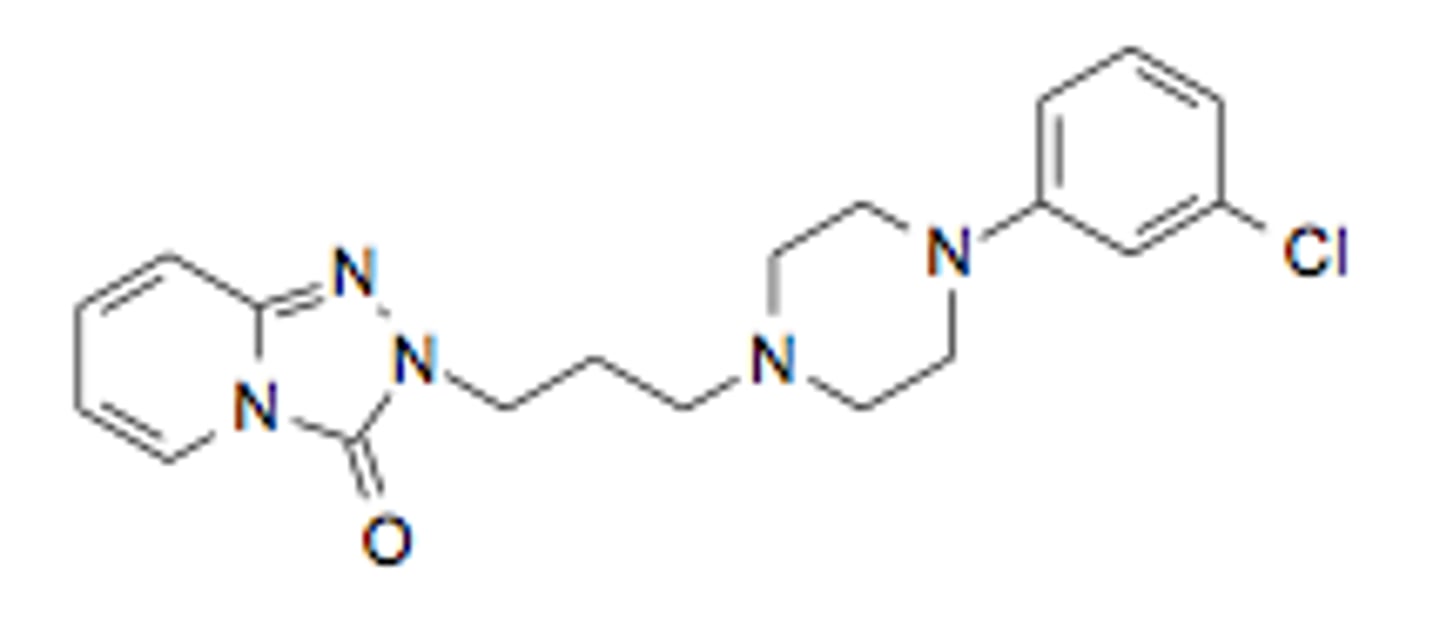
trazodone
- SARI
- potent 5-HT2A antagonist and weak SERT inhibitor
- metabolized in liver by CYP3A4 to produce 4 major metabolites
- metabolite mCCP is a psychoactive drug of the phenylpiperazine class; metabolized by CYP2D6
- mCPP possesses significant affinity for 5-HT receptors and SERT, acts as an agonist at most 5-HT receptors, but may contribute to side effect profile of trazodone including blurred vision, dizziness, nausea, dry mouth, headache, somnolence, fatigue, etc.
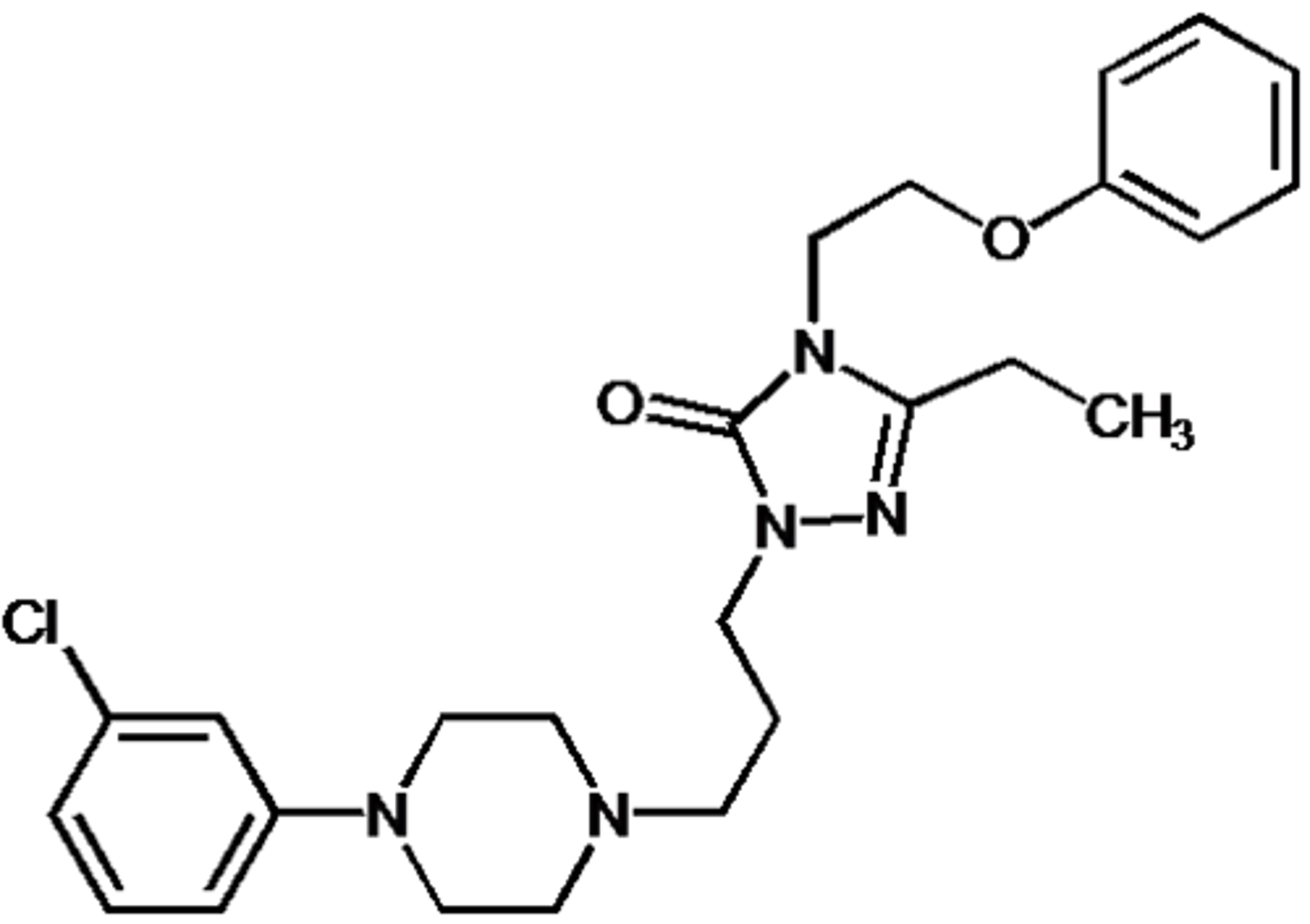
nefazodone
- SARI
- potent 5-HT2A antagonist and weak SERT inhibitor
5-HT1 receptor agonists
- 5-HT1A receptors expressed in the presynaptic neuron (B, autoreceptor) inhibit the reuptake of 5-HT
- 5-HT1A receptors expressed in other region (C) are involved in neuromodulation
- clinical significance = antianxiety, antidepressant, anti-aggressive, anti-craving, anti-cataleptic, and neuroprotective properties
- less effective than SSRI
- commonly used as add-ons to other antidepressants such as SSRIs
azapirones
- 5-HT1 receptor agonist
- structural feature = long-chain
- metabolized in liver to the common metabolite pyrimidinylpiperazine
- buspirone
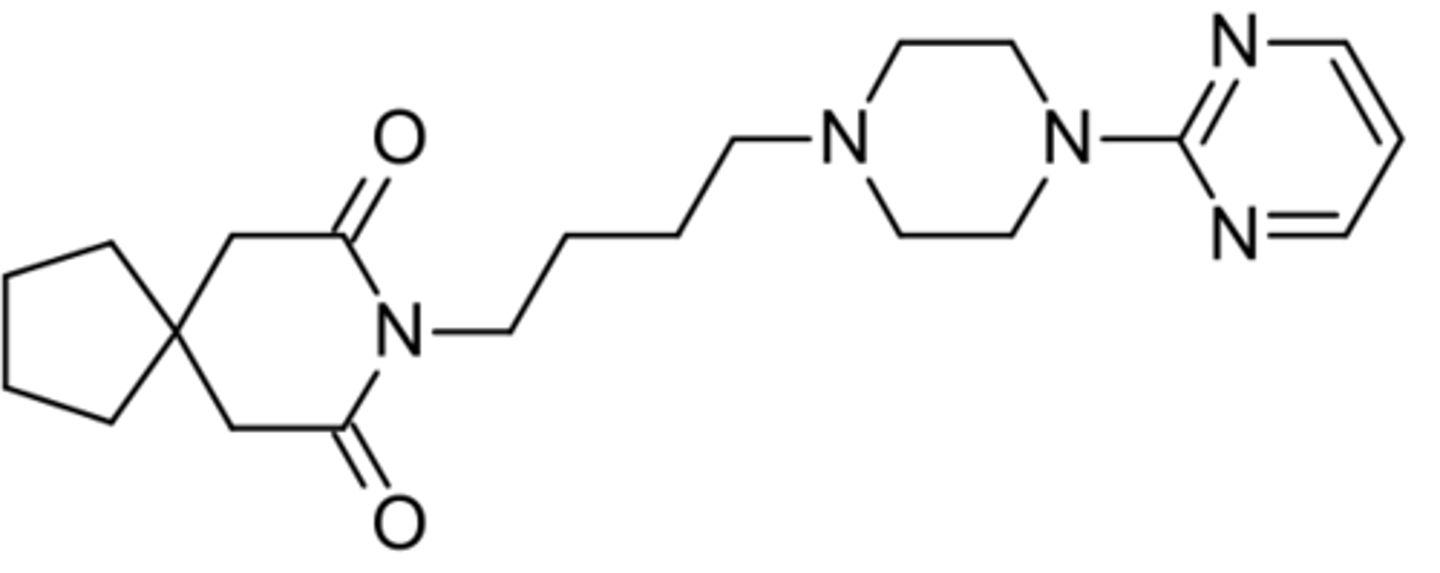
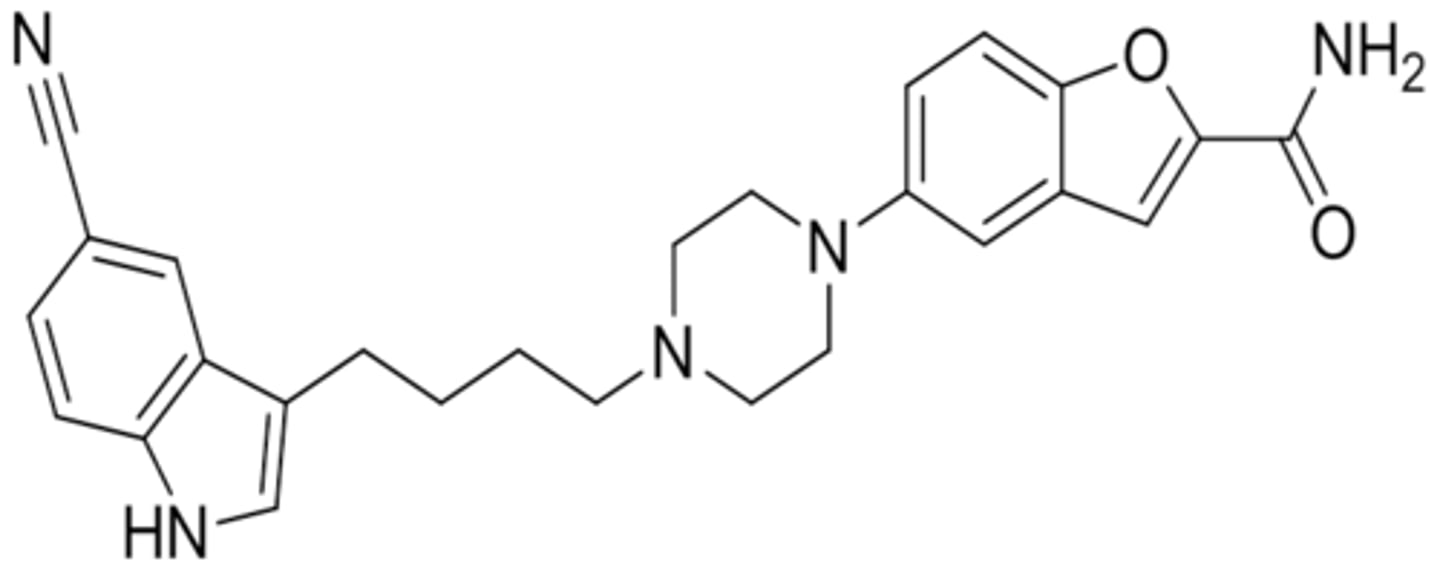
vilazodone
- partial agonist at 5-HT1A
- inhibits SERT and also inhibits NET and DAT
- major metabolite is hydroxyvilazodone
schizophrenia
- a serious mental illness that affects how a person thinks, feels, and behaves
- symptoms fall into 3 categories = psychotic, negative, and cognitive
- typically diagnosed in the late teen years to the early 30s and tends to emerge earlier in males than females
psychotic symptoms of schizophrenia
- altered perceptions, abnormal thinking, and odd behaviors
negative symptoms of schizophrenia
- loss of motivation, diminished feelings of pleasure in everyday life, difficulty showing emotions, etc.
cognitive symptoms of schizophrenia
- problems in attention, concentration, and memory
the dopamine (DA) hypothesis of schizophrenia
- in 1963 Dr. Carlsson proposed that disturbed and hyperactive dopaminergic signal transduction, particularly in the prefrontal region, results in schizophrenia symptoms
- based on the discovery and clinical effects of two dopamine receptor D2 antagonists (chlorpromazine and haloperidol)
- resulted in the development of typical/1st generation antipsychotic agents (D2 antagonists)
4 dopaminergic pathways in brain
- mesocortical pathway = prefrontal cortex, cognitive function (schizophrenia)
- mesolimbic pathway = nucleus accumbens, reward (addiction)
- nigrostriatal pathway = striatum, voluntary movement (Parkinson's)
- tuberoinfundibular pathway = pituitary, secretion of prolactin (milk)
1st generation antipsychotics
- D2 antagonists associated with extrapyramidal side effects (EPS)
- EPS = drug-induced movement disorders with symptoms including dystonia, akathisia, and parkinsonism (muscle contractions, motor restlessness, rigidity, etc.)
2nd generation antipsychotics
- stimulated by the discovery of clozapine in 1963 which binds to dopamine receptor (D2) as well as serotonin receptor (5-HT)
- potently antagonize the 5-HT2A receptor while possessing less affinity for D2 receptors than typical antipsychotic agents
- antagonists at 5-HT2A and D2 or antagonists at 5-HT2A and partial agonist at D2
- generally have antipsychotic efficacy with lower potential for EPS side effects
limitations of the DA model of psychosis
- exact molecular mechanism of psychosis has not been completely revealed
- does not explain the psychotomimetic effects of some drugs that do not activate D2 receptor such as LSD (potent 5-HT2A receptor agonist), phencyclidine, and ketamine
- NTs dopamine, serotonin, and GABA may all have effects of psychotic disorders
phencyclidine and ketamine
- NMDA glutamate receptor antagonists
- indirectly act to stimulate DA availability by decreasing the glutamate-mediated tonic inhibition of DA release
biochemical processes of DA
- biosynthesis = phenylalanine > tyrosine > L-dopa > dopamine
- DA is stored in vesicle (presynaptic) and released to synapse after triggered by an electrical stimulation
- binds to dopamine receptors to transduce signal or is taken up by DAT to stop signal then metabolized or restored in vesicle
dopamine receptors
- D1, D2, D3, D4, D5 subtypes are all GPCRs (two families)
- D1-like family coupled to Gs protein = D1 and D5
- D2-like family coupled to Gi protein = D2, D3, D4
D2 receptor
- main receptor for most antipsychotic drugs
- postsynaptic D2 = classic GPCR
- presynaptic D2 = autoreceptor that regulates the levels of DA in the synaptic cleft which activates presynaptic D2 and inhibits DA release
1st generation (typical) antipsychotics
- direct interaction with postsynaptic D2 type receptor
- D2 antagonists (most potent/greatest affinity at D2)
- many drugs also have appreciable affinity at other receptors (low selectivity D2 ligands)
- associated with higher risk of EPS (a movement disorder including continuous muscle contractions, motor restlessness, rigidity, etc.)
- representative type of drugs = phenothiazine (major) and thioxanthene
phenothiazine antipsychotics SAR
- containing phenothiazine core
- crystal structure partially overlaps with dopamine
- on the A ring an electronegative group (Cl, CF3, SCH3) is responsible for the asymmetry to the molecules
- electronegative group attracts the amine side chain toward the ring containing the electronegative atom
- electronegative group is crucial to activity (phenothiazines lacking the electronegative group at this position are inactive)
- side chain amine requires three carbons separating two nitrogen atoms (two-carbon side-chain separation does not have activity)
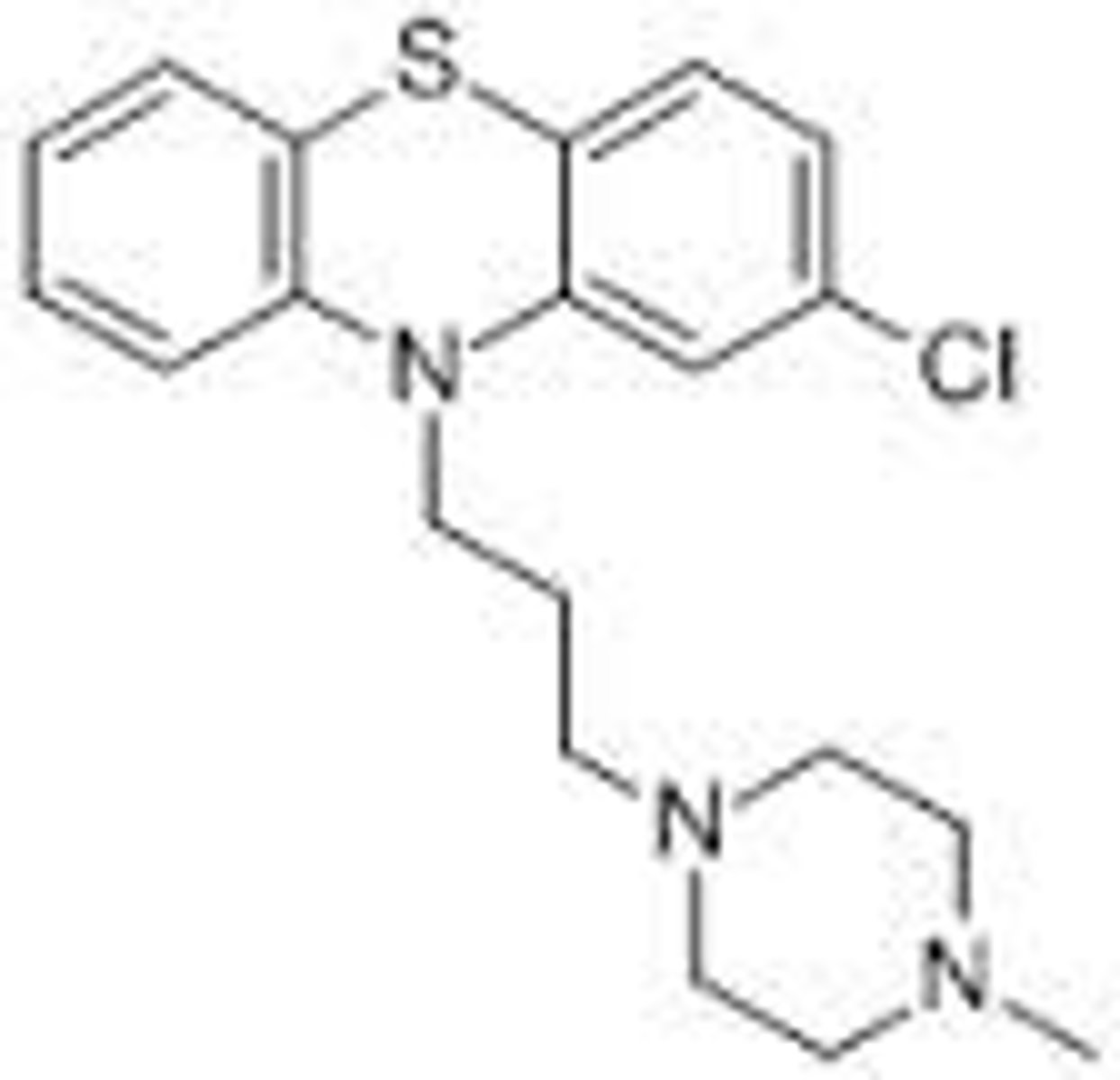
prochlorperazine
- 1st generation (typical) antipsychotic/phenothiazine
- used in the treatment of schizophrenia, psychosis, nausea, and migraines
- common side effects = sedation, EPS, and acute withdrawal syndrome if discontinued abruptly
thioxanthene antipsychotics SAR
- structurally related to phenothiazine
- containing an olefinic double bond between tricyclic ring and the side-chain
- exist in cis (Z) or trans (E) configurations
- cis isomer is 7-fold more active than trans isomer
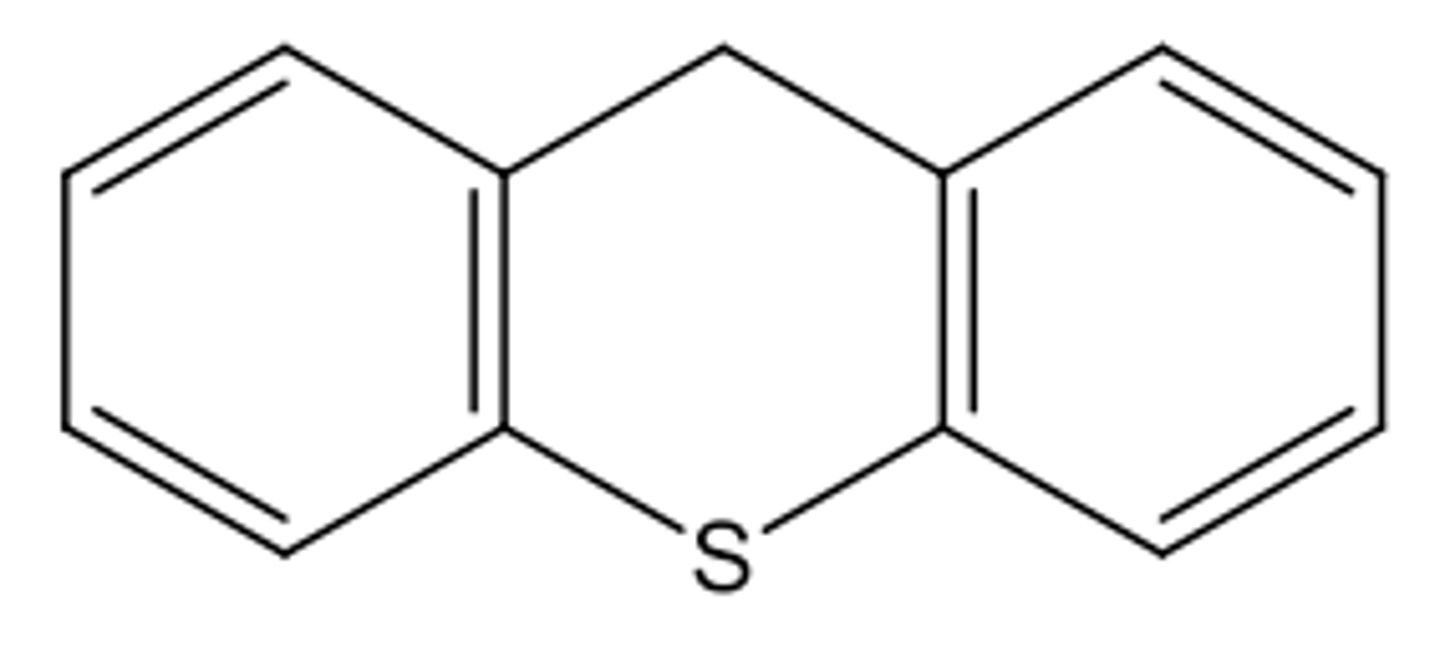
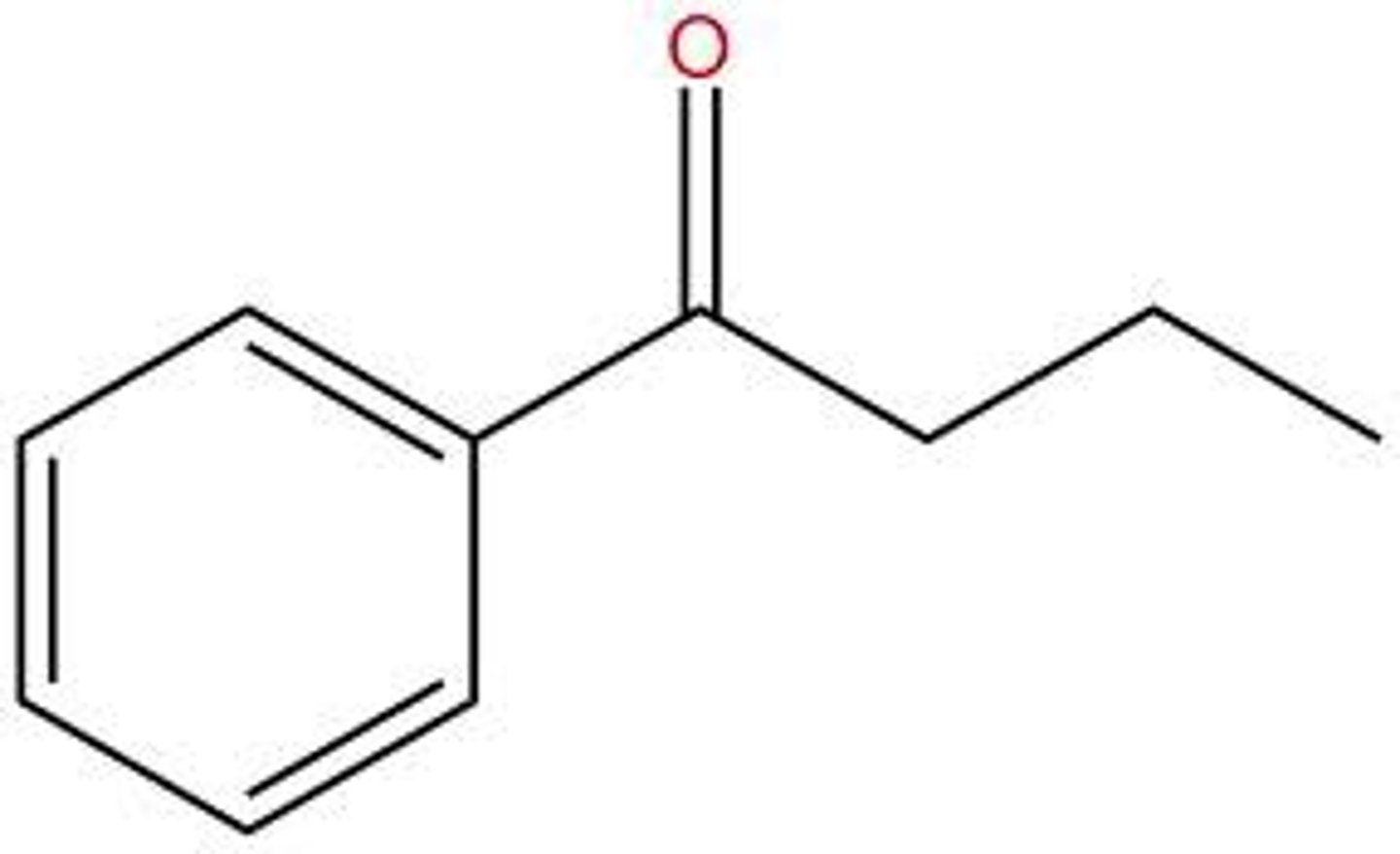
butyrophenone antipsychotics SAR
- phenone = aromatic ketone containing a phenyl group directly attached to a carbonyl group
- tertiary amino group attached at 4th carbon of the butyrophenone skeleton
- haloperidol (Haldol)
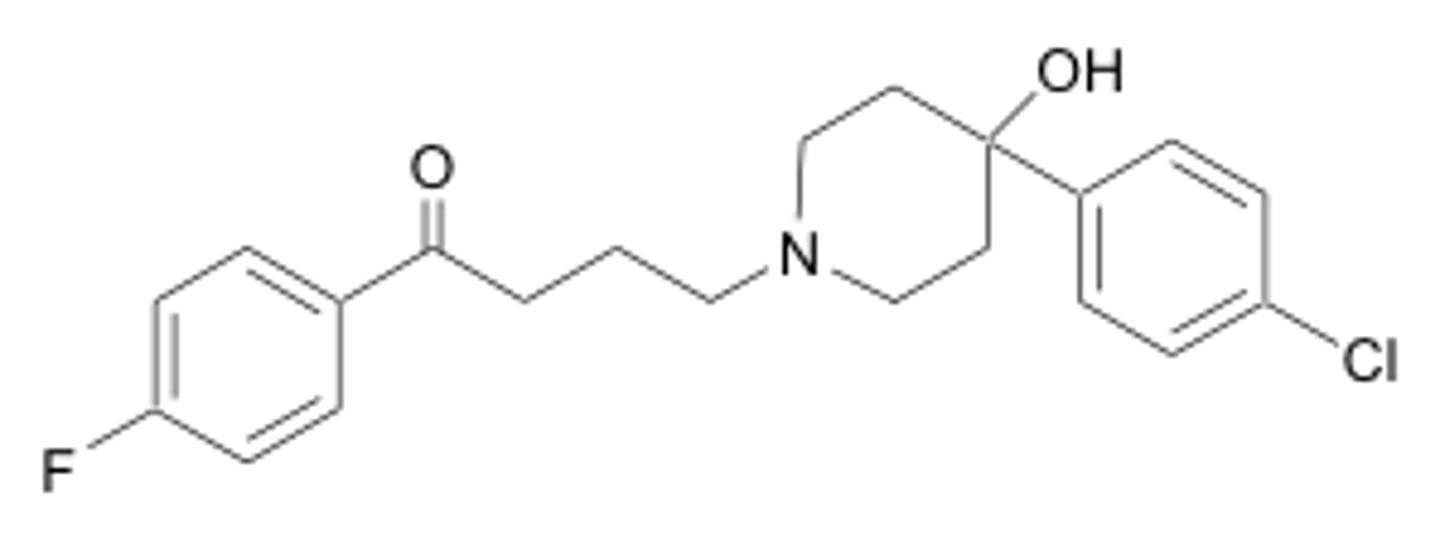
haloperidol (Haldol)
- 1st generation (typical) antipsychotic/butyrophenone
- discovered in 1958
- first antipsychotic medicine acting at D2 receptor
2nd generation (atypical) antipsychotics
- interact with D2-type receptor and 5-HT receptor and act as D2 antagonist/partial agonist and 5-HT2A antagonist
- many drugs also have appreciable affinity at other receptors (low selectivity D2 ligands)
- lower risk of EPS than 1st generation due to high affinity for 5-HT2A and reduced affinity for D2
- representative structures = dibenzodiazepine and long-chain arylpiperazine
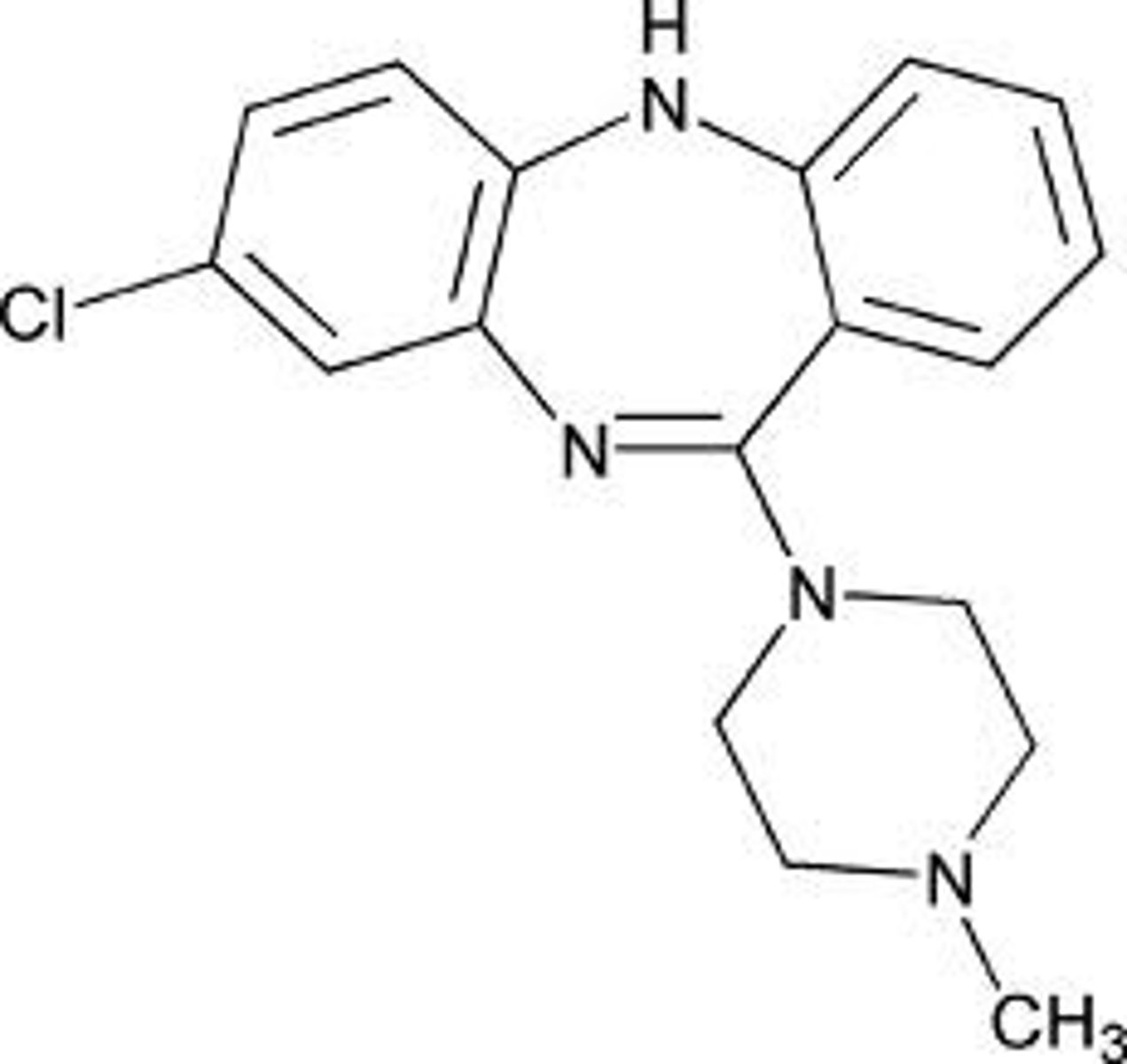
clozapine
- first atypical (2nd generation) antipsychotic (prototype)
- has minimum EPS and does not produce uncontrollable face and body movements (tardive dyskinesia) with long-term use
- effective in patients who do not respond to typical first-generation antipsychotics
- metabolized mainly by CYP1A2 and also by CYP3A4
- cigarette smoking induces activity of CYP1A; patients who smoke while taking clozapine have significantly lower serum levels of clozapine
- consume caffeine-containing beverages while taking clozapine increases EPS as caffeine is metabolized primarily by CYP1A
- significantly most effective!
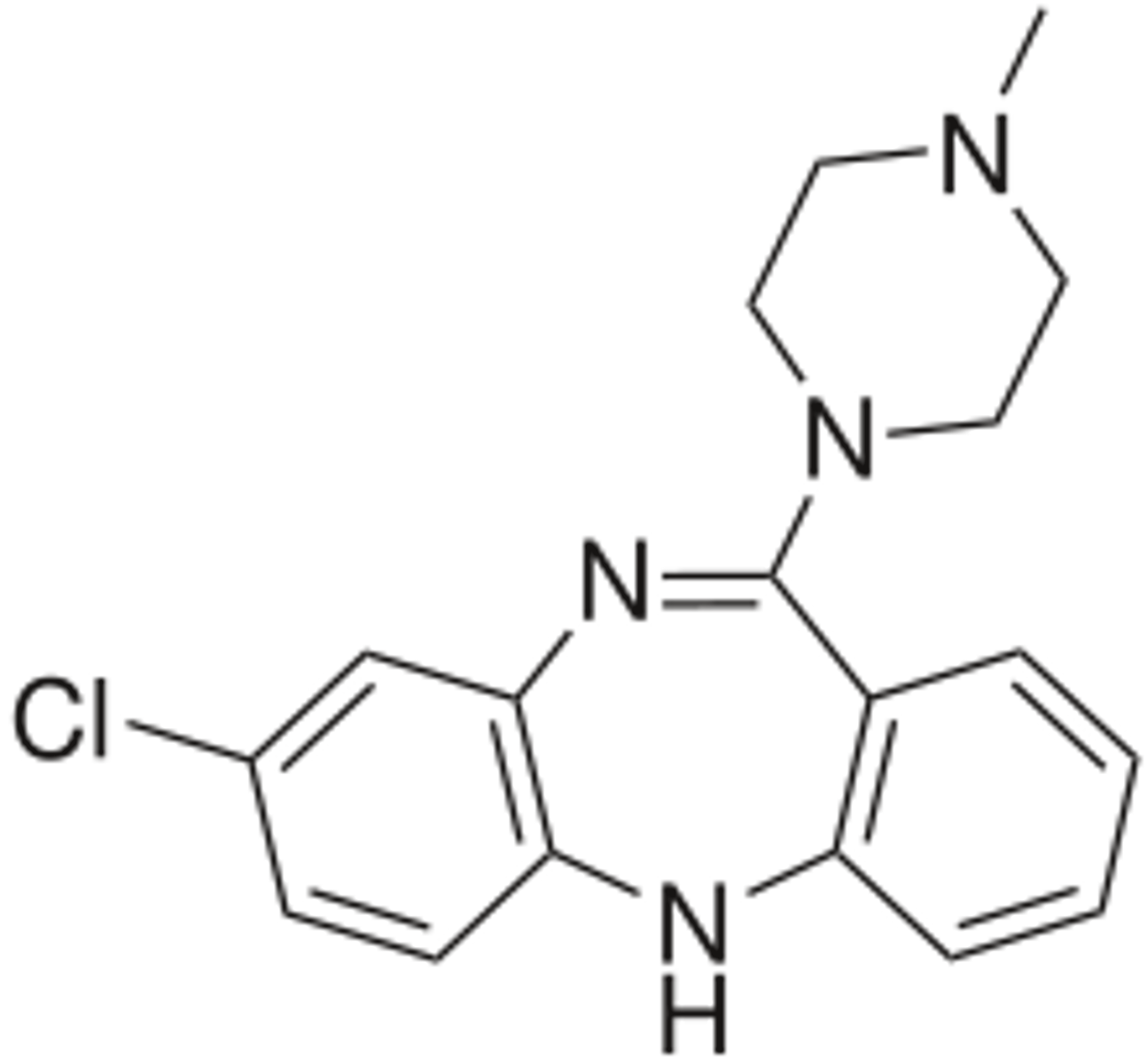
olanzapine
- 2nd generation atypical antipsychotic
- metabolized by CYP1A2 and UDPGT
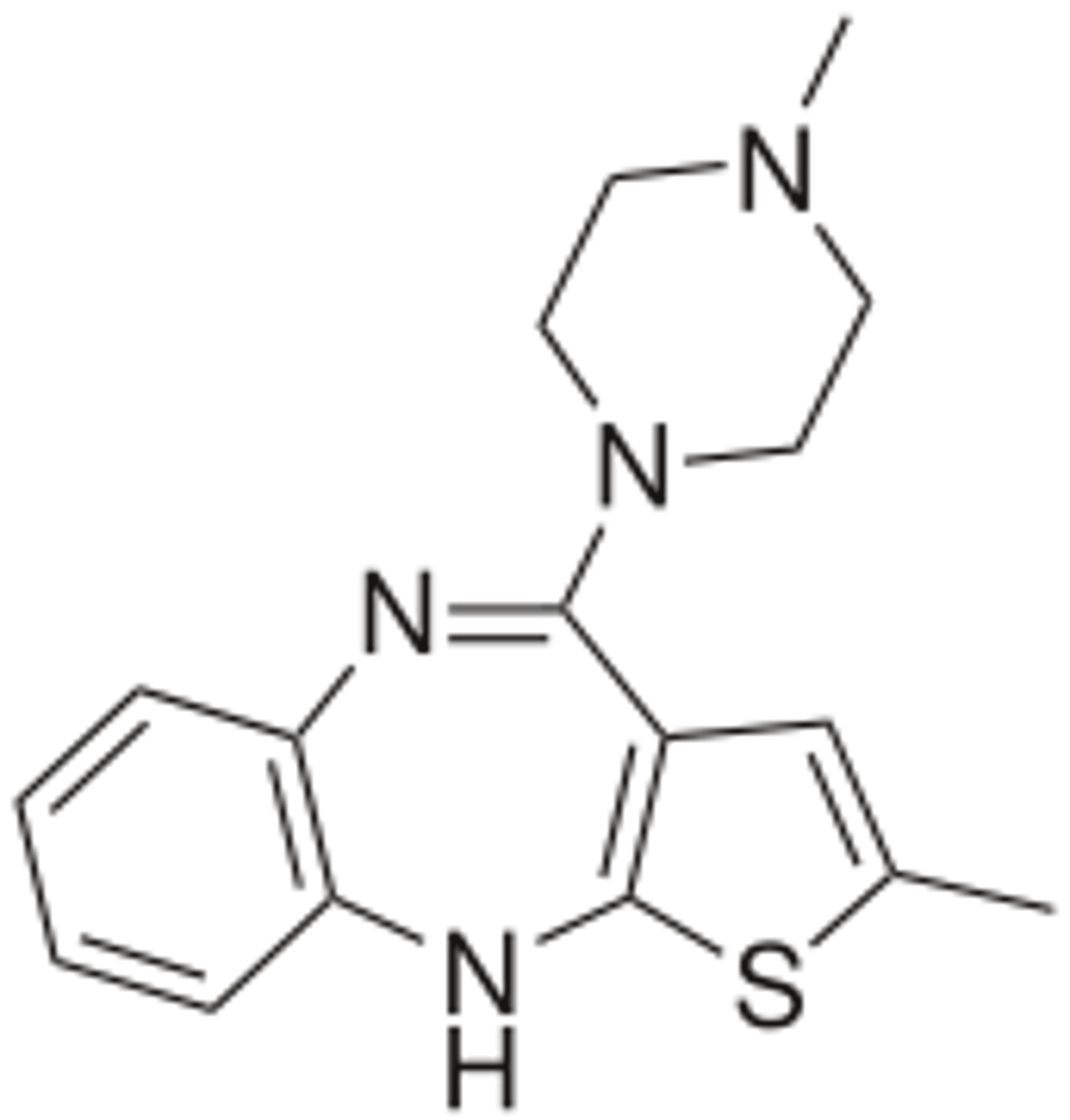
quetiapine
- 2nd generation atypical antipsychotic
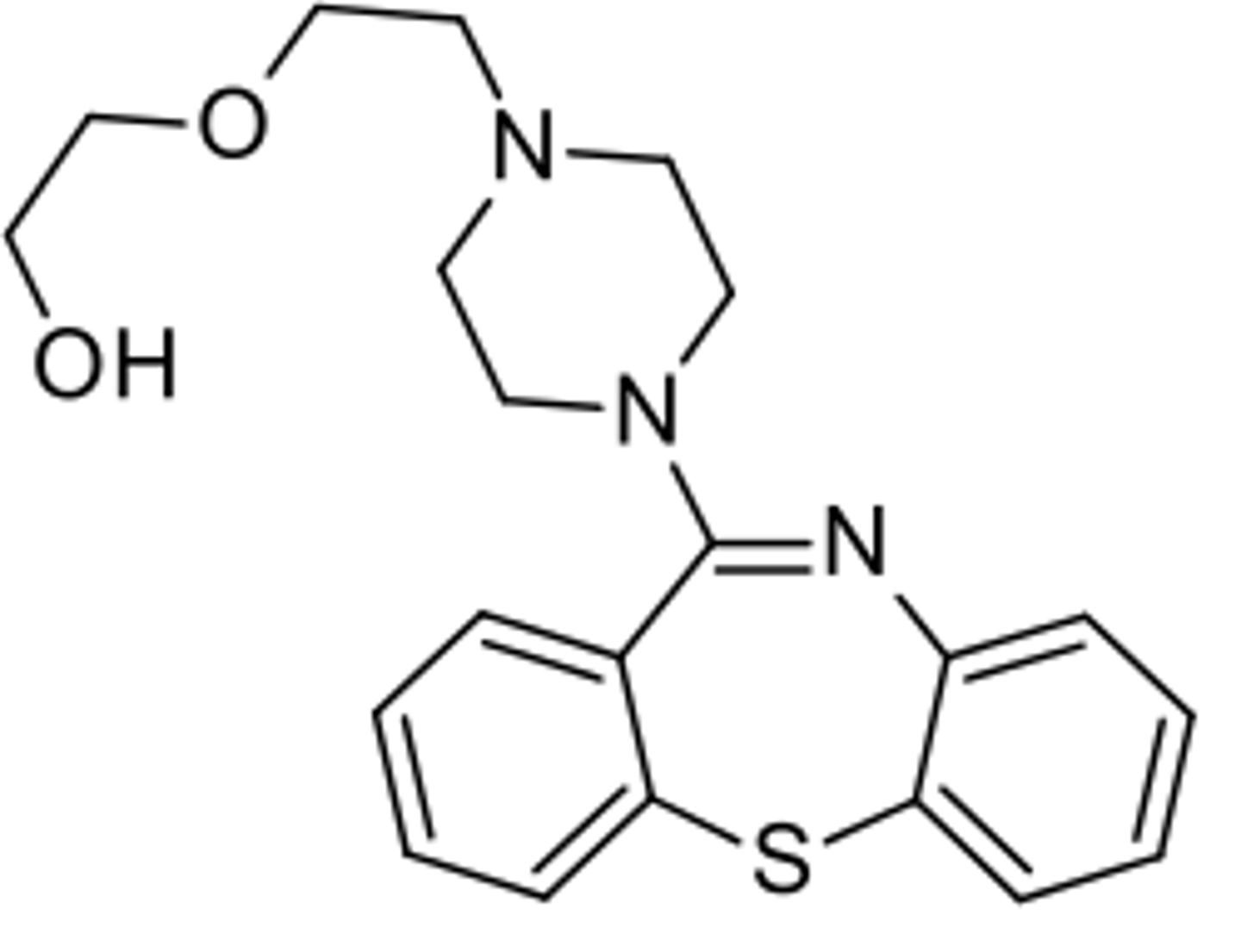
risperidone
- WHO's list of essential medicines as the safest and most effective medicines needed in a health system
- produces more EPS than most other atypical antipsychotics
- metabolized mainly by CYP2D6
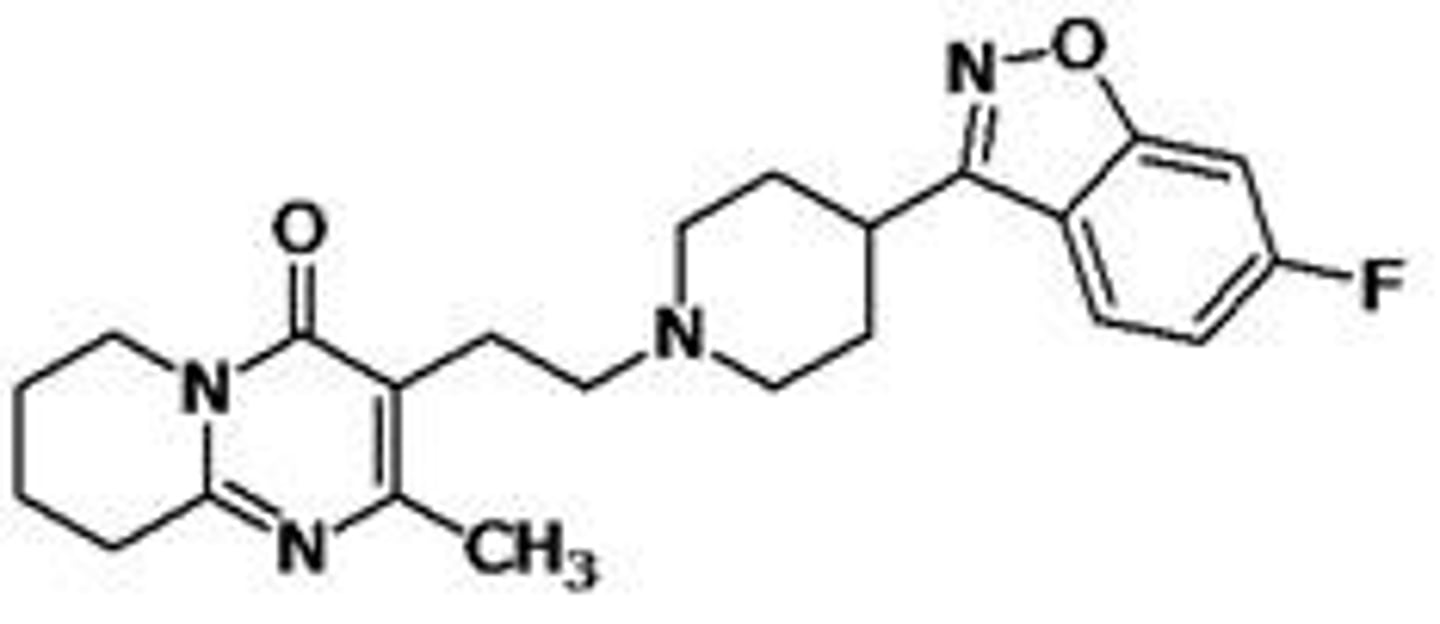
ziprasidone
- 2nd generation atypical antipsychotic
- metabolized mainly by CYP3A
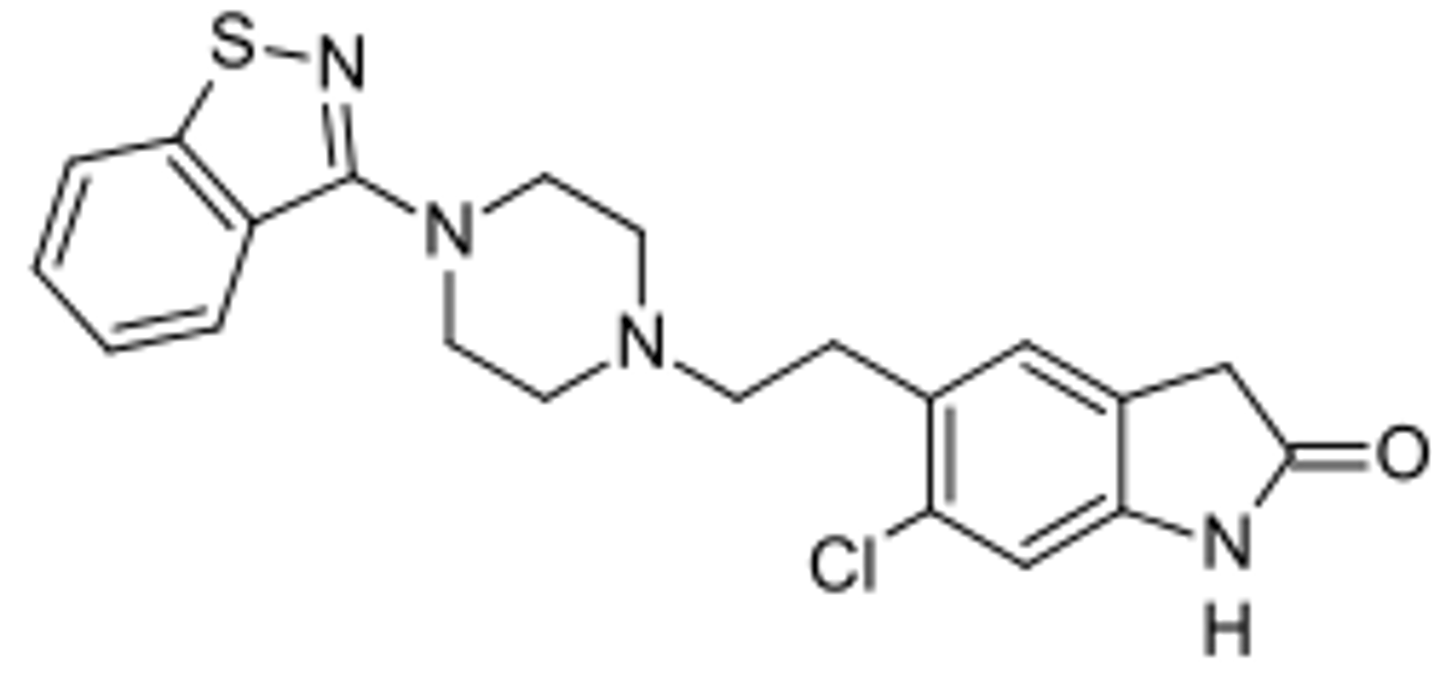
lurasidone
- 2nd generation atypical antipsychotic
- metabolized mainly by CYP3A
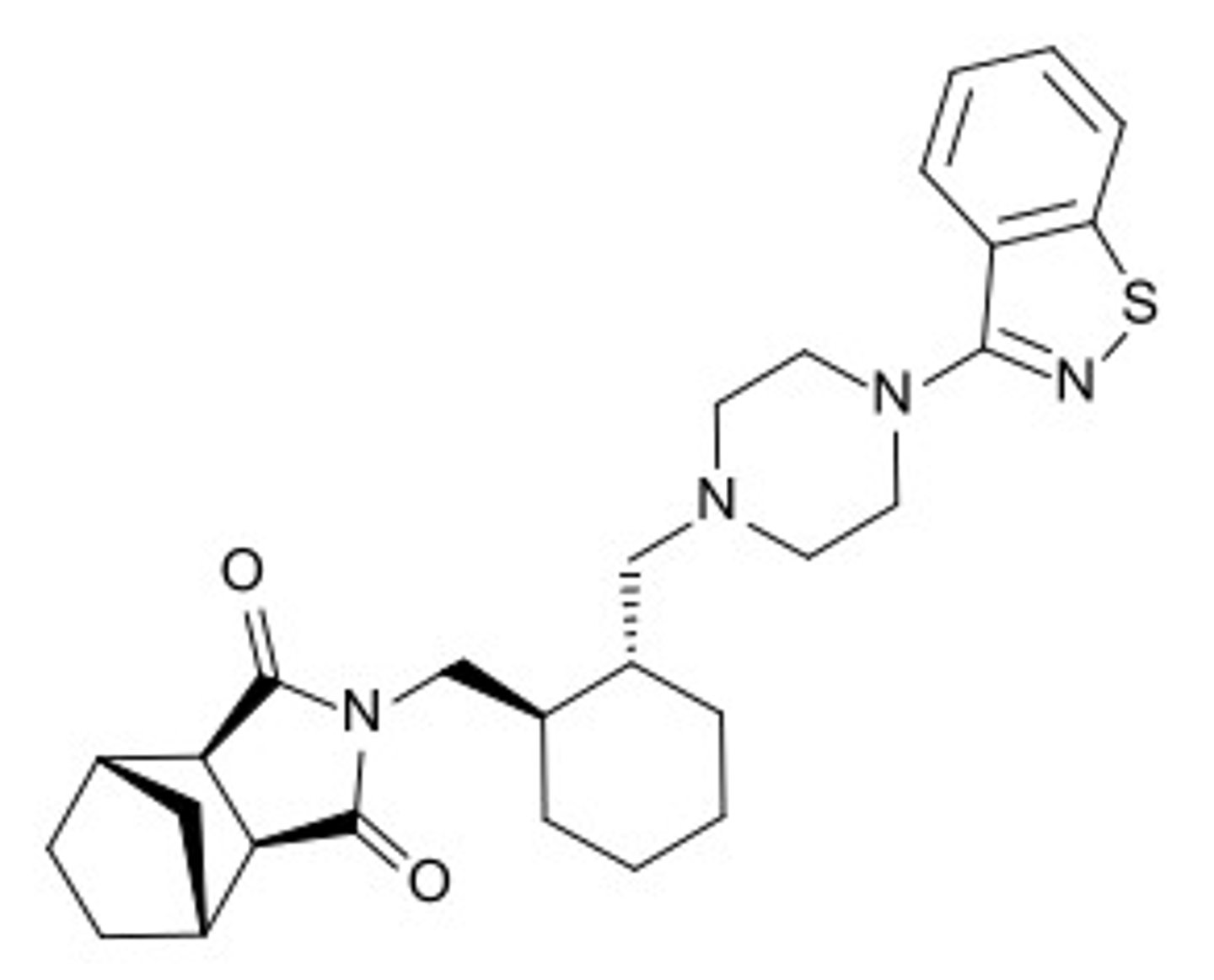
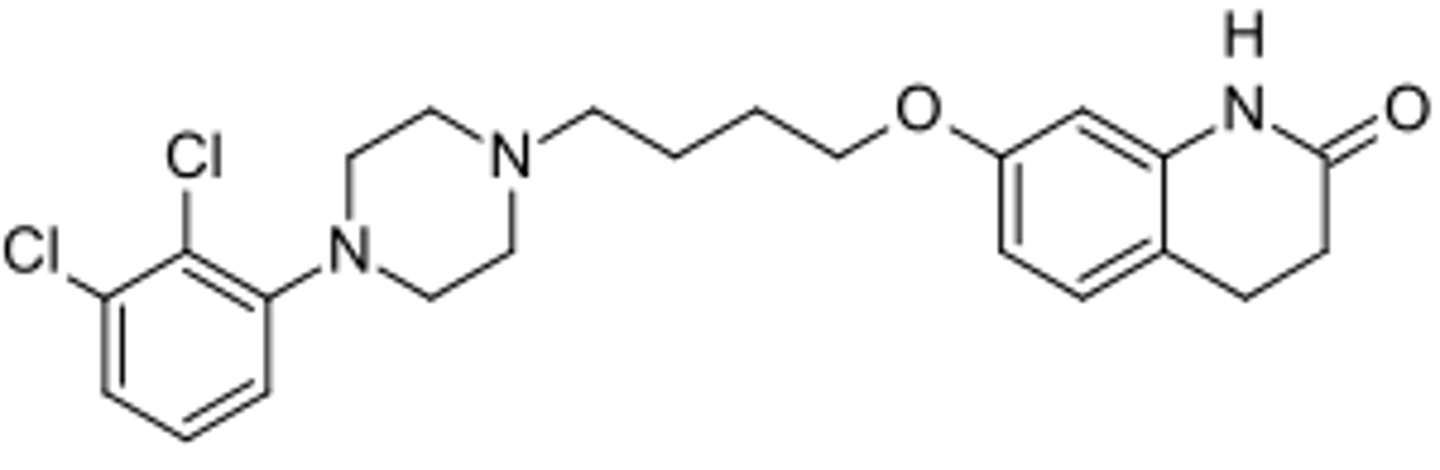
aripiprazole
- 2nd generation atypical antipsychotic