Cancer Therapies
1/96
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
97 Terms
what are the four pillars to cancer treatment?
surgery
radiation
chemotherapy
targeted therapy
how does direct DNA damage occur in radiation therapy?
DNA directly absorbs the energy from the radiation, which destabilizes chemical bonds in the bases or phosphate backbone, resulting in abasic sites, single-strand, and double-strand breaks
how does indirect DNA damage occur in radiation therapy?
radiation interacts with water (radiolysis of water), which generates highly reactive chemical species, especially hydroxyl radicals that can attack bases and the deoxyribose sugar, producing oxidized bases, particularly 8-oxo-guanine
what does radiation therapy do?
induces several types of DNA damage, with the most severe being double-strand breaks; can be caused by direct or indirect effects
what is interphase death?
cells stop dividing within hours of exposure to radiation due to severe damage to intracellular components including the DNA and proteins, leading to apoptosis
what are the two main mechanisms that cause cell death in response to the DNA damage caused by radiation treatment?
interphase death
proliferative death
why is proliferative death more common than interphase death?
proliferative death occurs when there is a lack of p53 expression and loss of p53 expression is common in many cancers
what is proliferative death?
as a result of mitotic catastrophe, where cells accumulate so much DNA damage that doesn’t get repaired (because of p53 mutation) or is repaired incorrectly (BRCA mutations, etc.)
accumulation of DNA damage prevents proliferation and eventually results in cell death
can occur up to several months after radiation therapy has finished
what is chemotherapy?
the use of drugs to stop the growth of cancer cells, either by killing the cells or by stopping them from dividing
what is neoadjuvant chemotherapy?
given before surgery to make surgical resection more effective
what is adjuvant chemotherapy?
given after surgery to eliminate residual cancer cells
what is radioresistance?
since radiation therapy relies on the accumulation of DNA damage to kill cancer cells, cancer cells can be resistant to radiation treatment if they have WT DNA repair proteins present → allows cancer cells to “keep up” with the DNA damage being caused by radiation and continuously fix it as it occurs
what has been used in recent years to help with radioresistance?
DNA-sequencing has been used on patient tumors to determine if they have mutated repair proteins, which would make them suitable candidates for radiation treatment
what are alkylating agents?
covalently add alkyl groups to DNA bases, typically guanine, resulting in impaired replication and transcription; can be monofunctional (one reactive group) or bifunctional (two reactive groups)
what are bifunctional alkylating agents?
can form crosslinks between two bases in the same DNA strand (intrastrand) crosslinks or on opposite strands (interstrand) crosslinks
why are interstrand crosslinks the deadliest?
they physically link two strand of DNA together which prevents separation of DNA during transcription, preventing transcription of nearby genes
prevents strands from being separated during replication → when replication fork reaches site of an interstrand crosslink, the replication fork collapses, resulting in double-strand breaks and ultimately death of the cell when the damage is severe enough
what are antimetabolites?
chemical that inhibits the use of a metabolite; typically similar in structure to the metabolite they inhibit, thus working through competitive inhibition
what is the main type of antimetabolite used?
base analogs
what are base analogs?
mimic DNA and RNA bases and get utilized by the cell instead of the actual base
what does the antimetabolite 5-FU (base analog of uracil) do?
once in the cell it gets converted into several metabolites: FdUMP, FdUTP, FUTP
what is the FdUTP mechanism?
FdUTP gets incorporated into DNA in place of a thymine nucleotide. when FdUTP is incorporated, its recognized as damage and removed with base-excision repair. since there is so much FdUTP in the cell and little to no thymine (FdUMP blocked it from being made) the cell just replaces FdUTP with another FdUTP → results in a futile cycle of removal and repair that ultimately results in DNA strand breaks and cell death
what is FUTP?
acts like FdUTP but is incorporated into the RNA instead of the DNA, blocking mRNA processing and translation of the mRNA
what is the FdUMP mechanism?
thymidylate (TS) is an enzyme that catalyses the conversion of dUMP to dTMP (thymidylate), this reaction provides the sole de novo source of thymidylate, which gets converted into dTTP → therefore, if you inhibit this reaction, thymine nucleotides are no longer generated; FdUMP binds to nucleotide-binding site of thymidylate synthase and forms a stable complex, blocking access of dUMP to nucleotide-binding site and inhibiting dTMP synthesis
what are microtubule targeting agents?
drugs can either stabilize or destabilize microtubules to kill the cell. this is possible because during mitosis we use microtubules to separate sister chromatids
if you stabilize microtubules with a drug, you cant shorten the microtubules to pull the sister chromatids apart
if you destabilize the microtubules, you can never form the mitotic spindle to begin with, also resulting in sister chromatids not being separated
what is platinum-based chemotherapy?
induce covalent modifications of DNA that create adducts that aren’t easily removed by repair machinery
what is cisplatin?
the first platinum-based chemotherapy discovered and generates covalent crosslinks, typically intrastrand crosslinks, between adjacent guanine nucleotides
what is paclitaxel?
a microtubule stabilizing drug that impairs microtubule dynamic instability → results in stiff, rigid microtubules that don’t connect to the kinetochore properly → results in spindle assembly checkpoint remaining active → when spindle assembly checkpoint signal sustained for too long, cell ultimately undergoes apoptosis (occasionally, cells escape mitosis abnormally rather than dying immediately, but this results in chromosome segregation errors that end up killing the cell later on)
what are topoisomerases?
as the DNA double helix is unwound, it becomes overwound and tangled, which creates mechanical stress that can block polymerases and stall chromosome separation → topoisomerases solve this problem by cutting DNA, allowing it to relax and untangle, the resealing it
why are cancer cells especially dependent on topoisomerases?
because they divide frequently and must replicate large amounts of DNA efficiently
what are the three mechanisms that topoisomerase inhibitors utilize?
suppressing enzyme activity by catalytic inhibitors
activating enzyme activity toward the bona fide or a surrogate substrate
topoisomerase poisoning - turning the enzyme into a poisonous agent that is toxic to cells (most common)
how does doxorubicin and other topoisomerase poisons work?
the drug binds to topoisomerase and the DNA, trapping the protein on the DNA. this trapping occurs after topoisomerase has already cut the DNA, and blocks the topoisomerase from religating the DNA → this turns topoisomerase into a DNA damaging agent that cuts the DNA without rejoining it
what is acquired resistance?
resistance arises after treatment due to a new mutation that was selected for
what is intrinsic/primary resistance?
there was already a drug resistant population present at the start of treatment
what are the major drivers of multi-drug resistance?
ATP transporters
what are ATP transporters?
transmembrane proteins that can pump numerous types of drugs, hormones, lipids, etc. across the membrane due to the presence of multiple ligand binding sites that are capable of binding a wide range of molecules
how do ABC transporters contribute to multi-drug resistance?
they can recognize a drug when it enters the cell and immediately pump it back out into the extracellular space
why are ABC transporters difficult to target therapeutically?
ABC transporters are essential for drug metabolism in the liver and inhibiting ABC transporters alters the dosing of other drugs given in combination
what are targeted therapies?
drugs designed against a specific molecular target, usually a protein
why are tumor suppressors not good targets for targeted therapies?
most drugs inhibit their targets rather than activate them
why are mutator genes not good targets for targeted therapies?
at the time of treatment the genome would have already been destabilized, and reactivating mutator genes would provide little benefit at that point in cancer progression
why are transcription factors generally considered undruggable?
due to their structure which contains three domains (DNA binding domain, transcriptional activation domain, and regulatory domain)
DNA domain is typically polar, which makes it challenging to get drugs to bind to it, and there is significant overlap in the 3D structure of the DNA binding domain between transcription factors, making it difficult to have high specificity.
transcriptional activation domain is highly disordered so there is generally no druggable regions
can transcription factors ever be druggable?
occasionally some transcription factors will have a druggable pocket in their regulatory domains, allowing a small subset of transcription factors to potentially have drugs designed against them
what is the target of most drugs?
oncogenes (which can be inhibited with small molecule drugs to shut off their function)
why is specificity of a drug to its target important?
the less specific a drug is to the target, the more likely you are to have side effects from that drug, as it hits other proteins and pathways
what drives the specificity of a drug to its target?
the 3D shape of the protein - ideally, a target protein will have a deep catalytic cleft that allows a drug to form multiple, noncovalent bonds (similar to substrate binding enzyme)
why are protein-protein interactions hard to target therapeutically?
because of the large points of contain between two proteins, which are much larger than what can be disrupted by a small molecule inhibitor due to their being so many points of contact between the proteins
what is pharmacodynamics?
biochemical and physiological effects of a drug (the mechanism of action) - what the drug does to the body
what is pharmacokinetics?
overall process of absorption, disruption, metabolism, and excretion of the drug (ADME) - what the body does to the drug
what establishes the therapeutic window of a drug?
the max tolerated dose info coupled with the pharmacogenetic, pharmacodynamic, and pharmacokinetic data
what is the therapeutic window?
range of concentrations that the drug will be effective at
below the therapeutic window → wont see any benefit
above the therapeutic window → toxic effects
what is overall survival (OS)?
gold standard - how long the patient lives from start of treatment
what is progression-free survival (PFS)?
length of time patient lives without the cancer getting worse
what is disease-free survival (DFS)?
time patients remain cancer free (typically used after curative treatment like surgery)
what is objective response rate (ORR)?
% of patients whose tumors shrink by a defined amount
what is duration of response (DoR)?
how long a tumor remains diminished after treatment
how is drug performance measured in clinical trials?
overall survival (OS)
progression-free survival (PFS)
disease-free survival (DFS)
objective response rate (ORR)
duration of response (DoR)
what is the predominant class of proteins targeted by drugs?
kinases - numerous oncogenes are kinases (MEK, ERK, PI3K, AKT, FAK, Src, VEGFR, and other receptor tyrosine kinases, etc)
why are kinases ideal targets for the development of small molecule drugs?
they have defined catalytic domains with an ATP binding pocket that is generally amenable to inhibition with small molecule drugs
why are epigenetic modifiers ideal drug targets
due to presence of multiple, varied functional domains with catalytic clefts suitable for drug binding, and they provide a mechanism for both suppressing oncogenes and activating tumor suppressors in certain contexts
how do epigenetic modifiers work to reactivate BRCA1?
a loss of function mutation in BRCA1 can’t be rescued with a small molecule drug but if BRCA1 is epigenetically silenced through promoter methylation, the DNA methyltransferases responsible for that methylation can be targeted therapeutically, reversing the methylation and reactivating BRCA1
what is rational drug design?
a drug is designed to bind to a specific part of your target; requires knowledge of the 3D structure of your target so that a drug can be intentionally built to interact with some part of that target and minimize off target effects
how are drugs designed through high throughput screening?
thousands of chemicals can be screened at once to see if they kill cells or inhibit a specific target you’re interested in. if a chemical is shown to work even moderately well, it can then be modified/altered to improve its binding to the intended target
what studies must be done before a clinical trial?
tested in cell culture, followed by animal models - usually 1 rodent and 1 non-rodent species → can proceed if it appears effective in animal models with no obvious signs of serious side effects
when picking a drug target, what is the biased approach?
relies on pre-existing data that suggests a protein has an important role in cancer progression
when picking a drug target, what is the unbiased approach?
looks at all (or many) possible targets to determine which one is ideal, without consideration of pre-existing data; increased in popularity due to use of CRISPR which allows you to simultaneously look at hundreds of genes and identify genetic vulnerabilities in cells
what are phase III clinical trials?
same overall design as phase II except even more patients enrolled → want to determine if the efficacy seen in phase II is statistically significant. for drug to get FDA approval, must outperform the current standard of care (even if it works the trial will fail if it doesnt outperform)
what is the goal of phase I clinical trials?
to determine if the drug is safe (not as concerned about effectiveness) - no control group because everyone is getting the drug
in a phase I clinical trial, do you care if the drug works in the sense of curing the disease?
no - you dont have enough people in the trial to determine that anyways from a statistical standpoint, but you do want to make sure the drug works in the sense that its actually getting into the cancer cells and inhibiting the target (pharmodynamic and pharmokinetic studies important aspects of phase I clinical trial)
how big are phase I clinical trials?
small, 20-100 individuals
how are phase I clinical trials done?
dose escalation studies where patients get gradually increasing amounts of drug to determine the max tolerated dose
how does vinblastine and other microtubule destabilizing drugs work?
binds to free tubulin and slows the rate of tubulin addition onto growing microtubules - if the rate of tubulin addition is slowed then the tubulin dimers at the free end of the microtubule will hydrolyze their GTP before the next dimers are added which leaves you with GDP-tubulin at the end → these GDP containing dimers associate less tightly, resulting in depolymerization
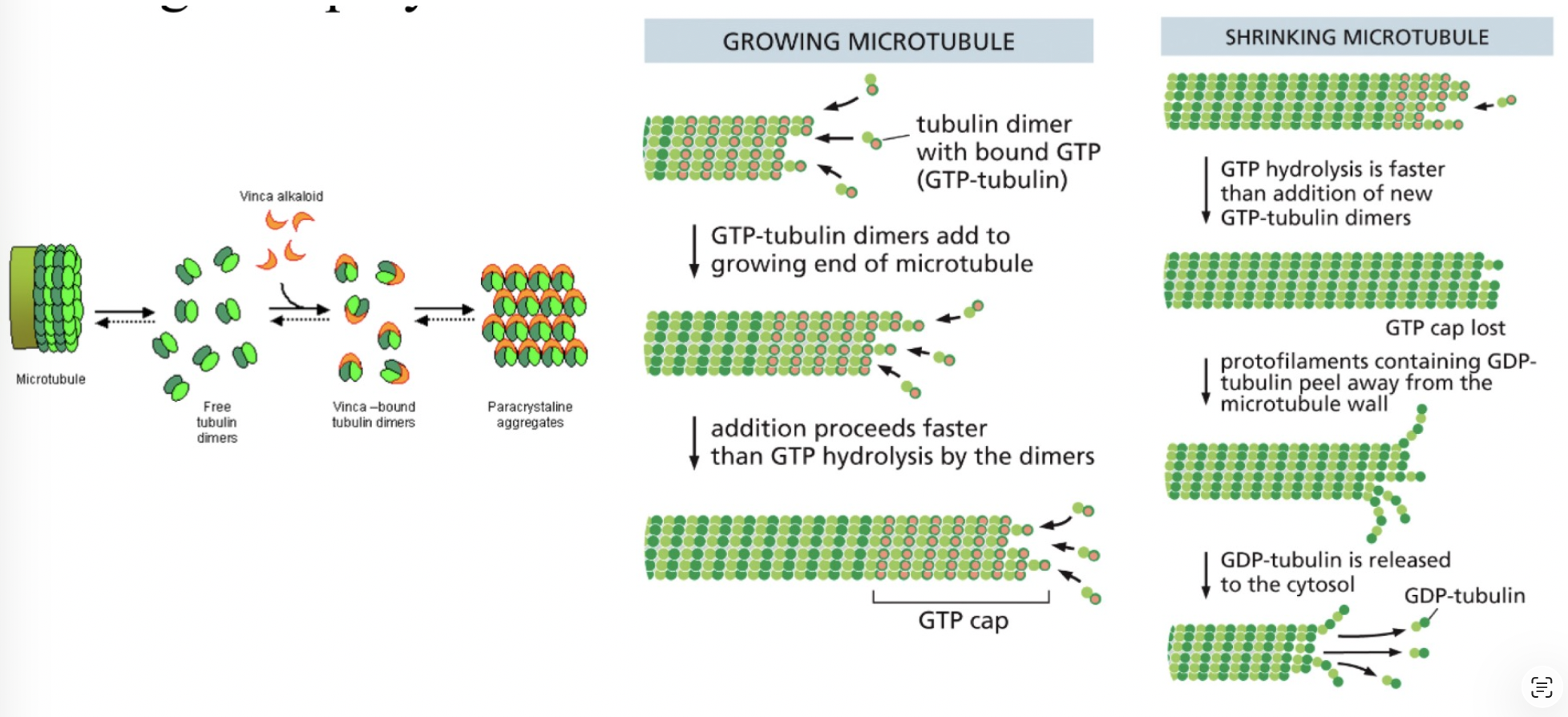
what is the role of free tubulin dimers in microtubule growth?
free tubulin dimers contain GTP that get hydrolyzed to GDP when the dimer is added to a growing microtubule. when microtubule growth proceeds rapidly, tubulin dimers are added to the end of the microtubule faster than the GTP they carry is hydrolyzed. this results in the end of a rapidly growing microtubule being composed entirely of GTP-tubulin dimers, which form a “GTP cap”. dimers with GTP bind more tightly to their neighbors in the microtubule than dimers that have GDP, and they pack together more efficiently, so the microtubule will continue to grow. but, the assembly also promotes hydrolysis of the GTP bound to the subunits, and this happens because the incoming subunit functions as a GAP for the subunit by completing its nucleotide-active site.
how big are phase II clinical trials?
200-300 individuals
what is the overall goal of phase II clinical trials?
now starting to look at drug efficacy - can it actually improve survival?
what is a placebo control group and in what phase is it initially utilized?
placebo should mimic the drug in how its administered as well as its appearance and frequency of administration. first utilized in phase II clinical trials
what is the purpose of the placebo control group?
helps determine if any improvement in patient outcomes are due to the drug, or some other aspect of the clinical trial - especially important since patients in clinical trials typically do better, on average, than patients not in a clinical trial due to increased monitoring of the patients
is the placebo control group for deadly diseases like cancer just a sugar pill?
no - it is unethical to withold treatment and only provide patients with a placebo. therefore the placebo group is current standard care + the placebo, while the treatment group is the current standard of care + the drug being tested
for example: if patients normally receive radiation + cisplatin to treat a certain type of cancer, then in the clinical trial your control group would be radiation + cisplatin + placebo, and your treatment group would be radiation + cisplatin + experimental drug
what are some important aspects of clinical trials?
avoiding bias (selection and experimental): selection bias is when the control and treatment groups differ in some important way, and experimental bias is when researchers or patients behave differently because they know who is getting what treatment
randomization: dont want all of the healthiest patients in the treatment group
allocation concealment: dont want patients to know what group they end up in (control or treatment)
blinding: preventing experimenters who administer treatment from knowing whats being administered
what is a phase IV clinical trial?
after market observation after the drug is approved to monitor for any long term outcomes or rare side-effects not identified in earlier trials
what is chromic myelogenous leukemia (CML)?
cancer of the blood-forming stem cells in the bone marrow that results in unregulated growth of myeloid cells in the bone marrow, which then leads to an accumulation of myeloblasts in the blood; disease often begins in a relatively slow-growing chronic phase, but if untreated it can progress to an accelerated phase and then a more aggressive blast crisis
what occurs in the chronic phase of CML?
CML cells are still able to differentiate fairly normally, so the blood and bone marrow contain increased numbers of more mature myeloid cells, especially granulocytes and their precursors. since cells are still maturing to a large extent, the disease often progresses more slowly at this stage, and some patients may initially have only mild symptoms or be diagnosed from an abnormal blood count. even though there are too many cells, they are not completely blocked in an immature state
what occurs in the blast crisis in CML?
disease has evolved into a much more aggressive phase. at this point, additional mutations and genomic changes have usually occurred, and the leukemic cells lose the ability to differentiate properly. as a result, large numbers of very immature cells called blasts build up in the bone marrow and blood. these blasts crowd out normal blood cell production, which can lead to anemia, infection risk, and bleeding problems
what is the philadelphia chromosome?
caused by reciprocal translocation between chromosomes 9 and 22 → results in formation of fusion gene between Bcr and Abl, which become one continuous gene → Bcr-Abl fusion protein is highly active, oncogenic kinase that phosphorylates and activates numerous oncogene pathways including Ras/MAPK, PI3K/AKT, JAK/STAT, and Myc
how was gleevec designed?
using rational drug design to bind to the ATP binding pocket in the kinase domain, locking Bcr-Abl in an inactive conformation and preventing it from phosphorylating target proteins
how much did gleevec improve survival in CML patients?
from roughly 8% before the drug to over 90% with the drug
how do patients develop resistance to gleevec?
gene amplification where the cells produce more Bcr-Abl than the drug can handle
point mutations in the Bcr-Abl gene that impairs drug binding (additional drugs have been developed that inhibit the mutant Bcr-Abl forms of the protein however)
what does the activation of EGFR lead to?
signaling through Ras/MAPK, JAK/STAT, Rho/Rac, and Src
what is the EGFR family of receptors?
family of receptor tyrosine kinases that is also referred to as the Erb-B family, for the erythroblastosis tumor virus, which encodes an aberrant form of the human EGF receptor (ErbB). also referred to as the HER family for Human EGF Receptors
what two forms are there of EGFR?
membrane-bound form
soluble forms that lack the transmembrane and intracellular domains - can be generated by either proteolytic cleavage of the membrane-associated receptors or by alternative splicing
what can EGFR form?
homo- or heterodimers with family members, as well as with other RTKs like PDGF, HGF, and IGF-1
what are the three main mechanisms that cells can develop resistance to EGFR inhibitors?
mutations in EGFR
activation of alternate survival pathways
cell state changes
how can the therapeutic index of future cancer drugs be improved?
target-driven therapeutic index
context-driven therapeutic index
what is target-driven therapeutic index?
target is present in cancer cells but not normal cells - challenging because it requires targeting mutant proteins which dont always have a significant enough structural change for a drug to differentiate between mutant and WT
what is context-driven therapeutic index?
changes in tumor create conditions that make the target essential for cancer cells but not normal cells. can be due to changes that are intrinsic to the cancer cells (ex: through epigenetic changes or mutations to other genes) or due to extrinsic factors such as microenvironmental changes
what is PARP’s role in DNA replication?
PARP interacts with and stimulates activity of multiple proteins that are involved in DNA replication, but it's particularly important for when things go wrong during replication. PARP mainly helps with replication stress caused by single-strand breaks, unligated Okazaki fragments, and stalled replication forks. Broadly, it does this by PARylating either itself or nearby proteins, and that PARylation acts as a signal to recruit DNA repair proteins.
what is PARP’s role in the DNA damage response?
PARP senses DNA damage and binds to it → activates PARP → PARylates target proteins, including itself. this recruits and activates several repair proteins involved in single- and double- strand break repair, including non-homologous end joining
homologous recombination repair used in double-strand breaks